
Mycotoxin Biotransformation by Native and Commercial Enzymes: Present and Future Perspectives


 ,
, 

Abstract
:1. Introduction
Methods for Mycotoxins Reduction
2. Mycotoxin Biotransformation by Enzymes
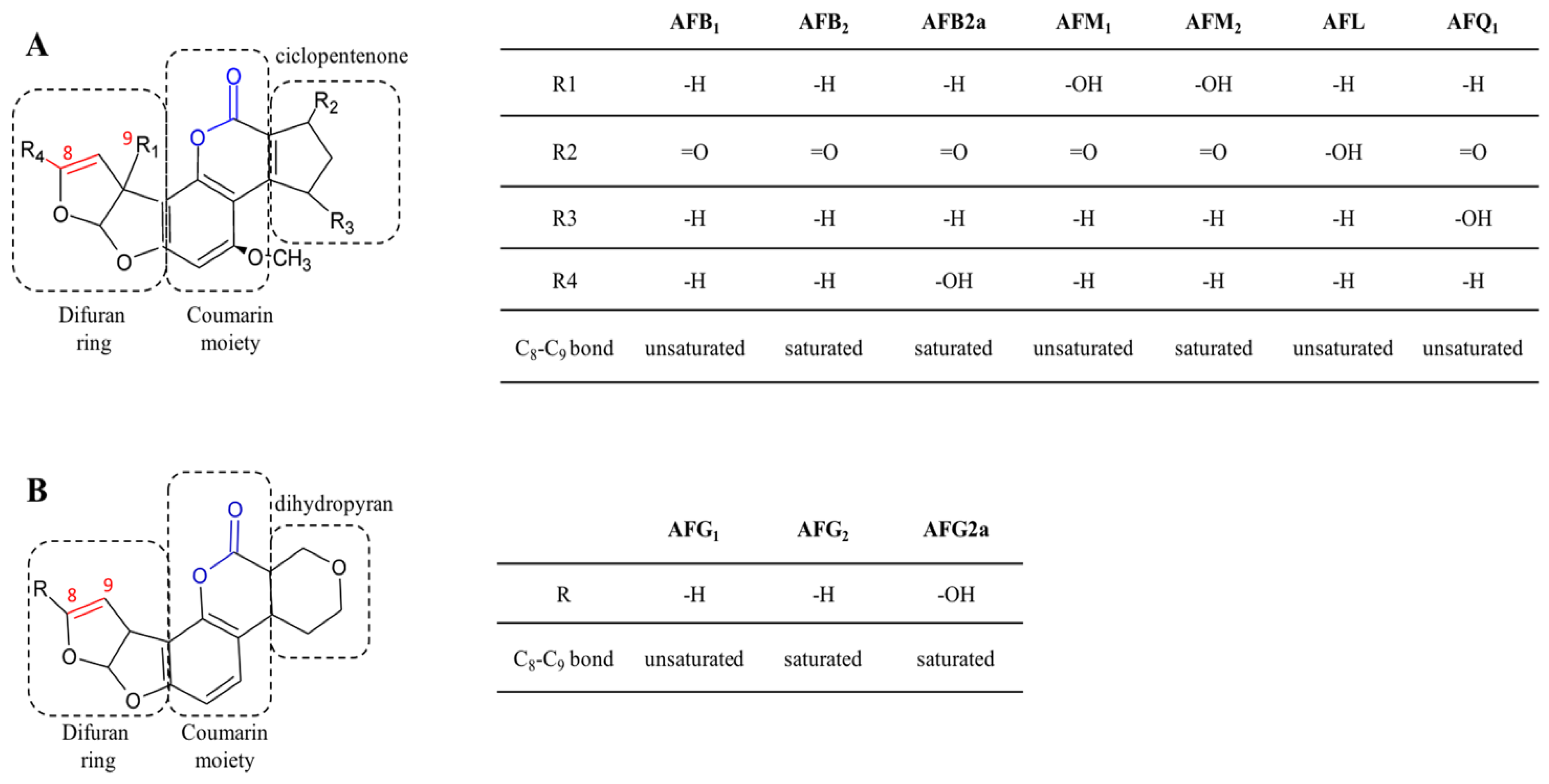
2.1. Aflatoxins
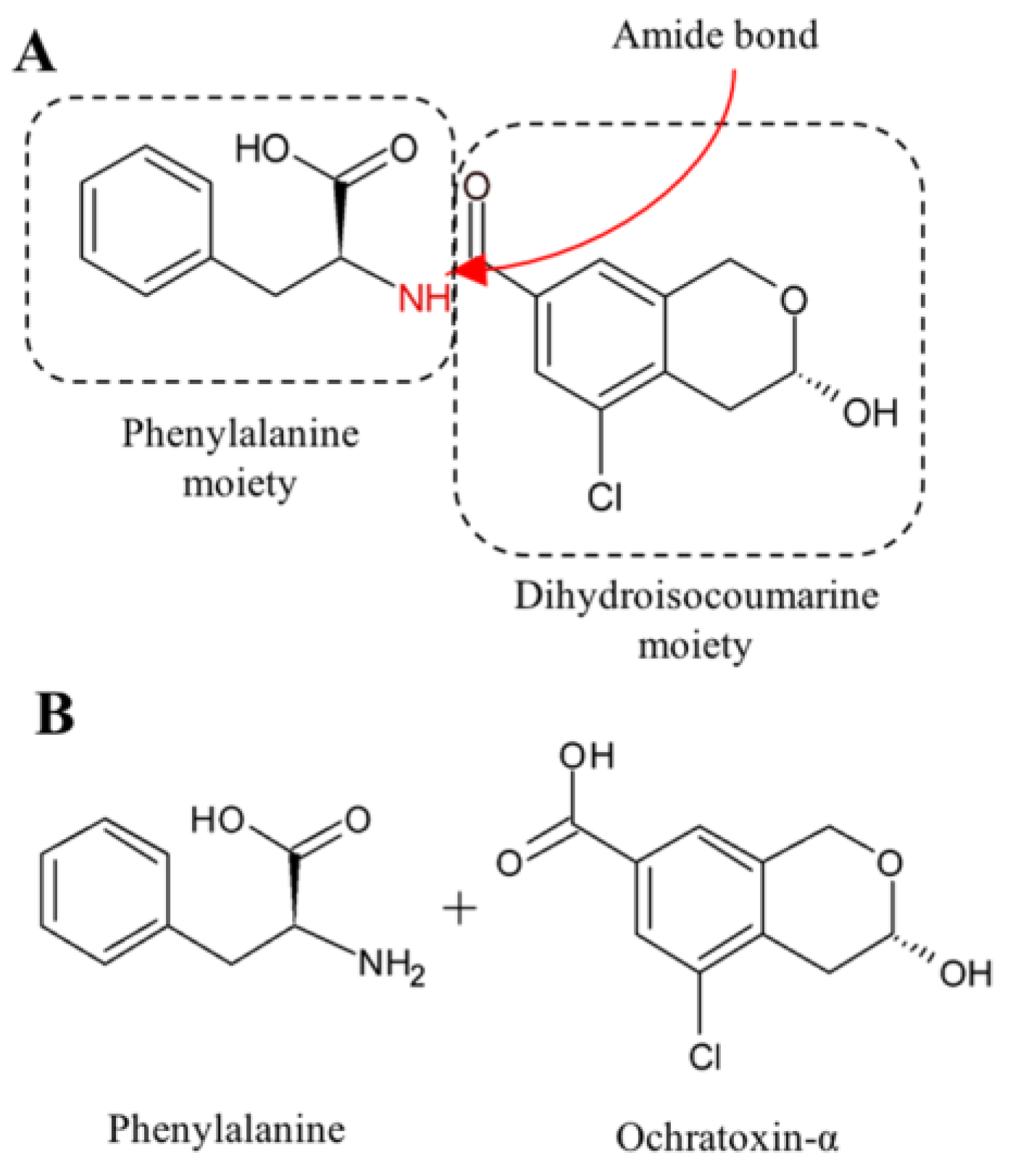
2.2. Ochratoxin A
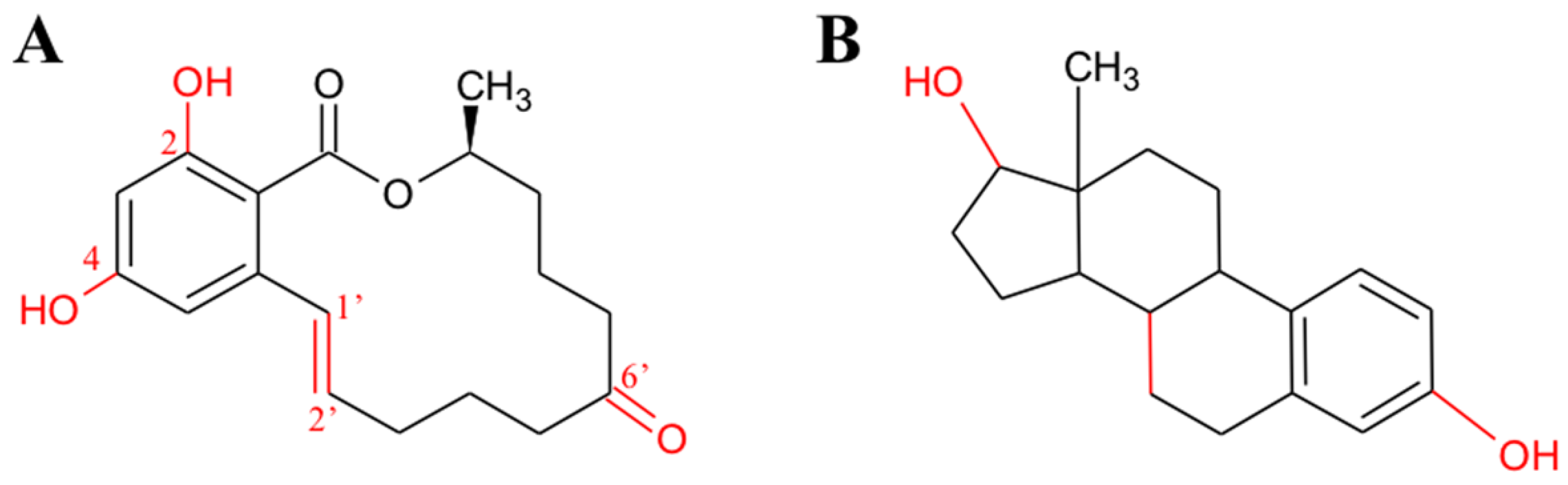
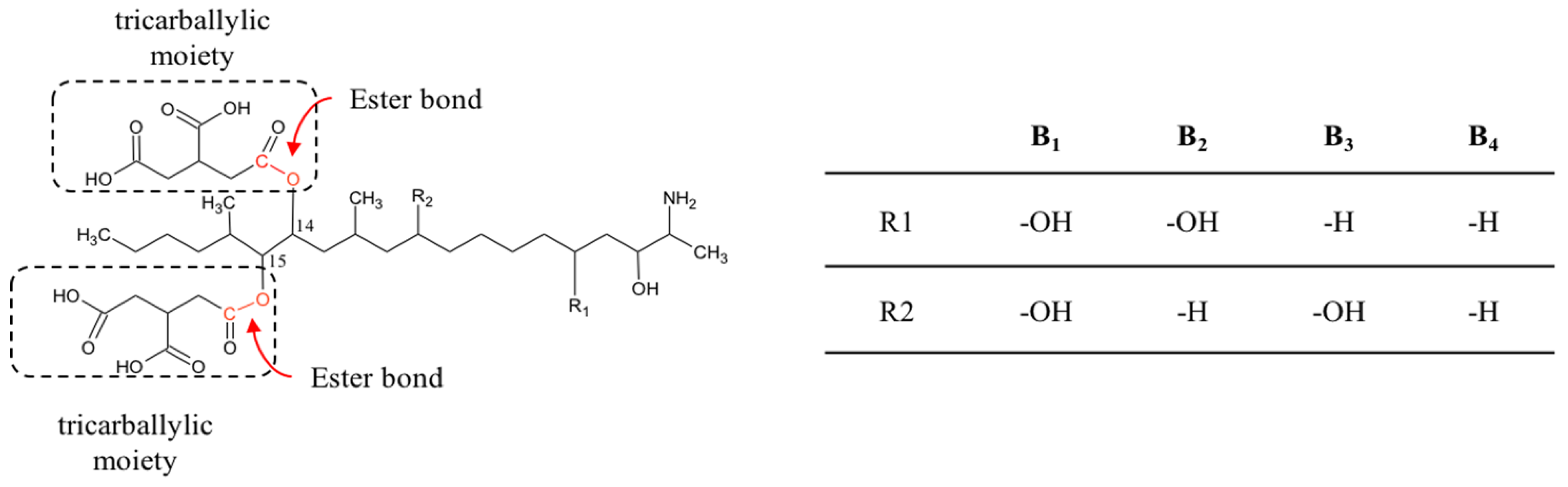
2.3. Fumonisins
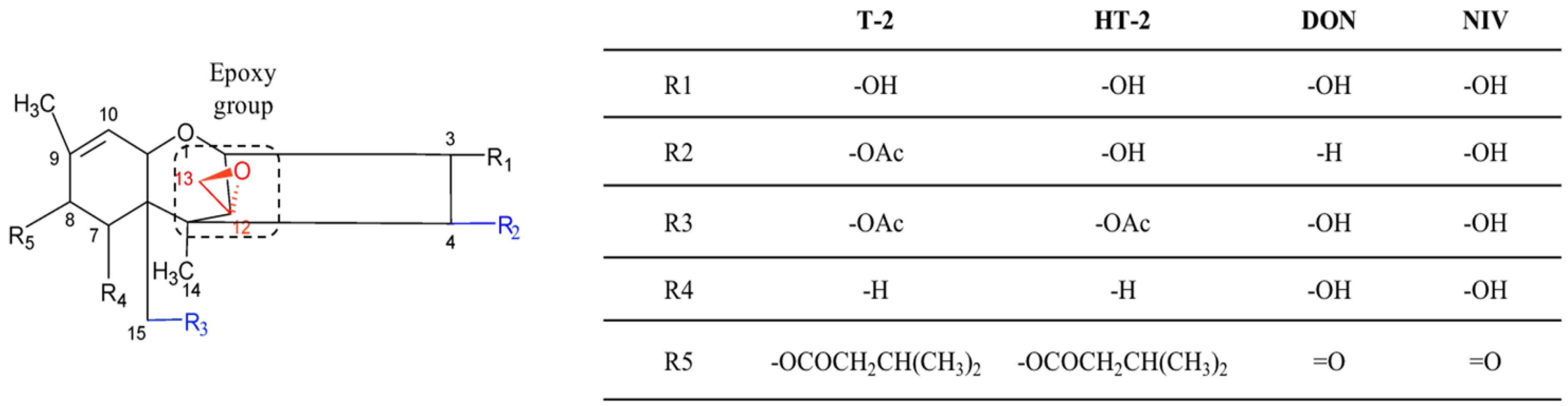
2.4. Trichothecenes
2.5. Zearalenone
3. Potentialities and Limitations of Mycotoxin Degrading Enzymes in Food, Feed, and Bioenergy
3.1. Food
3.2. Feed
3.3. Bioenergy
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1H-NMR | Proton Nuclear Magnetic Resonance |
| AFB1 | aflatoxin B1 |
| AFD1 | aflatoxin D1 |
| AFL | aflatoxicol |
| AFM1 | aflatoxin M1 |
| AFO | aflatoxin oxidase enzyme |
| AFQ1 | aflatoxin Q1 |
| AFs | aflatoxins |
| AP1 | aminopentol 1 |
| CPA | carboxypeptidase A |
| CPY | carboxypeptidase Y |
| CT | coated and tough |
| DDGS | distiller’s grains and soluble |
| DE | degrading enzyme |
| DOE | Design of experiments |
| DON | deoxynivalenol |
| DPs | degradation products |
| EDC | endocrine disrupting chemical |
| EFSA | European Food Safety Authority |
| ER | estrogen receptors |
| FB1 | fumonisin B1 |
| GAPs | Good Agricultural Practices |
| HFB | hydrolyzed FB1 |
| HR-ESI-MS | High Resolution electrospray ionization mass spectrometry |
| HT-2 | HT-2 toxin |
| IARC | International Agency of Research on Cancer |
| LCs | laccases |
| MADE | Myxobacteria Aflatoxin Degrading Enzyme |
| MnPs | Manganese Peroxidases |
| NIV | nivalenol |
| OTA | ochratoxin A |
| OTα | ochratoxin α |
| Phe | phenylalanine molecule |
| Prx | peroxiredoxin |
| T-2 | T-2 toxin |
| ZEN | zearalenone |
| α-ZAL | α-zearalanol |
| α-ZEL | α-zearalenol |
| β-ZAL | β-zearalanol |
| β-ZEL | β-zearalenol |
References and Notes
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Logrieco, A.F.; Mulé, G.; Moretti, A.; Bottalico, A. Toxigenic Fusarium species and mycotoxins associated with maize ear rot in Europe. Eur. J. Plant. Pathol. 2002, 108, 597–609. [Google Scholar]
- Bryden, W.L. Mycotoxins in the food chain: Human health implications. Asia Pac. J. Clin. Nutr. 2007, 16, 95–101. [Google Scholar] [PubMed]
- Shephard, G.S. Impact of mycotoxins on human health in developing countries. Food Addit. Contam. 2008, 25, 146–151. [Google Scholar]
- Sharma, R.P. Immunotoxicity of mycotoxins. J. Dairy Sci. 1993, 76, 892–897. [Google Scholar] [CrossRef]
- Zain, M.E. Impact of mycotoxins on humans and animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef]
- The International Agency for Research on Cancer (IARC). Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 2002; Volume 82, pp. 1–556. [Google Scholar]
- The International Agency for Research on Cancer (IARC). Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 1993; Volume 56, pp. 489–521. [Google Scholar]
