An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
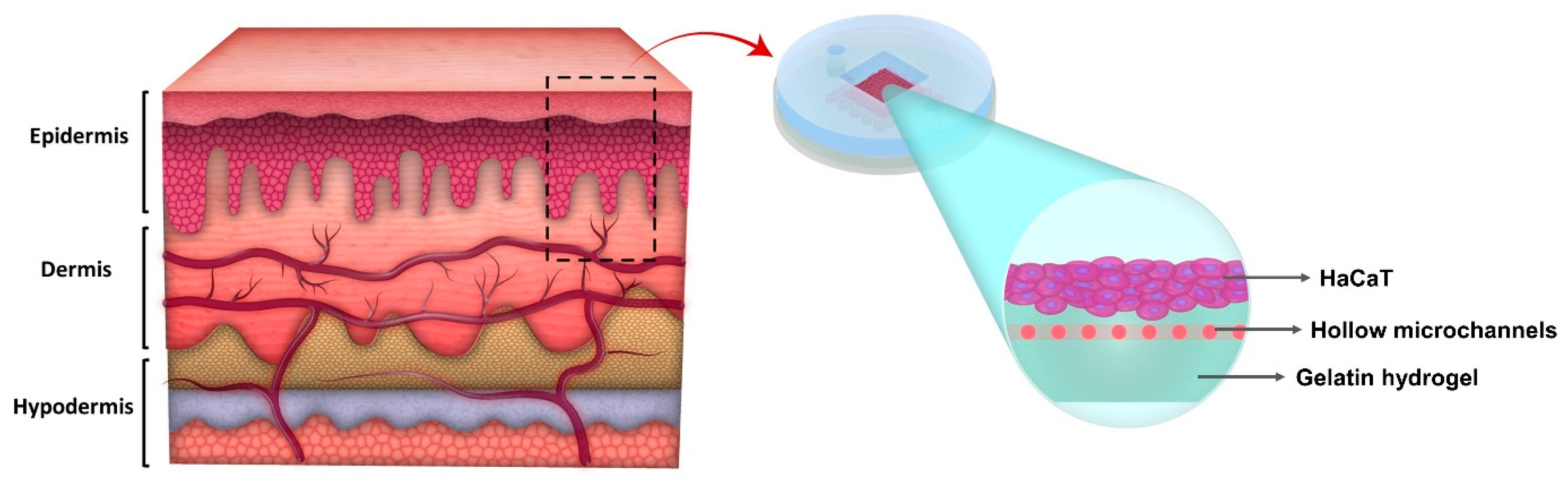
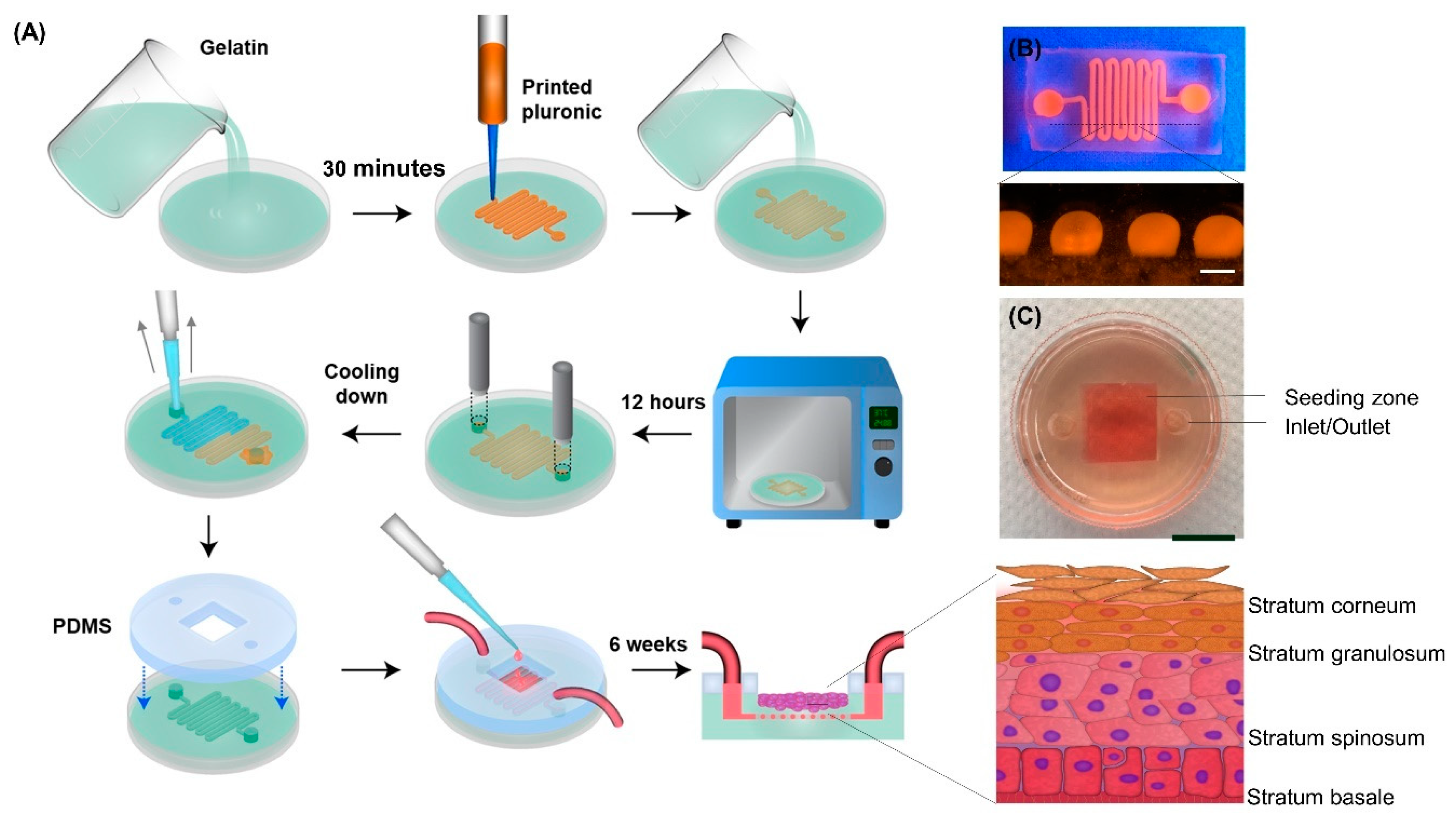
2.1. Model Development
2.2. Gelatin Hydrogel Characterization
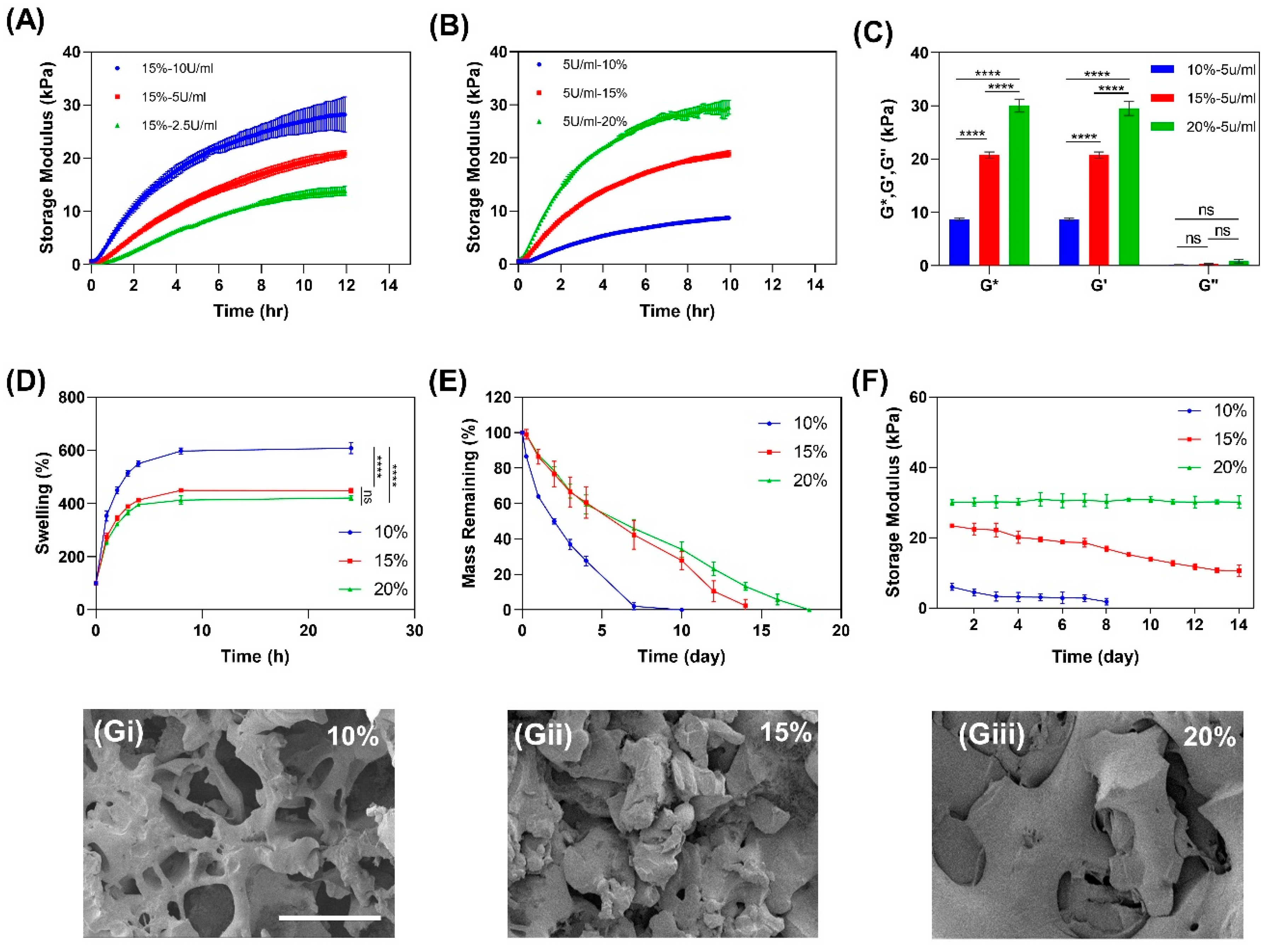
2.2.1. Mechanical Properties of Gelatin
2.2.2. Swelling Ratio
2.2.3. In Vitro Enzymatic Degradation
2.2.4. Mechanical Stability of Gelatin Hydrogel in Culture
2.2.5. Scanning Electron Microscopy of Gelatin Hydrogel
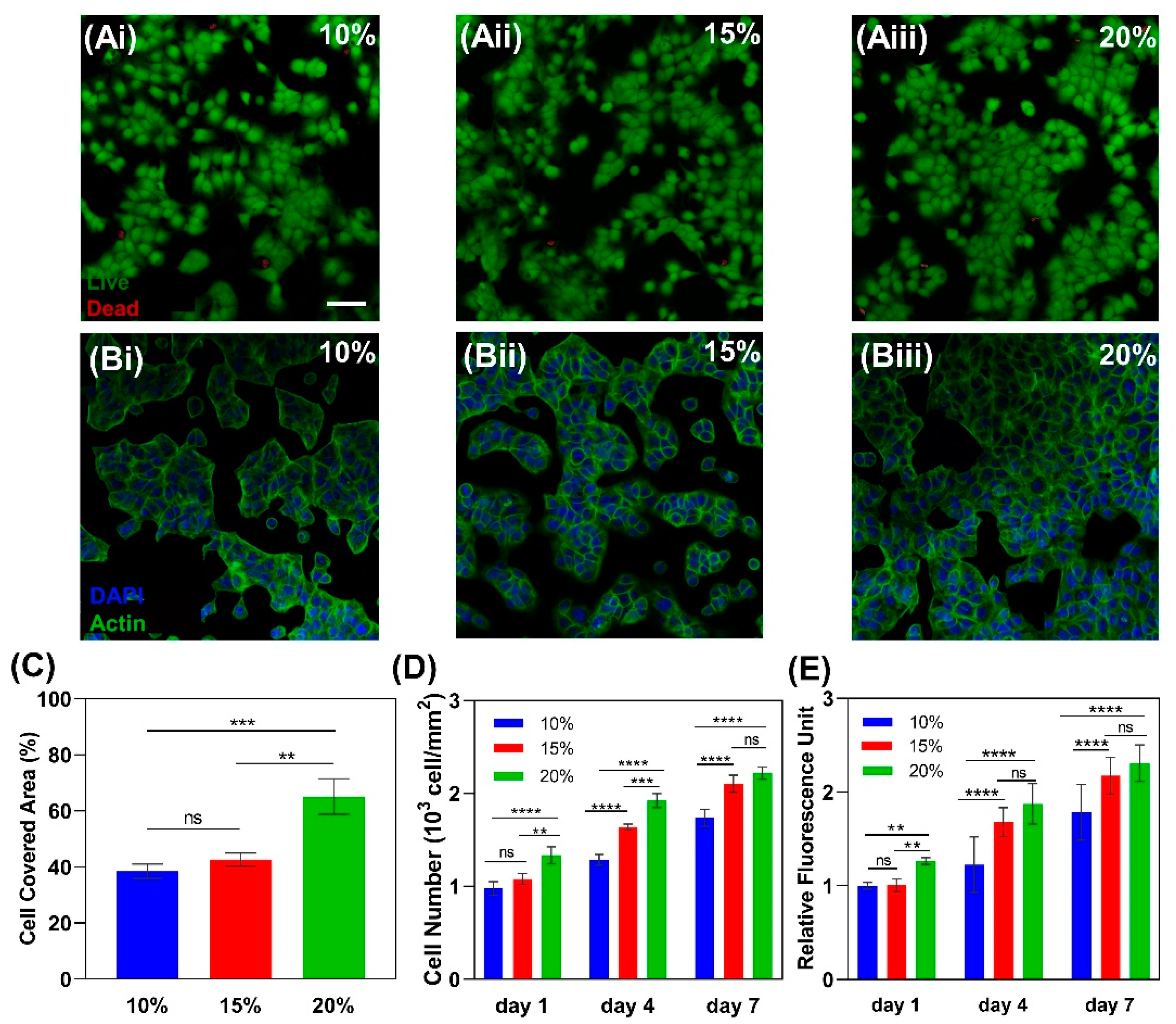
2.2.6. Cytocompatibility and Cell Attachment to Gelatin
2.3. Evaluating the Model Function
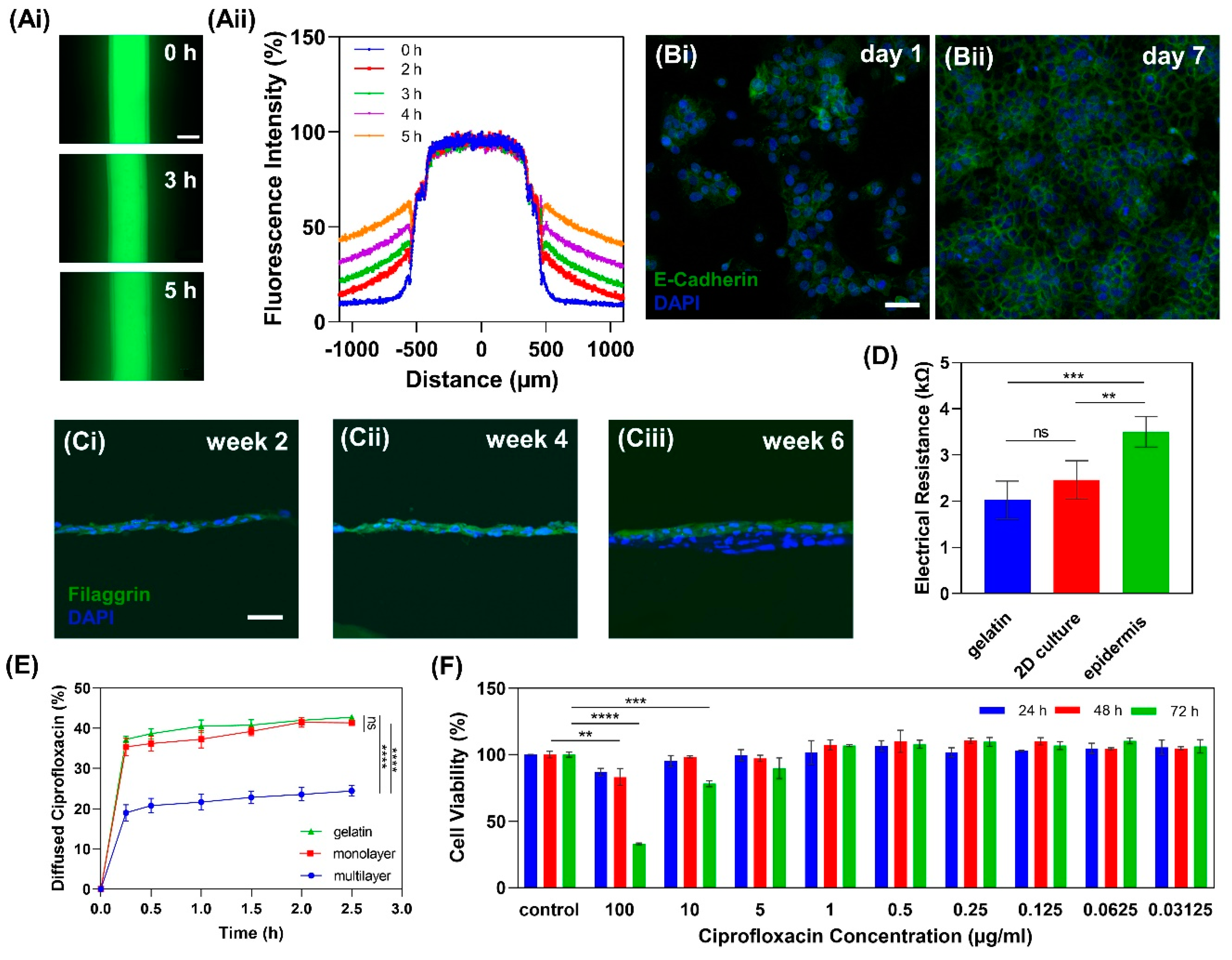
2.3.1. Gelatin Hydrogel Permeability
2.3.2. Cell Tight Junction Analysis
2.3.3. Multilayer Epidermis Formation
2.3.4. In Vitro Epidermis Barrier Function
2.3.5. Drug Cytotoxicity Test
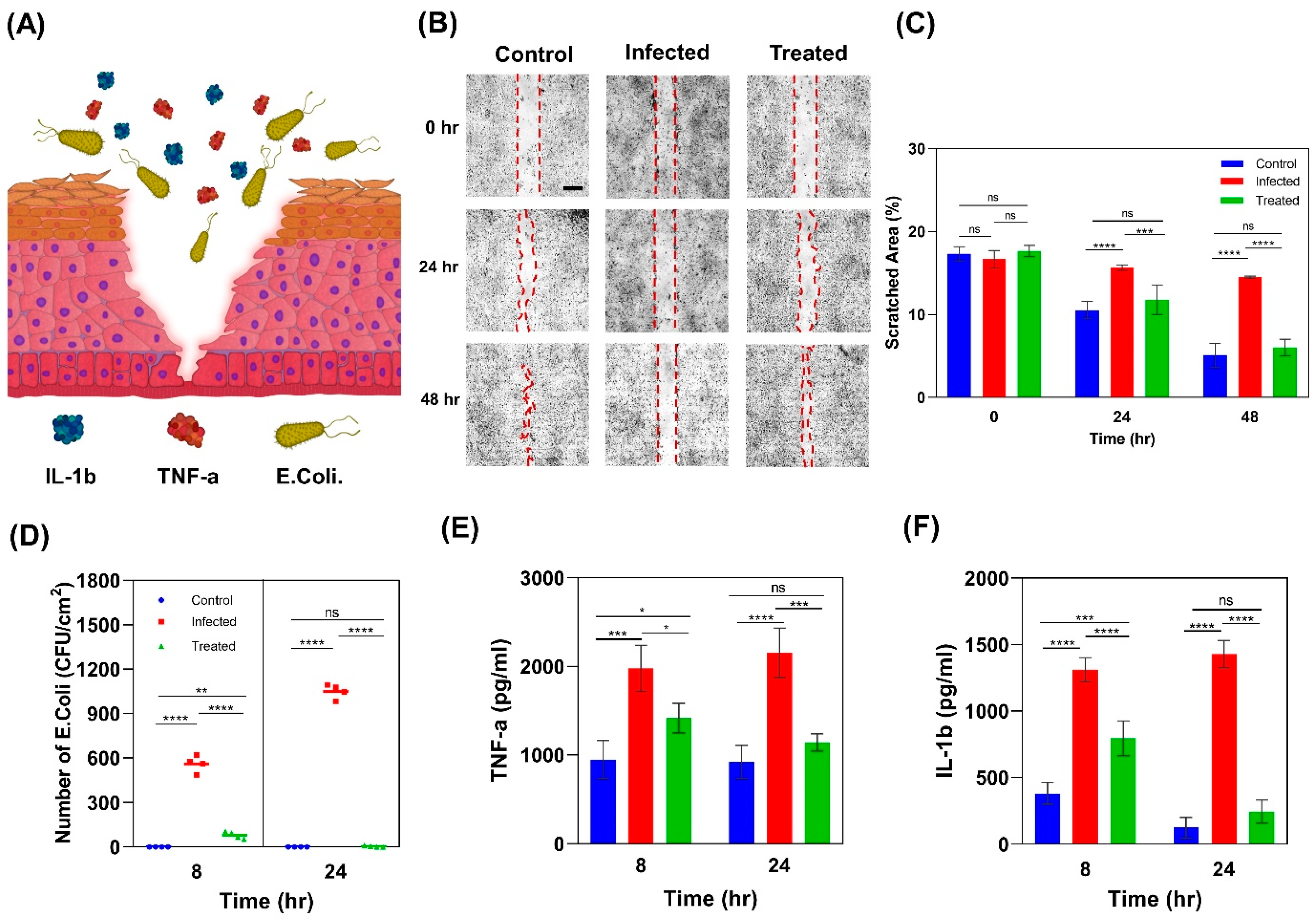
2.4. Wound Infection Modeling
2.4.1. Scratch Wound Healing Assay
2.4.2. Colony Forming Unit Counting
2.4.3. Pro-Inflammatory Response
3. Experimental Section
3.1. Preparation of Gelatin Hydrogel
3.2. Mechanical Properties Measurement
3.3. Swelling Ratio
3.4. In Vitro Enzymatic Degradation
3.5. Mechanical Stability of Gelatin Hydrogel in Culture
3.6. Scanning Electron Microscopy
3.7. Cell Attachment and Cell Number
3.8. Cell Morphology
3.9. Cell Proliferation
3.10. Model Development
3.11. Gelatin Hydrogel Permeability
3.12. Cell Tight Junction Analysis
3.13. In Vitro Epidermis Model Development
3.14. Protein Expression of Developed Epidermis Model
3.15. In Vitro Epidermis Electrical Resistance
3.16. In Vitro Epidermis Drug Permeability
3.17. Drug Cytotoxicity Test
3.18. Bacterial Study
3.19. Scratch Wound Healing Assay
3.20. Pro-Inflammatory Cytokine Analysis
3.21. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathes, S.H.; Ruffner, H.; Graf-hausner, U. The use of skin models in drug development. Adv. Drug Deliv. Rev. 2014, 69, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.F.; Sousa, A.; Barrias, C.C.; Bayat, A.; Granja, P.; Bartolo, P. Advances in bioprinted cell-laden hydrogels for skin tissue engineering. Biomanufacturing Rev. 2017, 2, 1. [Google Scholar] [CrossRef]
- Wufuer, M.; Lee, G.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016, 6, 37471. [Google Scholar] [CrossRef] [PubMed]
