The Applications of Lattice Light-Sheet Microscopy for Functional Volumetric Imaging of Hippocampal Neurons in a Three-Dimensional Culture System

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Rat Hippocampal Neurons for 3D Culture
2.2. Immunofluorescence Staining
2.3. Setup of Lattice Light-Sheet Microscopy
2.4. Voltage and Calcium Dye Labeling for Functional Imaging
2.5. Setup of Electrical Stimulation of Neurons
2.6. Image Processing and Analysis
3. Results
3.1. Characterization of Hippocampal Neurons in a 3D Culture System
3.2. Lattice Light-Sheet Microscopy (LLSM) for 3D-Cultured Neuron
3.3. Quantification of Voltage Responses at Different Subcellular Areas
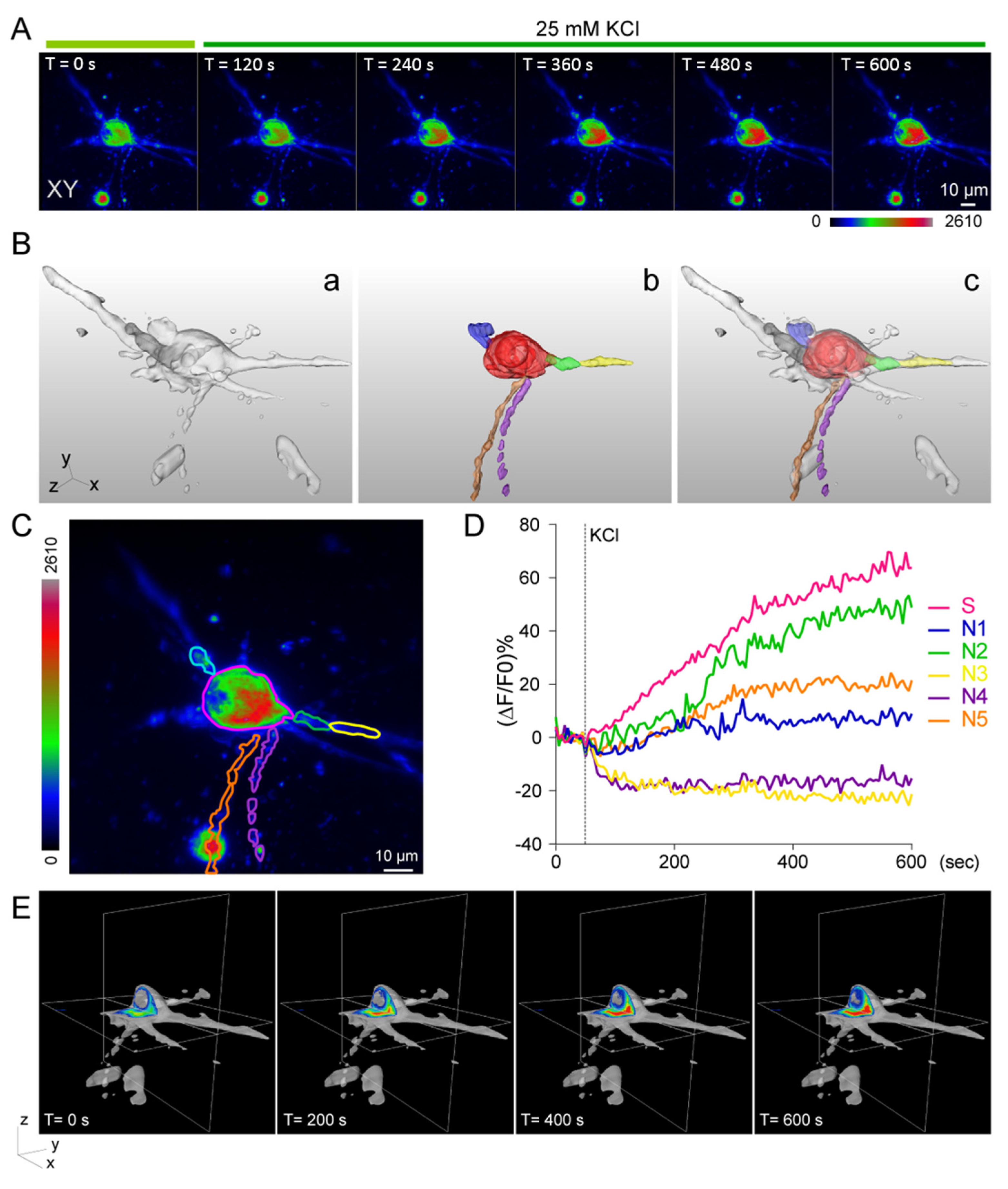
3.4. Calcium Imaging of Neuronal Activity under External Stimulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, M.; Zirath, H.; Ertl, P. Recent advances in microfluidic technologies for cell-to-cell interaction studies. Lab Chip 2018, 18, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.W.; Chueh, D.Y.; Chen, P. Investigation of size-dependent cell adhesion on nanostructured interfaces. J. Nanobiotechnol. 2014, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Chien, F.C.; Kuo, C.W.; Yang, Z.H.; Chueh, D.Y.; Chen, P. Exploring the formation of focal adhesions on patterned surfaces using super-resolution imaging. Small 2011, 7, 2906–2913. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Sarkar, S.; Peng, C.-C.; Kuo, C.W.; Chueh, D.-Y.; Wu, H.-M.; Liu, Y.-H.; Chen, P.; Tung, Y.-C. Study of oxygen tension variation within live tumor spheroids using microfluidic devices and multi-photon laser scanning microscopy. RSC Adv. 2018, 8, 30320–30329. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Kuhlenbeck, H. The Central Nervous System of Vertebrates: A General Survey of Its Comparative Anatomy with an Introduction to the Pertinent Fundamental Biologic and Logical Concepts; Karger Medical and Scientific Publishers: Basel, Switzerland, 1967. [Google Scholar]
- Hsiao, Y.-S.; Lin, C.-C.; Hsieh, H.-J.; Tsai, S.-M.; Kuo, C.-W.; Chu, C.-W.; Chen, P. Manipulating location, polarity, and outgrowth length of neuron-like pheochromocytoma (PC-12) cells on patterned organic electrode arrays. Lab Chip 2011, 11, 3674–3680. [Google Scholar] [CrossRef]
- Lu, C.H.; Hsiao, Y.S.; Kuo, C.W.; Chen, P. Electrically tunable organic bioelectronics for spatial and temporal manipulation of neuron-like pheochromocytoma (PC-12) cells. Biochim. Biophys. Acta. 2013, 1830, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.S.; Liao, Y.H.; Chen, H.L.; Chen, P.; Chen, F.C. Organic Photovoltaics and Bioelectrodes Providing Electrical Stimulation for PC12 Cell Differentiation and Neurite Outgrowth. ACS Appl. Mater. Interfaces 2016, 8, 9275–9284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.-C.; She, J.-W.; Wu, J.-G.; Chen, P.; Hsiao, Y.-S.; Yu, J. Poly(3,4-ethylenedioxythiophene) Polymer Composite Bioelectrodes with Designed Chemical and Topographical Cues to Manipulate the Behavior of PC12 Neuronal Cells. Appl. Mater. Interfaces 2019, 6, 1801576. [Google Scholar] [CrossRef]