- The International Agency for Research on Cancer (IARC). Chemical agents and related occupations. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 2012; Volume 100F, pp. 1–599. [Google Scholar]
- Alassane-Kpembi, I.; Schatzmayr, G.; Taranu, I.; Marin, D.; Puel, O.; Oswald, I.P. Mycotoxins co-contamination: Methodological aspects and biological relevance of combined toxicity studies. Crit. Rev. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, W.C. Interaction of fumonisin B(1) and aflatoxin B(1) in a short-term. Toxicology 2002, 171, 161–173. [Google Scholar] [CrossRef]
- Ji, C.; Fan, Y.; Zhao, L. Review on biological degradation of mycotoxins. Anim. Nutr. 2016, 2, 127–133. [Google Scholar]
- Gao, Y.N.; Wang, J.Q.; Li, S.L.; Zhang, Y.D.; Zheng, N. Aflatoxin M1 cytotoxicity against human intestinal Caco-2 cells is enhanced in the presence of other mycotoxins. Food Chem. Toxicol. 2016, 96, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Creppy, E.E.; Kane, A.; Bacha, H.; Steyn, P.S.; Roschenthaler, R.; Dirheimer, G. Influence of ochratoxin B on the ochratoxin A inhibition of phenylalanyl-tRNA formation In vitro and protein synthesis in hepatoma tissue culture cells. Toxicol. Lett. 1989, 45, 307–313. [Google Scholar] [PubMed]
- Bouslimi, A.; Bouaziz, C.; Ayed-Boussema, I.; Hassen, W.; Bacha, H. Individual and combined effects of ochratoxin A and citrinin on viability and DNA fragmentation in cultured Vero cells and on chromosome aberrations in mice bone marrow cells. Toxicology 2008, 251, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bouslimi, A.; Ouannes, Z.; El Golli, E.; Bouaziz, C.; Hassen, W.; Bacha, H. Cytotoxicity and oxidative damage in kidney cells exposed to the mycotoxins ochratoxin A and citrinin: Individual and combined effects. Toxicol. Mech. Method 2008, 18, 341–349. [Google Scholar]
- Klaric, M.S.; Zeljezic, D.; Rumora, L.; Peraica, M.; Pepeljnjak, S.; Domijan, A.M. A potential role of calcium in apoptosis and aberrant chromatin forms in porcine kidney PK15 cells induced by individual and combined ochratoxin A and citrinin. Arch. Toxicol. 2012, 86, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Knasmuller, S.; Cavin, C.; Chakraborty, A.; Darroudi, F.; Majer, B.J.; Huber, W.W.; Ehrlich, V.A. Structurally related mycotoxins ochratoxin A, ochratoxin B, and citrinin differ in their genotoxic activities and in their mode of action in human-derived liver (HepG2) cells: Implications for risk assessment. Nutr. Cancer 2004, 50, 190–197. [Google Scholar] [PubMed]
- Wu, F. Measuring the economic impacts of Fusarium toxins in animal feed. Anim. Feed Sci. Technol. 2007, 137, 363–374. [Google Scholar]
- Commission Regulation 2006/1881/EC of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Food Stuffs. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006R1881&from=en (accessed on 03 November 2016).