- Planz, V.; Lehr, C.; Windbergs, M. In vitro models for evaluating safety and ef fi cacy of novel technologies for skin drug delivery. J. Control. Release 2016, 242, 89–104. [Google Scholar] [CrossRef]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef]
- Kugelberg, E.; Norstro, T.; Petersen, T.K.; Duvold, T.; Andersson, D.I.; Hughes, D. Establishment of a Superficial Skin Infection Model in Mice by Using Staphylococcus aureus and Streptococcus pyogenes. Antimicrob. Agents Chemother. 2005, 49, 3435–3441. [Google Scholar] [CrossRef]
- Schaudinn, C.; Dittmann, C.; Jurisch, J.; Laue, M.; Günday-Türeli, N.; Blume-Peytavi, U.; Vogt, A.; Rancan, F. Development, standardization and testing of a bacterial wound infection model based on ex vivo human skin. PLoS ONE 2017, 12, e0186946. [Google Scholar] [CrossRef]
- Maboni, G.; Davenport, R.; Sessford, K.; Baiker, K.; Jensen, T.K.; Blanchard, A.; Wattegedera, S.; Entrican, G.; Totemeyer, S. A Novel 3D Skin Explant Model to Study Anaerobic Bacterial Infection. Front. Microbiol. 2017, 7, 404. [Google Scholar] [CrossRef]
- Andrade, T.A.M.; Aguiar, A.; Guedes, F.; Leite, M.; Caetano, G.; Coelho, E.B.; Das, P.; Frade, M. Ex vivo Model of Human Skin (hOSEC) as Alternative to Animal use for Cosmetic Tests. Procedia Eng. 2015, 110, 67–73. [Google Scholar] [CrossRef]
- Corzo-León, R.E.; Munro, C.A.; Maccallum, D.M. An ex vivo Human Skin Model to Study Superficial Fungal Infections. Front. Microbiol. 2019, 10, 1172. [Google Scholar] [CrossRef]
- De Breij, A.; Haisma, E.M.; Rietveld, M.; El Ghalbzouri, A.; Van Den Broek, P.J.; Dijkshoorn, L.; Nibbering, P.H. Three-dimensional human skin equivalent as a tool to study Acinetobacter baumannii colonization. Antimicrob. Agents Chemother. 2012, 56, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Randall, M.J.; Jüngel, A.; Rimann, M.; Wuertz-Kozak, K. Advances in the biofabrication of 3D skin in vitro: Healthy and pathological models. Front. Bioeng. Biotechnol. 2018, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Boelsma, E.; Verhoeven, M.C.; Ponec, M. Reconstruction of a Human Skin Equivalent Using a Spontaneously Transformed Keratinocyte Cell Line (HaCaT). J. Investig. Dermatol. 