- Hiruma, H.; Uemura, T.; Kimura, F. Neuronal Synchronization and Ionic Mechanisms for Propagation of Excitation in the Functional Network of Immortalized GT1-7 Neurons: Optical Imaging with a Voltage-Sensitive Dye. J. Neuroendocrinol. 1997, 9, 835–840. [Google Scholar] [CrossRef]
- Peterka, D.S.; Takahashi, H.; Yuste, R. Imaging voltage in neurons. Neuron 2011, 69, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Han, Z.; Platisa, J.; Wooltorton, J.R.; Cohen, L.B.; Pieribone, V.A. Single action potentials and subthreshold electrical events imaged in neurons with a fluorescent protein voltage probe. Neuron 2012, 75, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Shoham, D.; Glaser, D.E.; Arieli, A.; Kenet, T.; Wijnbergen, C.; Toledo, Y.; Hildesheim, R.; Grinvald, A. Imaging Cortical Dynamics at High Spatial and Temporal Resolution with Novel Blue Voltage-Sensitive Dyes. Neuron 1999, 24, 791–802. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef]
- Ulloa Severino, F.P.; Ban, J.; Song, Q.; Tang, M.; Bianconi, G.; Cheng, G.; Torre, V. The role of dimensionality in neuronal network dynamics. Sci. Rep. 2016, 6, 29640. [Google Scholar] [CrossRef]
- Anderson, H.E.; Fontaine, A.K.; Caldwell, J.H.; Weir, R.F. Imaging of electrical activity in small diameter fibers of the murine peripheral nerve with virally-delivered GCaMP6f. Sci. Rep. 2018, 8, 3219. [Google Scholar] [CrossRef]
- Johenning, F.W.; Theis, A.K.; Pannasch, U.; Ruckl, M.; Rudiger, S.; Schmitz, D. Ryanodine Receptor Activation Induces Long-Term Plasticity of Spine Calcium Dynamics. PLoS Biol. 2015, 13, e1002181. [Google Scholar] [CrossRef] [PubMed]
- Chamberland, S.; Yang, H.H.; Pan, M.M.; Evans, S.W.; Guan, S.; Chavarha, M.; Yang, Y.; Salesse, C.; Wu, H.; Wu, J.C.; et al. Fast two-photon imaging of subcellular voltage dynamics in neuronal tissue with genetically encoded indicators. eLife 2017, 6, e25690. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Schmidt, A.D.; Wittbrodt, J.; Stelzer, E.H. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 2008, 322, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z.; et al. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Pratiwi, F.W.; Wu, F.C.M.; Chen, P.; Chen, B.C. Recent Progress in Light Sheet Microscopy for Biological Applications. Appl. Spectrosc. 2018, 72, 1137–1169. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chen, Y.T.; Wang, J.Y.; Huang, Y.S.; Tai, C.Y. Postsynaptic Y654 dephosphorylation of beta-catenin modulates presynaptic vesicle turnover through increased n-cadherin-mediated transsynaptic signaling. Dev. Neurobiol. 2017, 77, 61–74. [Google Scholar] [CrossRef]