- Commission Recommendation 2013/165/EU of 27 March 2013 on the Presence of T-2 and HT-2 Toxin in Cereals and Cereal Products Text with EEA Relevance. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32013H0165&from=EN (accessed on 03 November 2016).
- Opinion of the Scientific Committee on Food (SCF) on Fusarium Toxins, Part 5: T-2 Toxin and HT-2 Toxin. 2001. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_catalogue_out88_en.pdf (accessed on 03 November 2016).
- Commission Recommendation 2006/576/EC of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006H0576&from=EN (accessed on 03 November 2016).
- Commission Recommendation 2006/583/EC of 17 August 2006 on the Prevention and Reduction of Fusarium Toxins in Cereals and Cereal Products. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006H0583&from=EN (accessed on 03 November 2016).
- Vanhoutte, I.; Audenaert, K.; De Gelder, L. Biodegradation of Mycotoxins: Tales from Known and Unexplored Worlds. Front. Microbiol. 2016, 7, 561. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, O.; Ehlbeck, J.; Hertel, C.; Habermeyer, M.; Roth, A.; Engel, K.H.; Holzhauser, T.; Knorr, D.; Eisenbrand, G. Opinion on the use of plasma processes for treatment of foods. Mol. Nutr. Food Res. 2013, 57, 920–927. [Google Scholar] [PubMed]
- Bong, J.P.; Kosuke, T.; Yoshiko, S.K.; Ik-Hwi, K.; Mi-Hee, L.; Dong-Wook, H.; Kie-Hyung, C.; Soon, O.H.; Jong-Chul, P. Degradation of mycotoxins using microwave-induced argon plasma at atmospheric pressure. Surf. Coat. Technol. 2007, 201, 5733–5737. [Google Scholar]
- Kriz, P.; Petr, B.; Zbynek, H.; Jaromir, K.; Pavel, O.; Petr, S.; Miroslav, D. Influence of plasma treatment in open air on mycotoxin content and grain nutriments. Plasma Med. 2015, 5, 145–158. [Google Scholar] [CrossRef]
- Herzallah, S.; Al Shawabkeh, K.; Al Fataftah, A. Aflatoxin decontamination of artificially contaminated feeds by sunlight, g-radiation, and microwave heating. J. Appl. Poult. Res. 2008, 17, 515–521. [Google Scholar]
- Fanelli, F.; Geisen, R.; Schmidt-Heydt, M.; Logrieco, A.F.; Mulè, G. Light regulation of mycotoxin biosynthesis: New perspectives for food safety. World Mycotoxin J. 2016, 9, 129–146. [Google Scholar] [CrossRef]
- Bretz, M.; Beyer, M.; Cramer, B.; Knecht, A.; Humpf, H.-U. Thermal degradation of the Fusarium mycotoxin deoxynivalenol. J. Agric. Food Chem. 2006, 54, 6445–6451. [Google Scholar] [CrossRef]
- Park, D.L.; Lee, L.S.; Price, R.L.; Pohland, A.E. Review of the decontamination of aflatoxins by ammoniation: Current status and regulation. J. Assoc. Off. Anal. Chem. 1988, 71, 685–703. [Google Scholar] [PubMed]
- Aiko, V.; Edamana, P.; Mehta, A. Decomposition and detoxification of aflatoxin B1 by lactic acid. J. Sci. Food Agric. 2016, 96, 1959–1966. [Google Scholar] [PubMed]
- Müller, H.M. Entgiftung von Mykotoxinen: II. Chemische Verfahren und Reaktion mit Inhaltsstoffen von Futtermitteln. Übers Tierernährg. 1983, 11, 47–80. [Google Scholar]
- Fouler, S.G.; Trivedi, A.B.; Kitabatake, N. Detoxification of citrinin and ochratoxin a by hydrogen peroxide. J. Assoc. Off. Agric. Chem. 1994, 77, 631–637. [Google Scholar]
- Maeba, H.; Takamoto, Y.; Kamimura, M.I.; Miura, T.O. Destruction and detoxification of aflatoxins with ozone. J. Food Sci. 1988, 53, 667–668. [Google Scholar] [CrossRef]
- Altug, T.; Youssef, A.E.; Marth, E.H. Degradation of aflatoxin B1 in dried figs by sodium bisulfite with or without heat, ultraviolet energy or hydrogen peroxide. J. Food Prot. 1990, 53, 581–628. [Google Scholar]
- Commission Regulation 2015/786/EU Defining Acceptability Criteria for Detoxification Processes Applied to Products Intended for Animal feed as Provided for in Directive 2002/32/EC of the European Parliament and of the Council. Available online: http://extwprlegs1.fao.org/docs/pdf/eur144560.pdf (accessed on 3 November 2016).