1999, 112, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Alberti, M.; Dancik, Y.; Wu, B.; Wu, R.; Feng, Z.; Ramasamy, S.; Bigliardi, P.L.; Bigliardi-qi, M.; Wang, Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 2018, 21, 326–340. [Google Scholar] [CrossRef]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Mohammadi, M.H.; Heidary Araghi, B.; Beydaghi, V.; Geraili, A.; Moradi, F.; Jafari, P.; Janmaleki, M.; Valente, K.P.; Akbari, M.; Sanati-Nezhad, A. Skin Diseases Modeling using Combined Tissue Engineering and Microfluidic Technologies. Adv. Healthc. Mater. 2016, 5, 2459–2480. [Google Scholar] [CrossRef]
- Pedde, R.D.; Mirani, B.; Navaei, A.; Styan, T.; Wong, S.; Mehrali, M.; Thakur, A.; Mohtaram, N.K.; Bayati, A.; Dolatshahi-Pirouz, A.; et al. Emerging Biofabrication Strategies for Engineering Complex Tissue Constructs. Adv. Mater. 2017, 29, 1606061. [Google Scholar] [CrossRef]
- Roger, M.; Fullard, N.; Costello, L.; Bradbury, S.; Markiewicz, E.; O’Reilly, S.; Darling, N.; Ritchie, P.; Määttä, A.; Karakesisoglou, I.; et al. Bioengineering the microanatomy of human skin. J. Anat. 2019, 234, 438–455. [Google Scholar] [CrossRef]
- Chaudhari, A.A.; Joshi, S.; Vig, K.; Sahu, R.; Dixit, S.; Baganizi, R.; Dennis, V.A.; Singh, S.R.; Pillai, S. A three-dimensional human skin model to evaluate the inhibition of Staphylococcus aureus by antimicrobial peptide-functionalized silver carbon nanotubes. J. Biomater. Appl. 2019, 33, 924–934. [Google Scholar] [CrossRef]
- Kim, B.S.; Gao, G.; Kim, J.Y.; Cho, D.-W. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Adv. Healthc. Mater. 2019, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Qu, X.; Zhu, J.; Ma, X.; Patel, S.; Liu, J.; Wang, P.; Lai, C.S.E.; Gou, M.; Xu, Y.; et al. Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomaterials 2017, 124, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Sheikholeslam, M.; Amini-Nik, S.; E E Wright, M.; Jeschke, M.G. Biomaterials for Skin Substitutes. Adv. Health Mater. 2017, 7, 1700897. [Google Scholar] [CrossRef] [PubMed]
- Bataillon, M.; Lelièvre, D.; Chapuis, A.; Thillou, F.; Autourde, J.B.; Durand, S.; Boyera, N.; Rigaudeau, A.S.; Besné, I.; Pellevoisin, C. Characterization of a new reconstructed full thickness skin model, t-skinTM, and its application for investigations of anti-aging compounds. Int. J. Mol. Sci. 2019, 20, 2240. [Google Scholar] [CrossRef] [PubMed]