- Fath, T.; Ke, Y.D.; Gunning, P.; Gotz, J.; Ittner, L.M. Primary support cultures of hippocampal and substantia nigra neurons. Nat. Protoc. 2009, 4, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, S.H.; D’Avanzo, C.; Hebisch, M.; Sliwinski, C.; Bylykbashi, E.; Washicosky, K.J.; Klee, J.B.; Brustle, O.; Tanzi, R.E.; et al. A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat. Protoc. 2015, 10, 985–1006. [Google Scholar] [CrossRef] [PubMed]
- Gee, K.R.; Brown, K.A.; Chen, W.N.U.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef]
- Johnson, P.L.; Smith, W.; Baynham, T.C.; Knisley, S.B. Errors Caused by Combination of Di-4 ANEPPS and Fluo3/4 for Simultaneous Measurements of Transmembrane Potentials and Intracellular Calcium. Ann. Biomed. Eng. 1999, 27, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Amira User Guide. Available online: https://www.fei.com/software/amira-user-guide/ (accessed on 11 September 2019).
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Brauninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Nat. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Stem Cell Models of Human Brain Development. Cell Stem Cell 2016, 18, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.R.; Ewald, A.J. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.H.; Tang, W.C.; Liu, Y.T.; Chang, S.W.; Wu, F.C.M.; Chen, C.Y.; Tsai, Y.C.; Yang, S.M.; Kuo, C.W.; Okada, Y.; et al. Lightsheet localization microscopy enables fast, large-scale, and three-dimensional super-resolution imaging. Commun. Biol. 2019, 2, 177. [Google Scholar] [CrossRef] [PubMed]
- Aerov, A.A. Why the water bridge does not collapse. Phys. Rev. E Stat. Nonlin. Soft Matter. Phys. 2011, 84, 036314. [Google Scholar] [CrossRef] [PubMed]
- Fluhler, E.; Burnham, V.G.; Loew, L.M. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry 1985, 24, 5749–5755. [Google Scholar] [CrossRef] [PubMed]
- Hassner, A.; Birnbaum, D.; Loew, L.M. Charge-shift probes of membrane potential. Synthesis. J. Org. Chem. 1984, 49, 2546–2551. [Google Scholar] [CrossRef]
- Peng, H.; Hawrylycz, M.; Roskams, J.; Hill, S.; Spruston, N.; Meijering, E.; Ascoli, G.A. BigNeuron: Large-Scale 3D Neuron Reconstruction from Optical Microscopy Images. Neuron 2015, 87, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Meijering, E.; Ascoli, G.A. From DIADEM to BigNeuron. Neuroinformatics 2015, 13, 259–260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gillette, T.A.; Brown, K.M.; Ascoli, G.A. The DIADEM metric: comparing multiple reconstructions of the same neuron. Neuroinformatics 2011, 9, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Maravall, M.; Mainen, Z.F.; Sabatini, B.L.; Svoboda, K. Estimating Intracellular Calcium Concentrations and Buffering without Wavelength Ratioing. Biophys. J. 2000, 78, 2655–2667. [Google Scholar] [CrossRef]
- Helmchen, F.; Imoto, K.; Sakmann, B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys. J. 1996, 70, 1069–1081. [Google Scholar] [CrossRef]