- Boudergue, C.; Burel, C.; Dragacci, S.; Favrot, M.; Fremy, J.; Massimi, C.; Prigent, P.; Debongnie, P.; Pussemier, L.; Boudra, H.; et al. Review of mycotoxin-detoxifying agents used as feed additives: mode of action, efficacy and feed/food safety. EFSA Support. Publ. 2009, 6. [Google Scholar] [CrossRef]
- Hassan, Y.I.; Zhou, T.; Bullerman, L.B. Sourdough lactic acid bacteria as antifungal and mycotoxin-controlling agents. Food Sci. Technol. Int. 2015, 22, 79–90. [Google Scholar]
- Hassan, Y.I.; Bullerman, L.B. Cell-surface binding of deoxynivalenol to Lactobacillus paracasei subsp. tolerans isolated from sourdough starter culture. JMBFS 2013, 2, 2323–2325. [Google Scholar]
- Böswald, C.; Engelhardt, G.; Vogel, H.; Wallnöfer, P.R. Metabolism of Fusarium mycotoxins zearalenone and deoxynivalenol by yeast strains of technological relevance. Nat. Toxins 1995, 3, 138–144. [Google Scholar]
- De Bellis, P.; Tristezza, M.; Haidukowski, M.; Fanelli, F.; Sisto, A.; Mulè, G.; Grieco, F. Biodegradation of ochratoxin A by bacterial strains isolated from vineyard soils. Toxins 2015, 7, 5079–5093. [Google Scholar] [CrossRef]
- Sato, I.; Ito, M.; Ishizaka, M.; Ikunaga, Y.; Sato, Y.; Yoshida, S.; Koitabashi, M.; Tsushima, S. Thirteen novel deoxynivalenol-degrading bacteria are classified within two genera with distinct degradation mechanisms. FEMS Microbiol. Lett. 2012, 327, 110–117. [Google Scholar] [PubMed]
- He, W.J.; Yuan, Q.S.; Zhang, Y.B.; Guo, M.W.; Gong, A.D.; Zhang, J.B.; Wu, A.B.; Huang, T.; Qu, B.; Li, H.P.; et al. Aerobic de-epoxydation of trichothecene mycotoxins by a soil bacterial consortium isolated using in situ soil enrichment. Toxins 2016, 8, 277. [Google Scholar]
- Essigmann, J.M.; Croy, R.G.; Nadzan, A.M.; Busby, W.F., Jr.; Reinhold, V.N.; Büchi, G.; Wogan, G.N. Structural identification of the major DNA adduct formed by aflatoxin B1 In vitro. Proc. Nat. Acad. Sci. USA 1977, 74, 1870–1874. [Google Scholar] [PubMed]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of aflatoxin carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 135–172. [Google Scholar] [PubMed]
- Schroeder, T.U.; Zweifel, P.; Sagelsdorff, U.; Friederich, J.; Luthy, C.; Schlatter, J. Ammoniation of aflatoxin-containing corn: Distribution, in vivo covalent deoxyribonucleic acid binding, and mutagenicity of reaction products. Agric. Food Chem. 1985, 33, 311–316. [Google Scholar] [CrossRef]
- Méndez-Albores, A.; Arámbula-Villa, G.; Loarca-Piña, M.G.; Castaño-Tostado, E.; Moreno-Martínez, E. Safety and efficacy evaluation of aqueous citric acid to degrade B-aflatoxins in maize. Food Chem. Toxicol. 2005, 43, 233–238. [Google Scholar] [PubMed]
- Motomura, M.; Toyomasu, T.; Mizuno, K.; Shinozawa, T. Purification and characterization of an aflatoxin degradation enzyme from Pleurotus ostreatus. Microbiol. Res. 2003, 158, 237–242. [Google Scholar] [PubMed]
- Liu, D.L.; Yao, D.S.; Liang, R.; Ma, L.; Cheng, W.Q.; Gu, L.Q. Detoxifcation of aflatoxin B1 by enzymes isolated from Armillariella tabescens. Food Chem. Toxicol. 1998, 36, 563–574. [Google Scholar]
- Chitrangada, D.; Mishra, H.N. In vitro degradation of aflatoxin B1 by horseradish peroxidase. Food Chem. 2000, 68, 309–313. [Google Scholar]
- Alberts, J.F.; Gelderblom, W.C.; Botha, A.; van Zyl, W.H. Degradation of aflatoxin B(1) by fungal laccase enzymes. Int. J. Food Microbiol. 2009, 30, 47–52. [Google Scholar]
- Novozymes A/S. Detoxification of Aflatoxin in Feed Products. World Patent 2009109607, 5 March 2009. [Google Scholar]
- Taylor, M.C.; Jackson, C.J.; Tattersall, D.B.; French, N.; Peat, T.S.; Newman, J.; Briggs, L.J.; Lapalikar, G.V.; Campbell, P.M.; Scott, C.; et al. Identification and characterization of two families of F420H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol. Microbiol. 2010, 78, 561–575. [Google Scholar] [PubMed]
- Yehia, R.S. Aflatoxin detoxification by manganese peroxidase purified from Pleurotus ostreatus. Braz. J. Microbiol. 2014, 45, 127–133. [Google Scholar] [PubMed]
- Zhao, L.H.; Guan, S.; Gao, X.; Ma, Q.G.; Lei, Y.P.; Bai, X.M.; Ji, C. Preparation, purification and characteristics of an aflatoxin degradation enzyme from Myxococcus fulvus ANSM068. J. Appl. Microbiol. 2011, 110, 147–155. [Google Scholar] [CrossRef]
- Loi, M.; Fanelli, F.; Zucca, P.; Liuzzi, V.C.; Quintieri, L.; Cimmarusti, M.T.; Monaci, L.; Haidukovski, M.; Logrieco, A.F.; Sanjust, E.; et al. Aflatoxin B1 and M1 degradation by Lac2 from Pleurotus pulmonarius and redox mediators. Toxins 2016, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Quintieri, L.; Liuzzi, V.C.; Haidukovski, M.; Logrieco, A.F.; Sanjust, E.; Fanelli, F.; Mulè, G. Aflatoxin M1 removal and biotechnological application of a laccase from Pleurotus eryngii for milk safety. Scienza e Tecnica Lattiero Casearia. In press.
- Cao, H.; Liu, D.; Mo, X.; Xie, C.; Yao, D. A fungal enzyme with the ability of aflatoxin B1 conversion: Purification and ESI-MS/MS identification. Microbiol. Res. 2011, 166, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.L.; Ma, L.; Gu, L.Q.; Liang, R.; Yao, D.S.; Chen, W.Q. Armillariella tabescens enzymatic detoxification of aflatoxin B1. Part III. Immobilized enzymatic detoxification. Ann. N. Y. Acad. Sci. 1998, 864, 592–599. [Google Scholar] [PubMed]
- Wu, Y.Z.; Lu, F.P.; Jiang, H.L.; Tan, C.P.; Yao, D.S.; Xie, C.F.; Liu, D.L. The furofuran-ring selectivity, hydrogen peroxide-production and low Km value are the three elements for highly effective detoxification of aflatoxin oxidase. Food Chem. Toxicol. 2015, 76, 125–131. [Google Scholar]
- Wang, J.; Ogata, M.; Hirai, H.; Kawagishi, H. Detoxification of aflatoxin B1 by manganese peroxidase from the white-rot fungus Phanerochaete sordida YK-624. FEMS Microbiol. Lett. 2011, 314, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Chitrangada, D.; Mishra, H.N. In vitro degradation of aflatoxin B1 in groundnut (Arachis hypogea) meal by horseradish peroxidase. Lebensm. Wiss. Technol. 2000, 33, 308–312. [Google Scholar]
- Viswanath, B.; Rajesh, B.; Janardhan, A.; Kumar, A.P.; Narasimha, G. Fungal laccases and their applications in bioremediation. Enzyme Res. 2014, 2014, 1–21. [Google Scholar]
- Banu, I.; Lupu, A.; Aprodu, I. Degradation of zearalenone by laccase enzyme. Sci. Study Res. 2013, 14, 79–84. [Google Scholar]
- Novozymes A/S. Detoxification of Aflatoxin in Feed Products. European Patent 2252163, 5 March 2009. [Google Scholar]
- Margot, J.; Bennati-Granier, C.; Maillard, J.; Blánquez, P.; Barry, D.A.; Holliger, C. Bacterial versus fungal laccase: Potential for micropollutant degradation. AMB Express 2013, 3, 63–77. [Google Scholar] [CrossRef]
- Guan, S.; Zhao, L.; Ma, Q.; Zhou, T.; Wang, N.; Hu, X.; Ji, C. In vitro efficacy of Myxococcus fulvus ANSM068 to biotransform aflatoxin B1. Int. J. Mol. Sci. 2010, 11, 4063–4079. [Google Scholar]
- Dirheimer, G.; Creppy, E.E. Mechanism of action of ochratoxin A. IARC Sci. Publ. 1991, 115, 171–186. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [PubMed]
- Vettorazzi, A.; van Delft, J.; López de Cerain, A. A review on ochratoxin A transcriptomic studies. Food Chem. Toxicol. 2013, 59, 766–783. [Google Scholar] [PubMed]
- Kőszegi, T.; Poór, M. Ochratoxin A: Molecular interactions, mechanisms of toxicity and prevention at the molecular Level. Toxins 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Abrunhosa, L.; Paterson, R.R.; Venâncio, A. Biodegradation of Ochratoxin A for Food and Feed Decontamination. Toxins 2010, 2, 1078–1099. [Google Scholar] [CrossRef]
- Pitout, M.J. The hydrolysis of Ochratoxin A by some proteolytic enzymes. Biochem. Pharmacol. 1969, 18, 485–491. [Google Scholar] [PubMed]
- Dridi, F.; Marrakchi, M.; Gargouri, M.; Saulnier, J.; Jaffrezic-Renault, N.; Lagarde, F. Comparison of carboxypeptidase Y and thermolysin for ochratoxin A electrochemical biosensing. Anal. Methods 2015, 7, 8954–8960. [Google Scholar]
- Stander, M.A.; Bornscheuer, U.T.; Henke, E.; Steyn, P.S. Screening of commercial hydrolases for the degradation of ochratoxin A. J. Agric. Food Chem. 2000, 48, 5736–5739. [Google Scholar] [PubMed]
- Abrunhosa, L.; Santos, L.; Venâncio, A. Degradation of ochratoxin A by proteases and by a crude enzyme of Aspergillus niger. Food Biotechnol. 2006, 20, 231–242. [Google Scholar]
- Danisco A/S. Food Additive Comprising an Amidase for Detoxifying Ochratoxin. World Patent 2012032472, 6 September 2011. [Google Scholar]
- Heinl, S.; Hartinger, D.; Moll, W.D.; Schatzmayr, G.; Grabherr, R. Identification of a fumonisin B1 degrading gene cluster in Sphingomonas sp. MTA144. New Biotechnol. 2009, 25, S61–S62. [Google Scholar] [CrossRef]
- Wang, E.; Norred, W.P.; Bacon, C.W.; Riley, R.T.; Merrill, A.H., Jr. Inhibition of sphingolipid biosynthesis by fumonisins: Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 1991, 266, 14486–14490. [Google Scholar] [PubMed]
- Merrill, A.H.; Wang, E.; Vales, T.R.; Smith, E.R.; Schroeder, J.J.; Menaldino, D.S.; Alexander, C.; Crane, H.M.; Xia, J.; Liotta, D.C.; et al. Fumonisin toxicity and sphingolipid biosynthesis. Adv. Exp. Med. Biol. 1996, 392, 297–306. [Google Scholar] [PubMed]
- Soriano, J.M.; González, L.; Catala, A.I. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Progr. Lipid Res. 2005, 44, 345–356. [Google Scholar] [CrossRef]
- Rheeder, J.P.; Marasas, W.F.O.; Vismer, H.F. Production of fumonisin analogs by Fusarium species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [PubMed]
- Aly, S.E.; Abdel-Galil, M.M.; Abdel-Wahhab, M.A. Application of adsorbent agents technology in the removal of aflatoxin B1 and fumonisin B1 from malt extract. Food Chem. Toxicol. 2004, 42, 1825–1831. [Google Scholar]
- Robinson, A.; Johnson, N.M.; Strey, A.; Taylor, J.F.; Marroquin-Cardona, A.; Mitchell, N.J.; Afriyie-Gyawu, E.; Ankrah, N.A.; Williams, J.H.; Wang, J.S.; et al. Calcium montmorillonite clay reduces urinary biomarkers of fumonisin B1 exposure in rats and humans. Food Addit. Contam. 2012, 29, 809–818. [Google Scholar]
- Pioneer Hi-Bred International, Inc. Fumonisin detoxification compositions and methods. U.S. Patent 5792931, 7 June 1995. [Google Scholar]
- Duvick, J. Prospects for reducing fumonisin contamination of maize through genetic modification. Environ. Health Persp. 2001, 109, 337–342. [Google Scholar] [CrossRef]
- Pioneer Hi-Bred International, Inc. Fumonisin-Detoxifying Enzymes. World Patent 1996006175, 11 August 1995. [Google Scholar]
- Duvick, J.; Rood, T.; Maddox, J.; Gilliam, J. Detoxification of mycotoxins in planta as a strategy for improving grain quality and disease resistance: Identification of fumonisin-degrading microbes from maize. In Molecular Genetics of Host-Specific Toxins in Plant Disease; Kohmoto, K., Yoder, O., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1998; pp. 369–381. [Google Scholar]
- Scientific Opinion on the safety and efficacy of fumonisin esterase (FUMzyme®) as a technological feed additive for pigs. EFSA J. 2014, 12, 3667.