- Mieremet, A.; Rietveld, M.; Van Dijk, R.; Bouwstra, J.; El Ghalbzouri, A.; Rietveld, M.M.; Van Dijk, M.R. Recapitulation of Native Dermal Tissue in a Full-Thickness Human Skin Model Using Human Collagens. Tissue Eng. Part A 2018, 24, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.E.L.; Tamamoto, K.A.; Nguyen, H.; Abbott, R.; Cairns, D.; Kaplan, D.L. 3D biomaterial matrix to support long term, full thickness, immuno-competent human skin equivalents with nervous system components. Biomater. 2019, 198, 194–203. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, R.; He, X.; Dai, H.; Betts, R.J.; Marionnet, C.; Bernerd, F.; Planel, E.; Wang, X.; Nocairi, H.; et al. Validation of a predictive method for sunscreen formula evaluation using gene expression analysis in a Chinese reconstructed full-thickness skin model. Int. J. Cosmet. Sci. 2019, 41, 147–155. [Google Scholar] [CrossRef]
- Zhao, X.; Lang, Q.; Yildirimer, L.; Lin, Z.Y.; Cui, W.; Annabi, N.; Ng, K.W.; Dokmeci, M.R.; Ghaemmaghami, A.M.; Khademhosseini, A. Photocrosslinkable Gelatin Hydrogel for Epidermal Tissue Engineering. Adv. Healthc. Mater. 2016, 5, 108–118. [Google Scholar] [CrossRef]
- Liu, Y.; Chan-Park, M.B. A biomimetic hydrogel based on methacrylated dextran-graft-lysine and gelatin for 3D smooth muscle cell culture. Biomaterials 2010, 31, 1158–1170. [Google Scholar] [CrossRef]
- Steen, P.E.V.D.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and Molecular Biology of Gelatinase B or Matrix Metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Boil. 2002, 37, 375–536. [Google Scholar] [CrossRef]
- Gläser, R.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schröder, J.M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2005, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Fazii, P.; Di Giulio, M.; Cellini, L. Bacterial isolates from infected wounds and their antibiotic susceptibility pattern: Some remarks about wound infection. Int. Wound J. 2015, 12, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Petkovšek, Ž.; Eleršič, K.; Gubina, M.; Žgur-Bertok, D.; Erjavec, M.S. Virulence potential of Escherichia coli isolates from skin and soft tissue infections. J. Clin. Microbiol. 2009, 47, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Nagoba, B.S.; Wadher, B.J.; Rao, A.K.; Kore, G.D.; Gomashe, A.V.; Ingle, A.B. A simple and effective approach for the treatment of chronic wound infections caused by multiple antibiotic resistant Escherichia coli. J. Hosp. Infect. 