
Modification of the Mycotoxin Deoxynivalenol Using Microorganisms Isolated from Environmental Samples

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Selection of Microbes in the Presence of High Concentrations of DON
2.2. Isolation of Individual DON Modifying Microbes
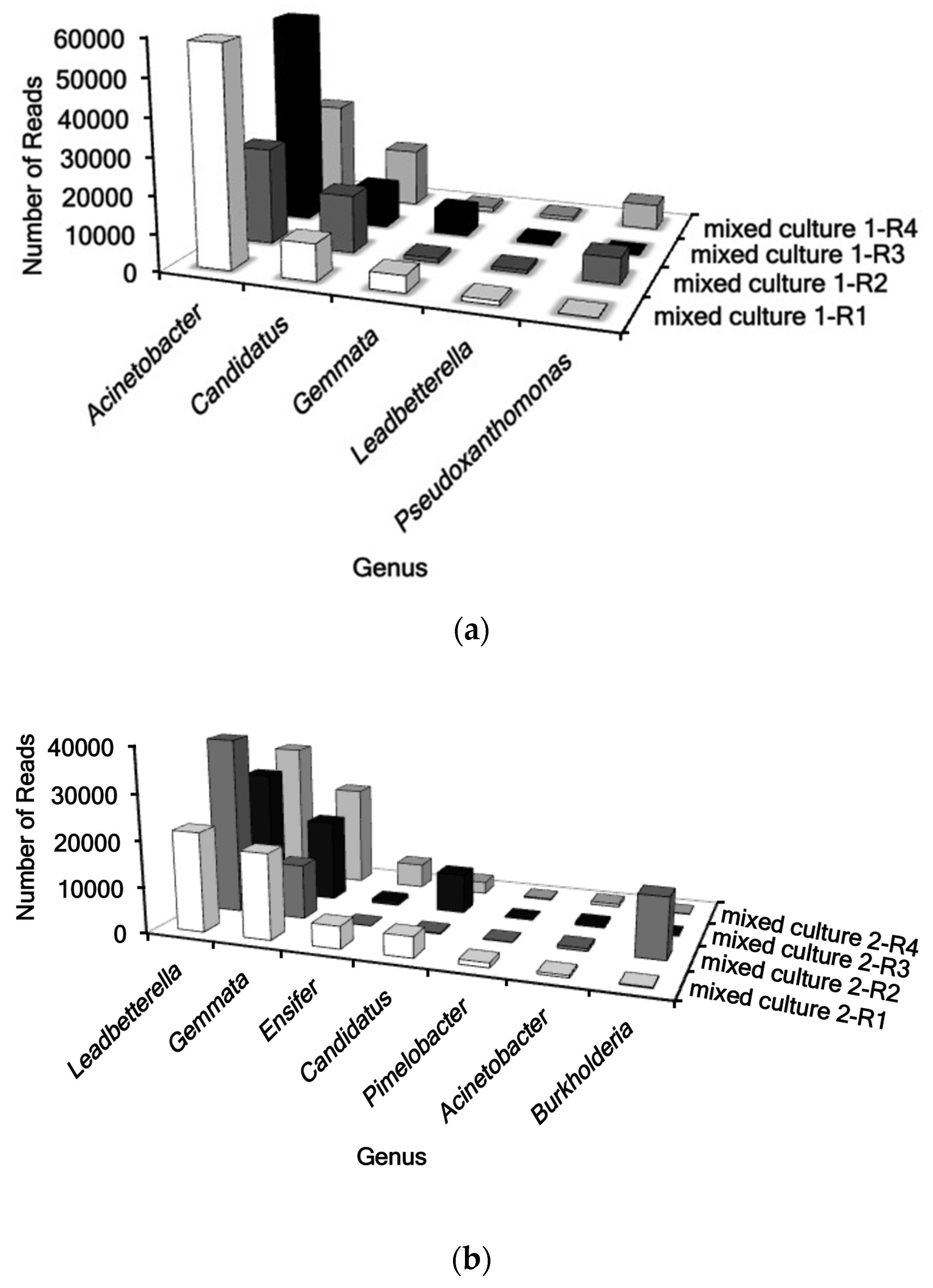
2.3. Identification of Mixed Cultures Using 16S Ribosomal Sequencing
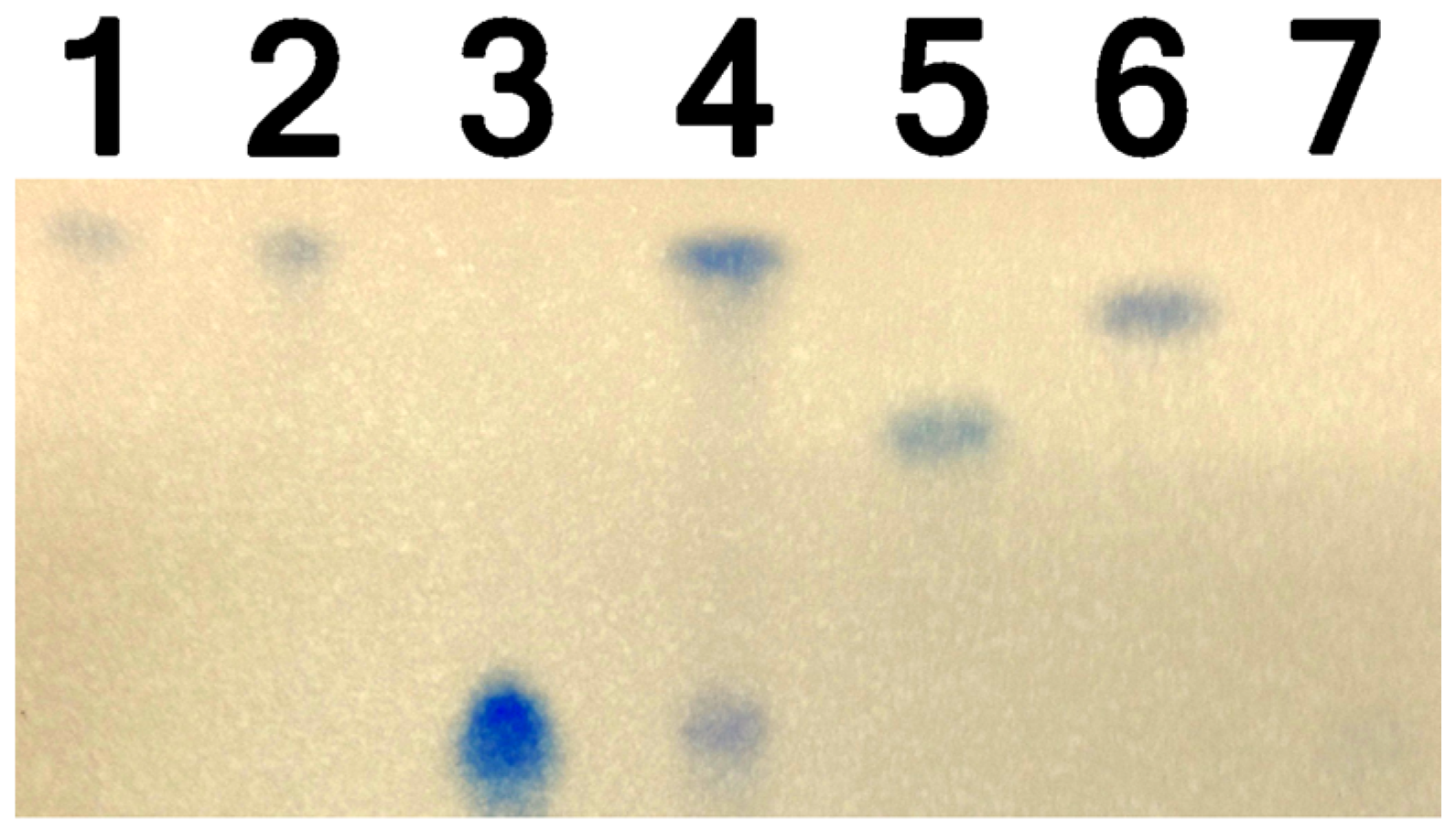
2.4. Thin Layer Chromatography to Identify DON Derivatives
2.5. Nuclear Magnetic Resonance to Identify Structure of DON Derivatives
2.6. DON Assays with Mixed Cultures with Naturally Contaminated Wheat Samples
3. Discussion
4. Materials and Methods
4.1. Field Collections
4.2. Selection of Microbes in the Presence of High Concentrations of DON
4.3. Isolation of Individual DON Modifying Microbes
4.4. Identification of Mixed Cultures Using 16S Ribosomal Sequencing
4.5. Thin Layer Chromatography to Identify DON Derivatives
4.6. Nuclear Magnetic Resonance to Identify Structure of DON Derivatives
4.7. DON Assays with Mixed Cultures with Naturally Contaminated Wheat
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sobrova, P.; Adam, V.; Vasatkova, A.; Beklova, M.; Zeman, L.; Kizek, R. Deoxynivalenol and its toxicity. Interdiscip. Toxicol. 2010, 3, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Placinta, C.; D’Mello, J.P.; Macdonald, A.M. A review of worldwide contamination of cereal grains and animal feed with Fusarium mycotoxins. Anim. Feed Sci. Technol. 1999, 78, 21–37. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. The Global Forum for Food Security and Nutrition; FAO: Rome, Italy, 2013. [Google Scholar]
- Zain, M.E. Impact of mycotoxins on humans and animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef]
- Fink-Grernmels, J. Mycotoxins: Their implications for human and animal health. Vet. Q. 1999, 21, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Pier, A.C. Major biological consequences of aflatoxicosis in animal production. J. Anim. Sci. 1992, 70, 3964–3967. [Google Scholar] [CrossRef] [PubMed]
- Pohland, A.E. Mycotoxins in review. Food Addit. Contam. 1993, 10, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.M. Trichothecenes in grains. Cereal Foods World 1990, 35, 661–666. [Google Scholar]