- Täubel, M. Isolierung und Charakterisierung von Mikroorganismen zur Biologischen Inaktivierung von Fumonisinen. Ph.D. Thesis, University of Natural Resources and Applied Life Sciences, Vienna, Austria, 2005. [Google Scholar]
- Hartinger, D.; Schwartz, H.; Hametner, C.; Schatzmayr, G.; Haltrich, D.; Moll, W.D. Enzyme characteristics of aminotransferase FumI of Sphingopyxis sp. MTA144 for deamination of hydrolyzed fumonisin B1. Appl. Microbiol. Biotechnol. 2011, 91, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Heinl, S.; Hartingerb, D.; Thamhesl, M.; Schatzmayrb, G.; Moll, W.D.; Grabherra, R. An aminotransferase from bacterium AtrichotheceneC 55552 deaminates hydrolyzed fumonisin B1. Biodegradation 2011, 22, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Method for the Production of an Additive for the Enzymatic Decomposition of Mycotoxins, Additive, and Use Thereof. U.S. Patent 8,703,460, 2008.
- Ueno, Y. Trichothecenes. In Chemical, Biological and Toxicological Aspects; Elsevier Scientific Publishers: Amsterdam, The Netherlands, 1983. [Google Scholar]
- Zöllner, P.; Mayer-Helm, B. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A 2006, 1136, 123–169. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.F. The trichothecenes and their biosynthesis. Prog. Chem. Org. Nat. Prod. 2007, 88, 63–130. [Google Scholar]
- Rocha, O.; Ansari, K.; Doohan, F.M. Effects of trichothecene mycotoxins on eukaryotic cells: A review. Food Addit. Contam. 2005, 22, 369–378. [Google Scholar] [CrossRef]
- Schuhmacher-Wolz, U.; Heine, K.; Schneider, K. Report on Toxicity Data on Trichothecene Mycotoxins HT-2 and T-2 Toxins. CT/EFSA/CONTAM/2010/03. Available online: http://www.efsa.europa.eu/en/scdocs/doc/65e.pdf (accessed on 10 November 2016).
- Miller, J.D. Aspects of the ecology of Fusarium toxins in cereals. Adv. Exp. Med. Biol. 2002, 504, 19–27. [Google Scholar]
- World Health Organization. WHO Food Additives Series: 47. Safety Evaluation of Certain Mycotoxins in Food. Prepared by the Fifty-Sixth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). 2001. Available online: http://www.inchem.org/documents/jecfa/jecmono/v47je01.htm (accessed on 10 November 2016).
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Wannemacher, R.W., Jr. Structure–function relationship of 12,13-epoxytrichothecene mycotoxins in cell culture: Comparison of whole animal lethality. Toxicon 1986, 24, 985–994. [Google Scholar] [CrossRef]
- Cundliffe, E.; Cannon, M.; Davies, J. Mechanism of inhibition of eukaryotic protein synthesis by trichothecene fungal toxins. Proc. Natl. Acad. Sci. USA 1974, 71, 30–34. [Google Scholar] [PubMed]
- Cundliffe, E.; Davies, J.E. Inhibition of initiation, elongation, and termination of eukaryotic protein synthesis by trichothecene fungal toxins. Antimicrob. Agents Chemother. 1977, 11, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, D.L. Trichothecenes in the environment: Relevance to human health. Toxicol. Lett. 2003, 20, 97–107. [Google Scholar]
- Karlovsky, P. Biological detoxification of the mycotoxin deoxynivalenol and its use in genetically engineered crops and feed additives. Appl. Microbiol. Biotechnol. 2011, 91, 491–504. [Google Scholar] [PubMed]
- Hassan, Y.I.; Zhu, H.; Zhu, Y.; Zhou, T. Beyond ribosomal binding-the increased polarity and aberrant molecular interactions of 3-epi-deoxynivalenol. Toxins 2016, 8, 261. [Google Scholar]
- He, J.W.; Hassan, Y.I.; Perilla, N.; Li, X.Z.; Boland, G.J.; Zhou, T. Bacterial epimerization as a route for deoxynivalenol detoxification: The influence of growth and environmental conditions. Front. Microbiol. 2016, 7, 572. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhou, T.; Young, J.C.; Boland, G.J.; Scott, P.M. Chemical and biological transformations for detoxification of trichothecene mycotoxins in human and animal food chains: A review. Trend Food Sci. Technol. 2010, 21, 67–76. [Google Scholar] [CrossRef]
- Ito, M.; Sato, I.; Ishizaka, M.; Yoshida, S.; Koitabashi, M.; Yoshida, S.; Tsushima, S. Bacterial cytochrome P450 system catabolizing the Fusarium toxin deoxynivalenol. Appl. Environ. Microbiol. 2013, 79, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Poppenberger, B.; Berthiller, F.; Lucyshyn, D.; Sieberer, T.; Schuhmacher, R.; Krska, R.; Kuchler, K.; Glössl, J.; Luschnig, C.; Adam, G. Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 2003, 278, 47905–47914. [Google Scholar] [CrossRef] [PubMed]
- Pierron, A.; Mimoun, S.; Murate, L.S.; Loiseau, N.; Lippi, Y.; Bracarense, A.P.; Schatzmayr, G.; He, J.W.; Zhou, T.; Moll, W.D.; et al. Microbial biotransformation of DON: Molecular basis for reduced toxicity. Sci. Rep. 2016, 6, 29105. [Google Scholar]
- Zinedine, A.; Soriano, J.M.; Molto, J.C.; Manes, J. Review on the toxicity, occurrence, metabolism, detoxification, regulations and intake of zearalenone: An oestrogenic mycotoxin. Food Chem. Toxicol. 2007, 45, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shier, W.T.; Shier, A.C.; Xie, W.; Mirocha, C.J. Structure-activity relationships for human estrogenic activity in zearalenone mycotoxins. Toxicon 2001, 39, 1435–1438. [Google Scholar] [CrossRef]
- Takahashi-Ando, N.; Kimura, M.; Kakeya, H.; Osada, H.; Yamaguchi, I. A novel lactonohydrolase responsible for the detoxification of zearalenone: Enzyme purification and gene cloning. Biochem. J. 2002, 365, 1–6. [Google Scholar]
- Takahashi-Ando, N.; Tokai, T.; Hamamoto, H.; Yamaguchi, I.; Kimura, M. Efficient decontamination of zearalenone, the mycotoxin of cereal pathogen, by transgenic yeasts through the expression of a synthetic lactonohydrolase gene. Appl. Microbiol. Biotechnol. 2005, 67, 838–844. [Google Scholar] [PubMed]
- Riken. Zearalenone-Detoxifying Enzyme Gene and Transformant Having the Gene Transferred Thereinto. World Patent 2003080842, 25 March 2003. [Google Scholar]
- Novozymes A/S. Detoxification of Feed Products. World Patent 2009109607, 5 March 2009. [Google Scholar]
- Yu, Y.; Wu, H.; Tang, Y.; Qiu, L. Cloning, expression of a peroxiredoxin gene from Acinetobacter sp. SM04 and characterization of its recombinant protein for zearalenone detoxification. Microbiol. Res. 2012, 167, 121–126. [Google Scholar] [PubMed]
- Tang, Y.; Xiao, J.; Chen, Y.; Yu, Y.; Xiao, X.; Yu, Y.; Wu, H. Secretory expression and characterization of a novel peroxiredoxin for zearalenone detoxification in Saccharomyces cerevisiae. Microbiol. Res. 2013, 168, 6–11. [Google Scholar]
- Altalhi, A.D.; El-Deeb, B. Localization of zearalenone detoxification gene(s) in pZEA-1 plasmid of Pseudomonas putida ZEA-1 and expressed in Escherichia coli. J. Hazard. Mater. 2009, 161, 1166–1172. [Google Scholar] [CrossRef]
- Patel, A.K.; Singhania, R.R.; Pandey, A. Novel enzymatic processes applied to the food industry. Curr. Opin. Food Sci. 2016, 7, 64–72. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Biores. Technol. 2002, 83, 1–11. [Google Scholar]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef]
- Baldino, L.; Cardea, S.; Reverchon, E. Supercritical assisted enzymatic membranes preparation, for active packaging applications. J. Memb. Sci. 2014, 453, 409–418. [Google Scholar]
- Regulation (EC) No 1332/2008 of the European Parliament and of the Council of 16 December 2008 on food enzymes and amending Council Directive 83/417/EEC, Council Regulation (EC) No 1493/1999, Directive 2000/13/EC, Council Directive 2001/112/EC and Regulation (EC) No 258/97.