2008, 69, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Hebeish, A.; El-Rafie, M.H.; EL-Sheikh, M.A.; Seleem, A.A.; El-Naggar, M.E. Antimicrobial wound dressing and anti-inflammatory efficacy of silver nanoparticles. Int. J. Biol. Macromol. 2014, 65, 509–515. [Google Scholar] [CrossRef]
- Maneerung, T.; Tokura, S.; Rujiravanit, R. Impregnation of silver nanoparticles into bacterial cellulose for antimicrobial wound dressing. Carbohydr. Polym. 2008, 72, 43–51. [Google Scholar] [CrossRef]
- Jawhara, S.; Mordon, S. In Vivo Imaging of Bioluminescent Escherichia coli in a Cutaneous Wound Infection Model for Evaluation of an Antibiotic Therapy. Antimicrob. Agents Chemother. 2004, 48, 3436–3441. [Google Scholar] [CrossRef]
- Lei, D.; Yang, Y.; Liu, Z.; Yang, B.; Gong, W.; Chen, S.; Wang, S.; Sun, L.; Song, B.; Xuan, H.; et al. 3D printing of biomimetic vasculature for tissue regeneration. Mater. Horizons 2019, 6, 1197–1206. [Google Scholar] [CrossRef]
- Flaten, G.E.; Rukavina, Z.; Engesland, A.; Filipović-Grčić, J.; Vanić, Ž.; Škalko-Basnet, N. In vitro skin models as a tool in optimization of drug formulation. Eur. J. Pharm. Sci. 2015, 75, 10–24. [Google Scholar] [CrossRef]
- Paulsen, S.J.; Miller, J.S. Tissue vascularization through 3D printing: Will technology bring us flow? Dev. Dyn. 2015, 244, 629–640. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Mandrycky, C.; Wang, Z.; Kim, K.; Kim, D.H. 3D bioprinting for engineering complex tissues. Biotechnol. Adv. 2016, 34, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M. Tissue Engineering for Artificial Organs: Regenerative Medicine, Smart Diagnostics and Personalized Medicine; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; Tissue Engineering of the Pancreas; pp. 553–573. [Google Scholar]
- Yang, G.; Xiao, Z.; Ren, X.; Long, H.; Qian, H. Enzymatically crosslinked gelatin hydrogel promotes the proliferation of adipose tissue-derived stromal cells. PeerJ 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Paguirigan, A.L.; Beebe, D.J. Protocol for the fabrication of enzymatically crosslinked gelatin microchannels for microfluidic cell culture. Nat. Protoc. 2007, 2, 1782–1788. [Google Scholar] [CrossRef]