- Desjardins, A.E. Fusarium Mycotoxins: Chemistry, Genetics, and Biology; American Phytopathological Society (APS Press): Paul, MN, USA , 2006. [Google Scholar]
- Schmale, D.G.; Munkvold, G.P. Mycotoxins in crops: A threat to human and domestic animal health. Plant Health Instr. 2009, 3, 340–353. [Google Scholar] [CrossRef]
- Tanaka, T.; Hasegawa, A.; Yamamoto, S.; Lee, U.S.; Sugiura, Y.; Ueno, Y. Worldwide contamination of cereals by the Fusarium mycotoxins nivalenol, deoxynivalenol, and zearalenone. 1. Survey of 19 countries. J. Agric. Food Chem. 1988, 36, 979–983. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Nganje, W.E.; Kaitibie, S.; Wilson, W.W.; Leistritz, F.L.; Bangsund, D.A. Economic Impacts of Fusarium Head Blight in Wheat and Barley: 1993–2001; Department of Agribusiness and Applied Economics, Agricultural Experiment Station, North Dakota State University: Fargo, ND, USA, 2004. [Google Scholar]
- Sato, N.; Ueno, A. Comparative toxicities of trichothecenes. In Mycotoxins in Human and Animal Health; Rodricks, J.V., Hesseltine, C.W., Mehlman, M.A., Eds.; Pathotox Publishers: Chicago, IL, USA; pp. 295–307.
- Ueno, Y.; Nakajima, M.; Sakai, K.; Ishii, K.; Sato, N. Comparative toxicology of trichothec mycotoxins: Inhibition of protein synthesis in animal cells. J. Biochem. 1973, 74, 285–296. [Google Scholar] [PubMed]
- Kimura, M.; Tokai, T.; Takahashi-Ando, N.; Ohsato, S.; Fujimura, M. Molecular and genetic studies of fusarium trichothecene biosynthesis: Pathways, genes, and evolution. Biosci. Biotechnol. Biochem. 2007, 71, 2105–2123. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P. Microbial Detoxification of Mycotoxins. J. Chem. Ecol. 2013, 39, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; He, J.; Gong, J. Microbial transformation of trichothecene mycotoxins. World Mycotoxin J. 2008, 1, 23–30. [Google Scholar] [CrossRef]
- Fuchs, E.; Binder, E.M.; Heidler, D.; Krska, R. Structural characterization of metabolites after the microbial degradation of type A trichothecenes by the bacterial strain BBSH 797. Food Addit. Contam. 2002, 19, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Völkl, A.; Vogler, B.; Schollenberger, M.; Karlovsky, P. Microbial detoxification of mycotoxin deoxynivalenol. J. Basic Microbiol. 