- Osma, J.F.; Toca-Herrera, J.L.; Rodrıguez-Couto, S. Uses of Laccases in the Food Industry. Enzym. Res. 2010, 2010, 918761. [Google Scholar] [CrossRef]
- Gassara-Chatti, F.; Brar, S.K.; Ajila, C.M.; Verma, M.; Tyagi, R.D.; Valero, J.R. Encapsulation of ligninolytic enzymes and its application in clarification of juice. Food Chem. 2013, 137, 18–24. [Google Scholar]
- Lettera, V.; Pezzella, C.; Cicatiello, P.; Piscitelli, A.; Giacobelli, V.G.; Galano, E.; Amoresano, A.; Sannia, G. Efficient immobilization of a fungal laccase and its exploitation in fruit juice clarification. Food Chem. 2016, 196, 1272–1278. [Google Scholar]
- Clickner, F.H.; Follwell, E.H. Application of ‘Protozyme’ (Aspergillus orizae) to poultry feeding. Poult. Sci. 1925, 5, 241–247. [Google Scholar] [CrossRef]
- Menezes-Blackburn, D.; Greiner, R. Enzymes used in animal feed: Leading technologies and forthcoming developments. In Functional Polymers in Food Science: From Technology to Biology, Volume 2: Food Processing; Cirillo, G., Spizzirri, U.G., Iemma, F., Eds.; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on Additives for use in Animal Nutrition. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32003R1831&from=EN (accessed on 17 November 2016).
- Beaman, K.R.; Lilly, K.G.S.; Gehring, C.K.; Turk, P.J.; Moritz, J.S. Influence of pelleting on the efficacy of an exogenous enzyme cocktail using broiler performance and metabolism. J. Appl. Poult. Res. 2012, 21, 744–775. [Google Scholar] [CrossRef]
- Novo Nordisk A/S. Enzyme-Containing Granules and Process for the Production Thereof. World Patent 1997039116, 14 April 1997. [Google Scholar]
- Xiros, C.; Topakas, E.; Christakopoulos, P. Hydrolysis and fermentation for cellulosic ethanol production. WIREs Energy Environ. 2013, 2, 633–654. [Google Scholar] [CrossRef]
- Kudanga, T.; Le Roes-Hill, M. Laccase applications in biofuels production: Current status and future prospects. Appl. Microbiol. Biotechnol. 2014, 98, 6525–6542. [Google Scholar]
- Jurado, M.; Prieto, A.; Martínez-Alcalá, A.; Martínez, A.T.; Martínez, M.J. Laccase detoxification of steam-exploded wheat straw for second generation bioethanol. Bioresour. Technol. 2009, 100, 6378–6384. [Google Scholar] [PubMed]
- Christy, P.M.; Divya, D.; Gopinath, L.R. A review on anaerobic decomposition and enhancement of biogas production through enzymes and microorganisms. Renew. Sustain. Energy Rev. 2014, 34, 167–173. [Google Scholar] [CrossRef]
- Parawira, W. Enzyme research and applications in biotechnological intensification of biogas production. Crit. Rev. Biotechnol. 2012, 32, 172–186. [Google Scholar]
- Effenberger, M.; Lebuhn, M.; Gronauer, A. Fermentermanagement-Stabiler Prozess bei NawaRo-Anlagen. Biogas Wandel 2007, 16, 99–105. [Google Scholar]
- Storm, I.M.L.D.; Sørensen, J.L.; Rasmussen, R.R.; Nielsen, K.F.; Thrane, U. Mycotoxins in silage. Stewart Posthar. Rev. 2008, 1–12. [Google Scholar] [CrossRef]
- Salati, S.; D’Imporzano, G.; Panseri, S.; Pasquale, E.; Adani, F. Degradation of aflatoxin b1 during anaerobic digestion and its effect on process stability. Int. Biodeter. Biodegrad. 2014, 94, 19–23. [Google Scholar]
- De Gelder, L.; Audenaert, K.; Willems, B.; Schelfhout, K.; De Saeger, S.; De Boevre, M. Biodegradability of Mycotoxins during Anaerobic Digestion, Abstract in De Saeger, S.; Audenaert, K.; Croubels, S. Report from the 5th International Symposium on Mycotoxins and Toxigenic Moulds: Challenges and Perspectives (MYTOX) Held in Ghent, Belgium, May 2016. Toxins 2016, 8, 146. [Google Scholar]
- Rintala, J.; Ahring, B.K. A two-stage thermophilic anaerobic process for the treatment of source sorted household solid waste. Biotechnol. Lett. 1994, 16, 1097–1102. [Google Scholar] [CrossRef]
- Binner, R.; Menath, V.; Huber, H.; Thomm, M.; Bischof, F.; Schmack, D.; Reuter, M. Comparative study of stability and half-life of enzymes and enzyme aggregates implemented in anaerobic biogas processes. Biomass Convers. Biorefin. 2011, 1, 1–8. [Google Scholar] [CrossRef]
- Dzuman, Z.; Stranska-Zachariasova, M.; Vaclavikova, M.; Tomaniova, M.; Veprikova, Z.; Slavikova, P.; Hajslova, J. Fate of Free and conjugated mycotoxins within the production of distiller’s dried grains with solubles (DDGS). J. Agric. Food Chem. 2016, 64, 5085–5092. [Google Scholar] [CrossRef]
- Wu, F.; Munkvold, G. Mycotoxins in ethanol co-products: Modeling economic impacts on the livestock industry and management strategies. J. Agric. Food Chem. 2008, 56, 3900–3911. [Google Scholar]
- Nathanail, A.V.; Gibson, B.; Han, L.; Peltonen, K.; Ollilainen, V.; Jestoi, M.; Laitila, A. The lager yeast Saccharomyces pastorianus removes and transforms Fusarium trichothecene mycotoxins during fermentation of brewer’s wort. Food Chem. 2016, 203, 448–455. [Google Scholar] [CrossRef]
- Boeira, L.; Bryce, J.; Stewart, G.; Flannigan, B. Inhibitory effect of Fusarium mycotoxins on growth of brewing yeasts. 1 zearalenone and fumonisin B1. J. Inst. Brew. 1999, 105, 366–375. [Google Scholar] [CrossRef]
- Commission Regulation (EU) No 68/2013 of 16 January 2013 on the Catalogue of Feed Materials. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32013R0068&from=EN (accessed on 17 November 2016).
- Feeds Regulations (SOR/83-593), Feeds act. Government of Canada, 1983. Available online: http://laws-lois.justice.gc.ca/PDF/SOR-83-593.pdf (accessed on 17 November 2016).
- Code of Federal Regulations (CFR), Title 21, Sections 573 Food Additives Permitted in Feed and Drinking Water of Animals. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=573&showFR=1 (accessed on 17 November 2016).
- Regulation (EC) No 1069/2009 of the European Parliament and of the Council of 21 October 2009 Laying down Health Rules as Regards Animal by-Products and Derived Products Not Intended for Human Consumption and Repealing Regulation (EC) No 1774/2002 (Animal by-products Regulation). Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32009R1069&from=EN (accessed on 17 November 2016).