- Suntornnond, R.; Tan, E.Y.S.; An, J.; Chua, C.K. A highly printable and biocompatible hydrogel composite for direct printing of soft and perfusable vasculature-like structures. Sci. Rep. 2017, 7, 16902. [Google Scholar] [CrossRef]
- Kolesky, D.B.; Homan, K.A.; Skylar-Scott, M.A.; Lewis, J.A. Three-dimensional bioprinting of thick vascularized tissues. Proc. Natl. Acad. Sci. 2016, 113, 3179–3184. [Google Scholar] [CrossRef]
- Kolesky, D.B.; Truby, R.L.; Gladman, A.S.; Busbee, T.A.; Homan, K.A.; Lewis, J.A. 3D Bioprinting of Vascularized, Heterogeneous Cell-Laden Tissue Constructs. Adv. Mater. 2014, 26, 3124–3130. [Google Scholar] [CrossRef]
- Ng, W.L.; Yeong, W.Y.; Naing, M.W. Cellular Approaches to Tissue-Engineering of Skin: A Review. J. Tissue Sci. Eng. 2015, 6, 150. [Google Scholar]
- Krishna, S.; Miller, L.S. Host–pathogen interactions between the skin and Staphylococcus aureus. Curr. Opin. Microbiol. 2012, 15, 28–35. [Google Scholar] [CrossRef]
- Yung, C.W.; Wu, L.Q.; Tullman, J.A.; Payne, G.F.; Bentley, W.E.; Barbari, T.A. Transglutaminase crosslinked gelatin as a tissue engineering scaffold. J. Biomed. Mater. Res. Part A 2007, 83, 1039–1046. [Google Scholar] [CrossRef]
- Kearney, S.P.; Khan, A.; Dai, Z.; Royston, T.J. Dynamic viscoelastic models of human skin using optical elastography. Phys. Med. Boil. 2015, 60, 6975–6990. [Google Scholar] [CrossRef] [PubMed]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Biomaterials Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef] [PubMed]
- Sundaramurthi, D.; Krishnan, U.M.; Sethuraman, S. Electrospun nanofibers as scaffolds for skin tissue engineering. Polym. Rev. 2014, 54, 348–376. [Google Scholar] [CrossRef]
- Cui, W.; Zhu, X.; Yang, Y.; Li, X.; Jin, Y. Evaluation of electrospun fibrous scaffolds of poly(dl-lactide) and poly(ethylene glycol) for skin tissue engineering. Mater. Sci. Eng. C 2009, 29, 1869–1876. [Google Scholar] [CrossRef]
- Schoop, V.M.; Fusenig, N.E.; Mirancea, N. Epidermal Organization and Differentiation of HaCaT Keratinocytes in Organotypic Coculture with Human Dermal Fibroblasts. J. Investig. Dermatol. 1999, 112, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; Sangiovanni, E.; Maggio, R.; Mattozzi, C.; Zava, S.; Corbett, Y.; Fumagalli, M.; Carlino, C.; Corsetto, P.A.; Scaccabarozzi, D.; et al. HaCaT Cells as a Reliable In Vitro Differentiation Model to Dissect the Inflammatory/Repair Response of Human Keratinocytes. Mediators Inflamm. 