2004, 44, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Shima, J.; Takase, S.; Takahashi, Y.; Iwai, Y.; Fujimoto, H.; Yamazaki, M.; Ochi, K. Novel detoxification of the trichothecene mycotoxin deoxynivalenol by a soil bacterium isolated by enrichment culture. Appl. Environ. Microbiol. 1997, 63, 3825–3830. [Google Scholar] [PubMed]
- He, J.W.; Bondy, G.S.; Zhou, T.; Caldwell, D.; Boland, G.J.; Scott, P.M. Toxicology of 3-epi-deoxynivalenol, a deoxynivalenol-transformation product by Devosia mutans 17-2-E-8. Food Chem. Toxicol. 2015, 84, 250–259. [Google Scholar] [CrossRef] [PubMed]
- He, J.W.; Yang, R.; Zhou, T.; Boland, G.J.; Scott, P.M.; Bondy, G.S. An epimer of deoxynivalenol: Purification and structure identification of 3-epi-deoxynivalenol. Food Addit. Contam. Part A 2015, 32, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Ikunaga, Y.; Sato, I.; Grond, S.; Numaziri, N.; Yoshida, S.; Yamaya, H.; Hiradate, S.; Hasegawa, M.; Toshima, H.; Ito, M.; et al. Nocardioides sp. strain WSN05-2, isolated from a wheat field, degrades deoxynivalenol, producing the novel intermediate 3-epi-deoxynivalenol. Appl. Microbiol. Biotechnol. 2011, 89, 419–427. [Google Scholar] [CrossRef] [PubMed]
- He, W.J.; Yuan, Q.S.; Zhang, Y.B.; Guo, M.W.; Gong, A.D.; Zhang, J.B.; Wu, A.B.; Huang, T.; Qu, B.; Li, H.P.; et al. Aerobic De-Epoxydation of Trichothecene Mycotoxins by a Soil Bacterial Consortium Isolated Using In Situ Soil Enrichment. Toxins 2016, 8, E277. [Google Scholar] [CrossRef] [PubMed]
- De Bellis, P.; Tristezza, M.; Haidukowski, M.; Fanelli, F.; Sisto, A.; Mulè, G.; Grieco, F. Biodegradation of Ochratoxin A by Bacterial Strains Isolated from Vineyard Soils. Toxins 2015, 7, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhou, T.; Gong, J.H.; Young, C.; Su, X.J.; Li, X.-Z.; Zhu, H.H.; Tsao, R.; Yang, R. Isolation of deoxynivalenol-transforming bacteria from the chicken intestines using the approach of PCR-DGGE guided microbial selection. BMC Microbiol. 2010, 10, 182–191. [Google Scholar] [CrossRef] [PubMed]