- Canepari, M.; Djurisic, M.; Zecevic, D. Dendritic signals from rat hippocampal CA1 pyramidal neurons during coincident pre- and post-synaptic activity: a combined voltage- and calcium-imaging study. J. Physiol. 2007, 580, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Deneke, V.E.; Di Talia, S. Chemical waves in cell and developmental biology. J. Cell Biol. 2018, 217, 1193–1204. [Google Scholar] [CrossRef]
- Kim, S.H.; Ryan, T.A. CDK5 serves as a major control point in neurotransmitter release. Neuron 2010, 67, 797–809. [Google Scholar] [CrossRef]
- Wickenden, A.D. Overview of Electrophysiological Techniques. Curr. Protoc. Pharmacol. 2000, 11, 11.1.1–11.1.17. [Google Scholar] [CrossRef]
- Obaid, A.L.; Loew, L.M.; Wuskell, J.P.; Salzberg, B.M. Novel naphthylstyryl-pyridium potentiometric dyes offer advantages for neural network analysis. J. Neurosci. Methods 2004, 134, 179–190. [Google Scholar] [CrossRef]
- Rohr, S.; Salzberg, B.M. Multiple site optical recording of transmembrane voltage (MSORTV) in patterned growth heart cell cultures: assessing electrical behavior, with microsecond resolution, on a cellular and subcellular scale. Biophys. J. 1994, 67, 1301–1315. [Google Scholar] [CrossRef]
- Preuss, S.; Stein, W. Comparison of two voltage-sensitive dyes and their suitability for long-term imaging of neuronal activity. PLoS ONE 2013, 8, e75678. [Google Scholar] [CrossRef] [PubMed]
- Fairless, R.; Beck, A.; Kravchenko, M.; Williams, S.K.; Wissenbach, U.; Diem, R.; Cavalie, A. Membrane potential measurements of isolated neurons using a voltage-sensitive dye. PLoS ONE 2013, 8, e58260. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Liu, Y.-T.; Lu, C.-H.; Lee, P.-Y.; Tsai, Y.-C.; Wu, J.-S.; Chen, P.; Chen, B.-C. The Applications of Lattice Light-Sheet Microscopy for Functional Volumetric Imaging of Hippocampal Neurons in a Three-Dimensional Culture System. Micromachines 2019, 10, 599. https://doi.org/10.3390/mi10090599
Chen C-Y, Liu Y-T, Lu C-H, Lee P-Y, Tsai Y-C, Wu J-S, Chen P, Chen B-C. The Applications of Lattice Light-Sheet Microscopy for Functional Volumetric Imaging of Hippocampal Neurons in a Three-Dimensional Culture System. Micromachines. 2019; 10(9):599. https://doi.org/10.3390/mi10090599
Chicago/Turabian StyleChen, Chin-Yi, Yen-Ting Liu, Chieh-Han Lu, Po-Yi Lee, Yun-Chi Tsai, Jyun-Sian Wu, Peilin Chen, and Bi-Chang Chen. 2019. "The Applications of Lattice Light-Sheet Microscopy for Functional Volumetric Imaging of Hippocampal Neurons in a Three-Dimensional Culture System" Micromachines 10, no. 9: 599. https://doi.org/10.3390/mi10090599
APA StyleChen, C.-Y., Liu, Y.-T., Lu, C.-H., Lee, P.-Y., Tsai, Y.-C., Wu, J.-S., Chen, P., & Chen, B.-C. (2019). The Applications of Lattice Light-Sheet Microscopy for Functional Volumetric Imaging of Hippocampal Neurons in a Three-Dimensional Culture System. Micromachines, 10(9), 599. https://doi.org/10.3390/mi10090599