2017, 2017, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, C.; Strandman, S.; Hui, E.; Montagnon, E.; Schmitt, C.; Henni, A.H.; Lerouge, S. Validation and application of a nondestructive and contactless method for rheological evaluation of biomaterials. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2016, 105, 2565–2573. [Google Scholar] [CrossRef]
- Hollister, S.J. Porous scaffold design for tissue engineering. Nat. Mater. 2005, 4, 518–524. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.; Luo, X.; Qiu, J.; Tang, C. Substrate stiffness regulates the proliferation, migration, and differentiation of epidermal cells. Burns 2012, 38, 414–420. [Google Scholar] [CrossRef]
- Tunggal, J.A.; Helfrich, I.; Schmitz, A.; Schwarz, H.; Günzel, D.; Fromm, M.; Kemler, R.; Krieg, T.; Niessen, C.M. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005, 24, 1146–1156. [Google Scholar] [CrossRef]
- Izaguirre, M.F.; Larrea, D.; Adur, J.F.; Zamboni, J.D.; Vicente, N.B.; Galetto, C.; Casco, V.H. Role of E-Cadherin in Epithelial Architecture Maintenance. Cell Commun. Adhes. 2010, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, A.; Sutherland, C.; Irvine, A.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.B.; Myiagi, S.; Nogales, C.G.; Campos, M.S.; Lage-Marques, J.L. Time- and concentration-dependent cytotoxicity of antibiotics used in endodontic therapy. J. Appl. Oral Sci. 2010, 18, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Gürbay, A.; Garrel, C.; Osman, M.; Richard, M.J.; Favier, A.; Hincal, F. Cytotoxicity in ciprofloxacin-treated human fibroblast cells and protection by vitamin E. Hum. Exp. Toxicol. 2002, 21, 635–641. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Werner, S.; French, L.E.; Beer, H.D. Interleukin-1, inflammasomes and the skin. Eur. J. Cell Biol. 2010, 89, 638–644. [Google Scholar] [CrossRef]
- Marcatili, A.; Ero, G.C.D.I.; Galdiero, M.; Folgore, A.; Petrillo, G. TNF-a, IL-la, IL-6 and lCAM-l expression in human keratinocytes stimulated in vitro with Escherichia coli heat-shock proteins. Microbiology 1997, 143, 45–53. [Google Scholar] [CrossRef]