- He, J.W.; Hassan, Y.I.; Perilla, N.; Li, X.Z.; Boland, G.J.; Zhou, T. Bacterial epimerization as a route for deoxynivalenol detoxification: The influence of growth and environmental conditions. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Karlovsky, P. Biological detoxification of the mycotoxin deoxynivalenol and its use in genetically engineered crops and feed additives. Appl. Microbiol. Biotechnol. 2011, 91, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J.V. Teixobactin, the first of a new class of antibiotics discovered by iChip technology? J. Antimicrob. Chemother. 2015, 70, 2679–2680. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wu, Z.; Wu, S.; Dai, Y.; Sun, C. Degradation of ochratoxin A by Bacillus amyloliquefaciens ASAG1. Food Addit. Contam. Part A 2015, 32, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Ito, M.; Ishizaka, M.; Ikunaga, Y.; Sato, Y.; Yoshida, S.; Koitabashi, M.; Tsushima, S. Thirteen novel deoxynivalenol-degrading bacteria are classified within two genera with distinct degradation mechanisms. FEMS Microbiol. Lett. 2012, 327, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, P.A.; McMaster, N.J.; Musser, R.; Schmale, D.G. Survey of mycotoxins in corn distillers’ dried grains with solubles from seventy-eight ethanol plants in Twelve states in the U.S. in 2011. Toxins 2014, 6, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Mirocha, C.J.; Kolaczkowski, E.; Xie, W.; Yu, H.; Jelen, H. Analysis of deoxynivalenol and its derivatives (batch and single kernel) using gas chromatography/mass spectrometry. J. Agric. Food Chem. 1998, 46, 1414–1418. [Google Scholar] [CrossRef]
- Takitani, S.; Asabe, Y.; Kato, T.; Suzuki, M.; Ueno, Y. Spectrodensitometric determination of trichothecene mycotoxins with 4-(p-nitrobenzyl) pyridine on silica gel thin-layer chromatograms. J. Chromatogr. A 1979, 172, 335–342. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Culture Sample | Replicate | DON (μg/mL) Analytical Rep 1 | DON (μg/mL) Analytical Rep 2 | Mean DON (μg/mL) |
|---|---|---|---|---|
| Mixed Culture 1-R1 | 1 | 0.16 | 0.16 | 0.16 |
| Mixed Culture 1-R2 | 2 | 2.48 | 2.72 | 2.6 |
| Mixed Culture 1-R3 | 3 | 3.12 | 3.04 | 3.08 |
| Mixed Culture 1-R4 | 4 | 2.72 | 2.68 | 2.7 |
| Mixed Culture 2-R1 | 1 | <0.2 | <0.2 | <0.2 |
| Mixed Culture 2-R2 | 2 | 4.32 | 3.8 | 4.06 |
| Mixed Culture 2-R3 | 3 | <0.2 | <0.2 | <0.2 |
| Mixed Culture 2-R4 | 4 | <0.2 | <0.2 | <0.2 |