- Sosa, M.A.; Chovau, S.; Van der Bruggen, B.; Espinosa, J. Ethanol production from corn contaminated with fumonisins: A preliminary economic analysis including novel processing alternatives. Ind. Eng. Chem. Res. 2013, 52, 7504–7513. [Google Scholar] [CrossRef]
- Liu, K. Chemical Composition of Distillers Grains, a Review. J. Agric. Food Chem. 2011, 59, 1508–1526. [Google Scholar] [CrossRef]
- Zhejiang University. A Method for Reducing the Content of Patulin in Apple Juice Concentrate. Chinese Patent 103859016, 26 February 2014. [Google Scholar]
- Pioneer Hi-Bred International, Inc. Beauvericin Detoxification Method Using Bacteria. U.S. Patent 6126934, 29 January 1998. [Google Scholar]
- Pioneer Hi-Bred International, Inc. Moniliformin detoxification compositions and methods. Canadian Patent 2272554, 12 November 1997. [Google Scholar]
- Liuzzi, V.C.; Fanelli, F.; Tristezza, M.; Haidukowski, M.; Picardi, E.; Manzari, C.; Lionetti, C.; Grieco, F.; Logrieco, A.F.; Thon, M.R.; et al. Transcriptional analysis of Acinetobacter sp. neg1 capable of degrading ochratoxin A. Front. Microbiol. 2017, 7, 2162. [Google Scholar]
- Fanelli, F.; Chiara, M.; Liuzzi, V.C.; Haidukowski, M.; Tristezza, M.; Manzari, C.; D’Erchia, A.M.; Pesole, G.; Horner, D.S.; Mulè, G. Draft genome sequence of Acinetobacter sp. neg1 capable of degrading ochratoxin A. FEMS Microbiol. Lett. 2015, 362, 7. [Google Scholar] [CrossRef]
- USDA Economic Research Service. Adoption of Genetically Engineered Crops in the U.S. USDA: Washington, DC, USA, 2011. Available online: https://www.ers.usda.gov/data-products/adoption-of-genetically-engineered-crops-in-the-us.aspx (accessed on 17 November 2016). [Google Scholar]
- Hohn, T.M.; Peters, C.; Salmeron, J. Trichothecene-Resistant Transgenic Plants. U.S. Patent 20020162136, 12 February 2002. [Google Scholar]
- Kimura, M.; Kaneko, I.; Komiyama, M.; Takatsuki, A.; Koshino, H.; Yoneyama, K.; Yamaguchi, I. Trichothecene 3-O-acetyltransferase protects both the producing organism and transformed yeast from related mycotoxins. J. Biol. Chem. 1998, 273, 1654–1661. [Google Scholar]
- Kimura, M.; Shingu, Y.; Yoneyama, K.; Yamaguchi, I. Features of Tri101, the trichothecene 3-O-acetyltransferase gene, related to the self-defense mechanism in Fusarium graminearum. Biosci. Biotech. Bioch. 1998, 62, 1033–1036. [Google Scholar] [CrossRef]
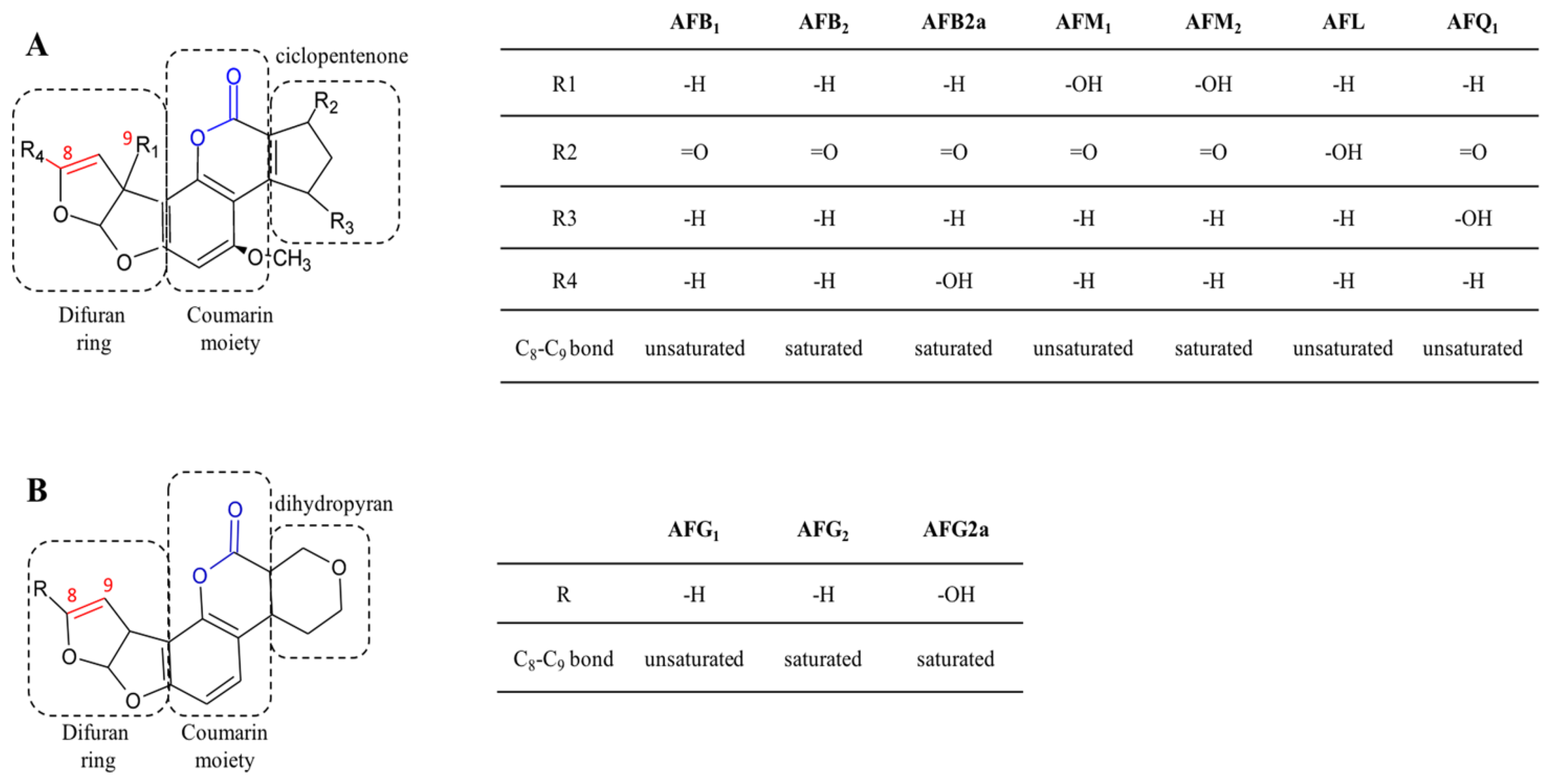
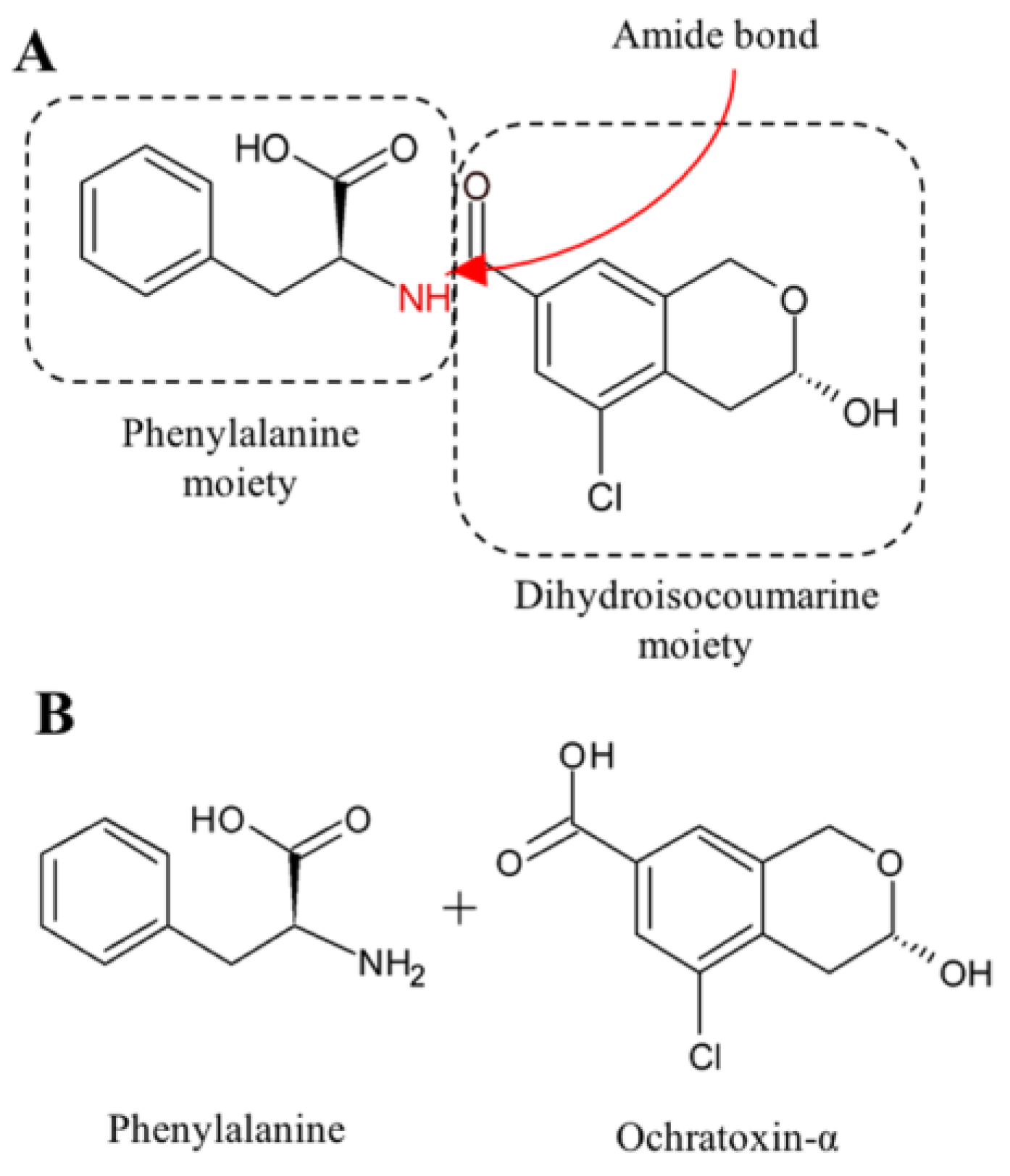
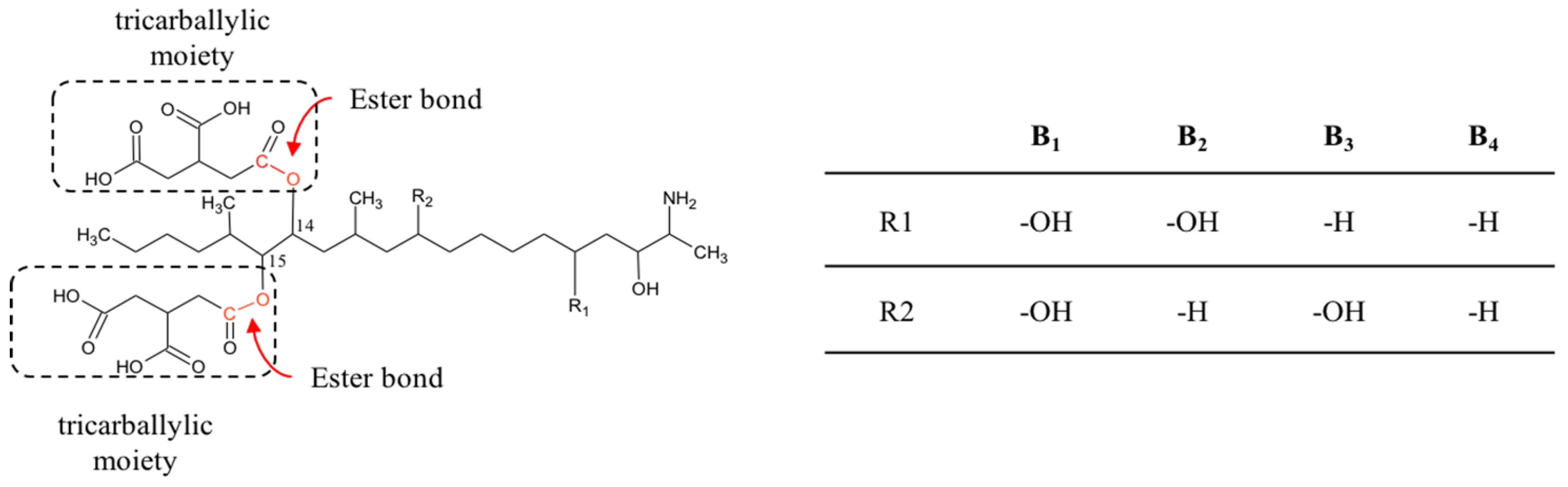
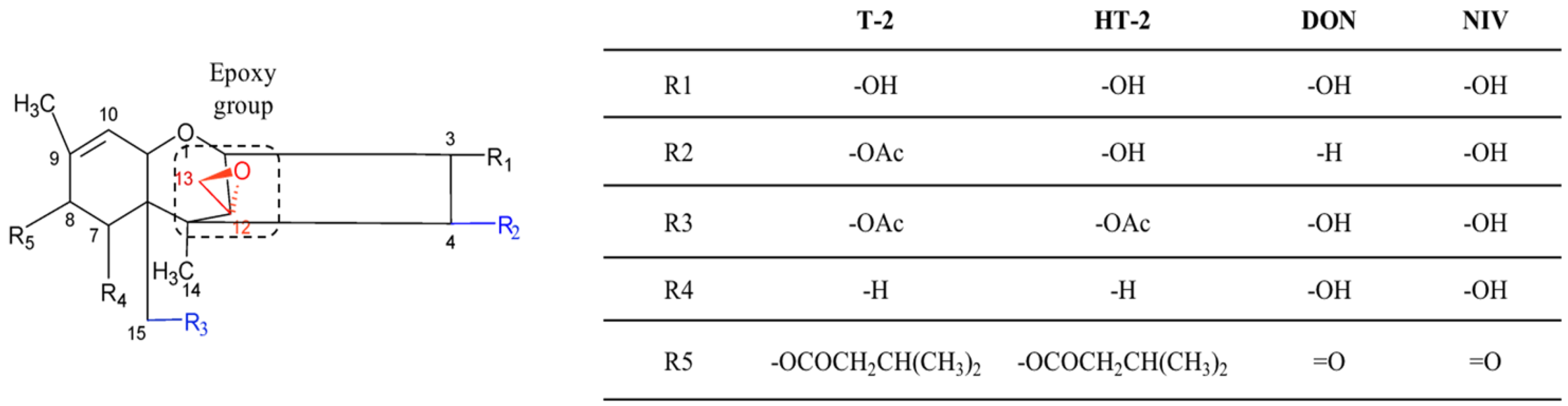

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Accession/EC | Producing Organism | AF Target | Toxin Concentration | In Vitro/In Matrix Degrading Conditions | In Vitro/In Matrix Degradation | Toxicity/Mutagenicity Test | Reference |
|---|---|---|---|---|---|---|---|---|
| aflatoxin oxidase enzyme (AFO) | EC 1.1 | Armillariella tabescens | AFB1 | 0.05 µg/mL |
|
|
| [51] |
| peroxidase | EC 1.11.1.7 | horseradish (Armoracia rusticana) | AFB1 | 312 µg/mL |
| 42.2% | reduced toxicity on Bacillus megaterium | [52] |
| 440 µg/mL |
| 41.1% | ||||||
| laccase | EC 1.10.3.2 | Trametes versicolor (commercial enzyme from Sigma-Aldrich, Missouri, U.S.) | AFB1 | 1.40 µg/mL |
| 87% | reduced mutagenicity on Salmonella typhimurium TA 100 | [53] |
| laccase | EC 1.10.3.2 | Streptomyces coelicor | AFB1 | 9.36 µg/mL |
| 100% | n.p. | [54] |
| F420H2-dependent reductases | E.C. 1.5.8 | Mycobacterium smegmatis | AFB1 AFB2 AFG1 AFG2 | n.p. | n.p. | n.p. | n.p. | [55] |