- Buommino, E.; De Filippis, A.; Parisi, A.; Nizza, S.; Martano, M.; Iovane, G.; Donnarumma, G.; Tufano, M.A.; De Martino, L. Innate immune response in human keratinocytes infected by a feline isolate of Malassezia pachydermatis. Vet. Microbiol. 2013, 163, 90–96. [Google Scholar] [CrossRef]
- Gupta, S.; Tang, C.; Tran, M.; Kadouri, D.E. Effect of Predatory Bacteria on Human Cell Lines. PLoS ONE 2016, 11, e0161242. [Google Scholar] [CrossRef]
- Holland, D.B.; Bojar, R.A.; Farrar, M.D.; Holland, K.T. Differential innate immune responses of a living skin equivalent model colonized by staphylococcus epidermidis or staphylococcus aureus. FEMS Microbiol. Lett. 2009, 290, 149–155. [Google Scholar] [CrossRef]
- Wang, B.; Ruiz, N.; Pentland, A.; Caparon, M. Keratinocyte proinflammatory responses to adherent and nonadherent group A streptococci. Infect. Immun. 1997, 65, 2119–2126. [Google Scholar] [CrossRef]
- Maas-szabowski, N.; Stärker, A.; Fusenig, N.E. Epidermal tissue regeneration and stromal interaction in HaCaT cells is initiated by TGF- α. J. Cell Sci. 2003, 116, 2937–2948. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahanshahi, M.; Hamdi, D.; Godau, B.; Samiei, E.; Sanchez-Lafuente, C.L.; Neale, K.J.; Hadisi, Z.; Dabiri, S.M.H.; Pagan, E.; Christie, B.R.; et al. An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response. Micromachines 2020, 11, 227. https://doi.org/10.3390/mi11020227
Jahanshahi M, Hamdi D, Godau B, Samiei E, Sanchez-Lafuente CL, Neale KJ, Hadisi Z, Dabiri SMH, Pagan E, Christie BR, et al. An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response. Micromachines. 2020; 11(2):227. https://doi.org/10.3390/mi11020227
Chicago/Turabian StyleJahanshahi, Maryam, David Hamdi, Brent Godau, Ehsan Samiei, Carla Liria Sanchez-Lafuente, Katie J. Neale, Zhina Hadisi, Seyed Mohammad Hossein Dabiri, Erik Pagan, Brian R. Christie, and et al. 2020. "An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response" Micromachines 11, no. 2: 227. https://doi.org/10.3390/mi11020227
APA StyleJahanshahi, M., Hamdi, D., Godau, B., Samiei, E., Sanchez-Lafuente, C. L., Neale, K. J., Hadisi, Z., Dabiri, S. M. H., Pagan, E., Christie, B. R., & Akbari, M. (2020). An Engineered Infected Epidermis Model for In Vitro Study of the Skin’s Pro-Inflammatory Response. Micromachines, 11(2), 227. https://doi.org/10.3390/mi11020227