| Mixed Culture 3-R1 | 1 | 3.92 | 3.8 | 3.86 |
| Mixed Culture 3-R2 | 2 | 3.92 | 3.76 | 3.84 |
| Mixed Culture 3-R3 | 3 | 4.08 | 3.8 | 3.94 |
| Mixed Culture 3-R4 | 4 | 3.28 | 3.64 | 3.46 |
| Control-R1 | 1 | 4.46 | 4.48 | 4.47 |
| Control-R2 | 2 | 4.48 | 4.28 | 4.38 |
| Control-R3 | 3 | 3.84 | 4.04 | 3.94 |
| Sample ID | Culture ID | Assay | Replicate | Starting DON (μg/mL) in Wheat | Final DON (μg/mL) in Wheat |
|---|---|---|---|---|---|
| 1 | Mixed Culture 1 | 1 | 1 | 7.10 | 5.08 |
| 2 | Mixed Culture 1 | 1 | 2 | 7.10 | <0.20 |
| 3 | Mixed Culture 1 | 1 | 3 | 7.10 | 0.08 |
| 4 | Mixed Culture 1 | 2 | 1 | 7.10 | 7.04 |
| 5 | Mixed Culture 1 | 2 | 2 | 7.10 | 7.76 |
| 6 | Mixed Culture 1 | 2 | 3 | 7.10 | 5.88 |
| 4.3 (mean) | |||||
| 7 | Mixed Culture 2 | 1 | 1 | 7.10 | 4.0 |
| 8 | Mixed Culture 2 | 1 | 2 | 7.10 | 6.4 |
| 9 | Mixed Culture 2 | 1 | 3 | 7.10 | 3.48 |
| 10 | Mixed Culture 2 | 2 | 1 | 7.10 | 7.56 |
| 11 | Mixed Culture 2 | 2 | 2 | 7.10 | 7.88 |
| 12 | Mixed Culture 2 | 2 | 3 | 7.10 | 7.28 |
| 6.1 (mean) | |||||
| Control | Control (no cultures) | 7.10 (mean) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, N.M.; McMaster, N.; Gantulga, D.; Soyars, C.; McCormick, S.P.; Knott, K.; Senger, R.S.; Schmale, D.G. Modification of the Mycotoxin Deoxynivalenol Using Microorganisms Isolated from Environmental Samples. Toxins 2017, 9, 141. https://doi.org/10.3390/toxins9040141
Wilson NM, McMaster N, Gantulga D, Soyars C, McCormick SP, Knott K, Senger RS, Schmale DG. Modification of the Mycotoxin Deoxynivalenol Using Microorganisms Isolated from Environmental Samples. Toxins. 2017; 9(4):141. https://doi.org/10.3390/toxins9040141
Chicago/Turabian StyleWilson, Nina M., Nicole McMaster, Dash Gantulga, Cara Soyars, Susan P. McCormick, Ken Knott, Ryan S. Senger, and David G. Schmale. 2017. "Modification of the Mycotoxin Deoxynivalenol Using Microorganisms Isolated from Environmental Samples" Toxins 9, no. 4: 141. https://doi.org/10.3390/toxins9040141
APA StyleWilson, N. M., McMaster, N., Gantulga, D., Soyars, C., McCormick, S. P., Knott, K., Senger, R. S., & Schmale, D. G. (2017). Modification of the Mycotoxin Deoxynivalenol Using Microorganisms Isolated from Environmental Samples. Toxins, 9(4), 141. https://doi.org/10.3390/toxins9040141