| Mn peroxidase | EC 1.11.1.7 | Pleurotus ostreatus | AFB1 | 0.31 µg/mL |
| 90% | n.p. | [56] |
| aflatoxin degradation enzyme | n.p. | Pleurotus ostreatus | AFB1 | 5 µg/mL |
| n.q. | n.p. | [50] |
| myxobacteria aflatoxin degrading enzyme (MADE) | n.p. | Myxococcus fulvus ANSM068 | AFB1 | 0.1 µg/mL |
| 72% with culture filtrates | n.p. | [57] |
| AFG1 | 97% | |||||||
| AFM1 | 96% | |||||||
| laccase (lac2) | EC 1.10.3.2 | Pleurotus pulmonarius (ITEM 17144) | AFB1 | 1 µg/mL |
| 90% | n.p. | [58] |
| AFM1 | 0.05 µg/mL | 100% | ||||||
| Ery4 | CAO79915.1/EC 1.10.3.2 | Pleurotus eryngii (PS419) | AFM1 | 0.05 µg/mL |
| 100% | n.p. | [59] |
|
| Enzyme | Producing Organism | Accession/EC | Toxin Concentration | Degrading Conditions | Degradation | Reference |
|---|---|---|---|---|---|---|
| carboxylesterase and aminotransferase | Sphingomonas sp. ATCC55552 | E.C. 3.1.1, E.C. 2.6.1 | 1000 µg/mL |
| 100% | [89] |
| carboxylesterase B and aminotransferase | Sphingopyxis sp. MTA144 | E.C. 3.1.1, E.C. 2.6.1/ FJ426269.1 | 3.6 µg/mL |
| 100% | [80] |
| fumonisin esterase | Sphingopyxis sp. MTA144 | E.C. 3.1.1.87 | 60 µg/mL |
| 100% conversion to HFB1 | [91] |
| Enzyme | Accession/EC | Producing Organism | Trichothecene Target | Toxin Concentration | Degrading Conditions | In Vitro Degradation | Toxicity-Mutagenicity Test | Reference |
|---|---|---|---|---|---|---|---|---|
| cytochrome P450 system (Ddna + Kdx + KdR) | E.C. 1.14 AB744215.1 AB744217.1 | Sphingomonas sp. strain KSM1 | DON | 99.86 µg/mL |
| 100% after 3 days | reduced phytotoxicity to wheat | [112] |
| NIV | 105.25 µg/mL | 100% after 5 days | ||||||
| UDP-glycosyltransferase | AC006282 | Arabidopsis thaliana | DON | n.p. | n.p. | n.p. | increased resistance in transgenic Arabidopsis | [113] |
| Enzyme | Producing Organism | EC | Toxin Concentration | Degrading Conditions | Degradation | Toxicity-Mutagenicity Test | Reference |
|---|---|---|---|---|---|---|---|
| laccase | Trametes versicolor (commercial enzyme from Sigma-Aldrich) | EC 1.10.3.2 | 6.2 × 10−4 µg/mL |
| up to 58 % | n.p. | [67] |
| laccase | Streptomyces coelicolor | EC 1.10.3.2 | 9.36 µg/mL |
| 100% | n.p. | [120] |
| lactono hydrolase | Clonostachys rosea | E.C. 3.1.1 | 2 µg/mL |
| 100% | n.p. | [117,118] |
| 2cys-peroxiredoxin | Acinetobacter sp. SM04 | EC 1.11.1.15 | 20 µg/mL |
| up to 95% | reduced MCF-7 cells proliferation by 75% | [121] |
| 1 µg/mL |
| up to 90% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loi, M.; Fanelli, F.; Liuzzi, V.C.; Logrieco, A.F.; Mulè, G. Mycotoxin Biotransformation by Native and Commercial Enzymes: Present and Future Perspectives. Toxins 2017, 9, 111. https://doi.org/10.3390/toxins9040111
Loi M, Fanelli F, Liuzzi VC, Logrieco AF, Mulè G. Mycotoxin Biotransformation by Native and Commercial Enzymes: Present and Future Perspectives. Toxins. 2017; 9(4):111. https://doi.org/10.3390/toxins9040111
Chicago/Turabian StyleLoi, Martina, Francesca Fanelli, Vania C. Liuzzi, Antonio F. Logrieco, and Giuseppina Mulè. 2017. "Mycotoxin Biotransformation by Native and Commercial Enzymes: Present and Future Perspectives" Toxins 9, no. 4: 111. https://doi.org/10.3390/toxins9040111
APA StyleLoi, M., Fanelli, F., Liuzzi, V. C., Logrieco, A. F., & Mulè, G. (2017). Mycotoxin Biotransformation by Native and Commercial Enzymes: Present and Future Perspectives. Toxins, 9(4), 111. https://doi.org/10.3390/toxins9040111