
Functional Genomic Analysis of Aspergillus flavus Interacting with Resistant and Susceptible Peanut

Abstract
:
1. Introduction
2. Results
2.1. RNA-seq and Transcriptome Profiles of the A. flavus

{kind=link}
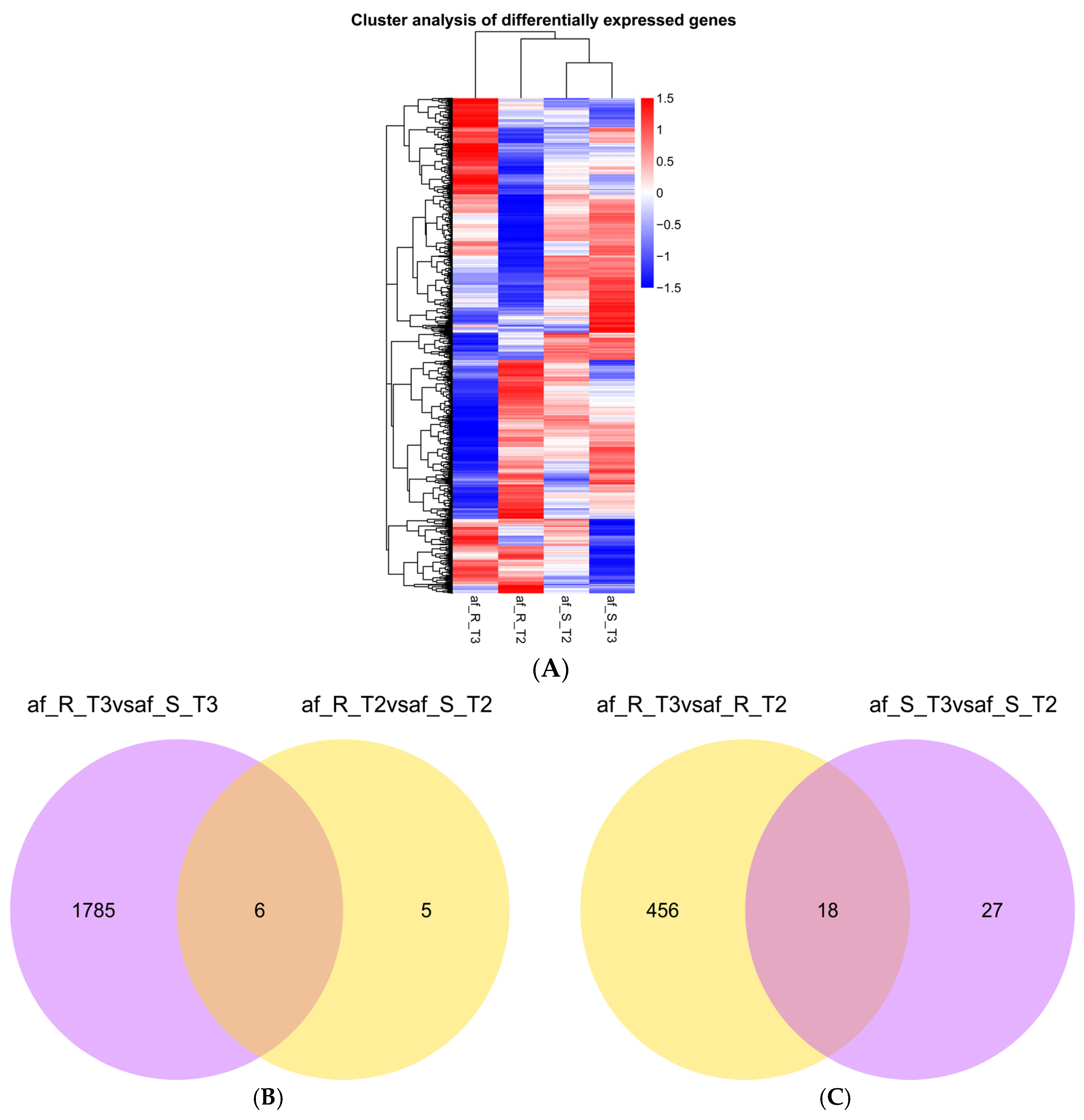{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Clean Bases (Gb) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) | rRNA (%) |
|---|---|---|---|---|---|---|---|---|
| af1_R_T1 | 57,599,682 | 55,753,432 | 5.58 | 0.03 | 97.25 | 91.78 | 45.18 | 2.07 |
| af2_R_T1 | 54,642,766 | 52,827,190 | 5.28 | 0.06 | 97.32 | 91.95 | 44.87 | 1.97 |
| af1_R_T2 | 65,478,414 | 63,861,410 | 6.38 | 0.06 | 97.65 | 92.74 | 44.57 | 2.43 |
| af2_R_T2 | 58,356,648 | 55,965,182 | 5.60 | 0.07 | 96.51 | 90.11 | 45.17 | 1.80 |
| af1_R_T3 | 67,147,990 | 63,523,784 | 6.36 | 0.09 | 95.81 | 88.30 | 50.08 | 3.80 |
| af2_R_T3 | 56,259,592 | 53,486,954 | 5.34 | 0.10 | 95.77 | 87.83 | 50.04 | 3.20 |
| af1_S_T1 | 59,822,028 | 58,024,886 | 5.80 | 0.06 | 97.27 | 91.85 | 44.23 | 1.77 |
| af2_S_T1 | 61,482,172 | 59,691,186 | 5.96 | 0.06 | 97.29 | 91.89 | 44.34 | 2.33 |
| af1_S_T2 | 55,081,582 | 52,844,526 | 5.28 | 0.08 | 96.26 | 89.17 | 45.10 | 1.57 |
| af2_S_T2 | 58,420,538 | 56,092,690 | 5.60 | 0.08 | 96.21 | 89.01 | 45.49 | 1.87 |
| af1_S_T3 | 56,907,234 | 54,518,226 | 5.46 | 0.08 | 96.20 | 89.00 | 45.75 | 2.47 |
| af2_S_T3 | 50,079,030 | 48,035,682 | 4.80 | 0.06 | 97.20 | 91.23 | 45.53 | 3.90 |
2.2. Identification of Differentially Expressed Genes
| Comparison | Number of DEGs | ||
|---|---|---|---|
| Up-Regulated | Down-Regulated | Total | |
| af_R_T2 vs. af_S_T2 | 9 | 2 | 11 |
| af_R_T3 vs. af_S_T3 | 647 | 1144 | 1791 |
| af_R_T3 vs. af_R_T2 | 317 | 157 | 474 |
| af_S_T3 vs. af_S_T2 | 34 | 11 | 45 |
| total | 1926 | ||
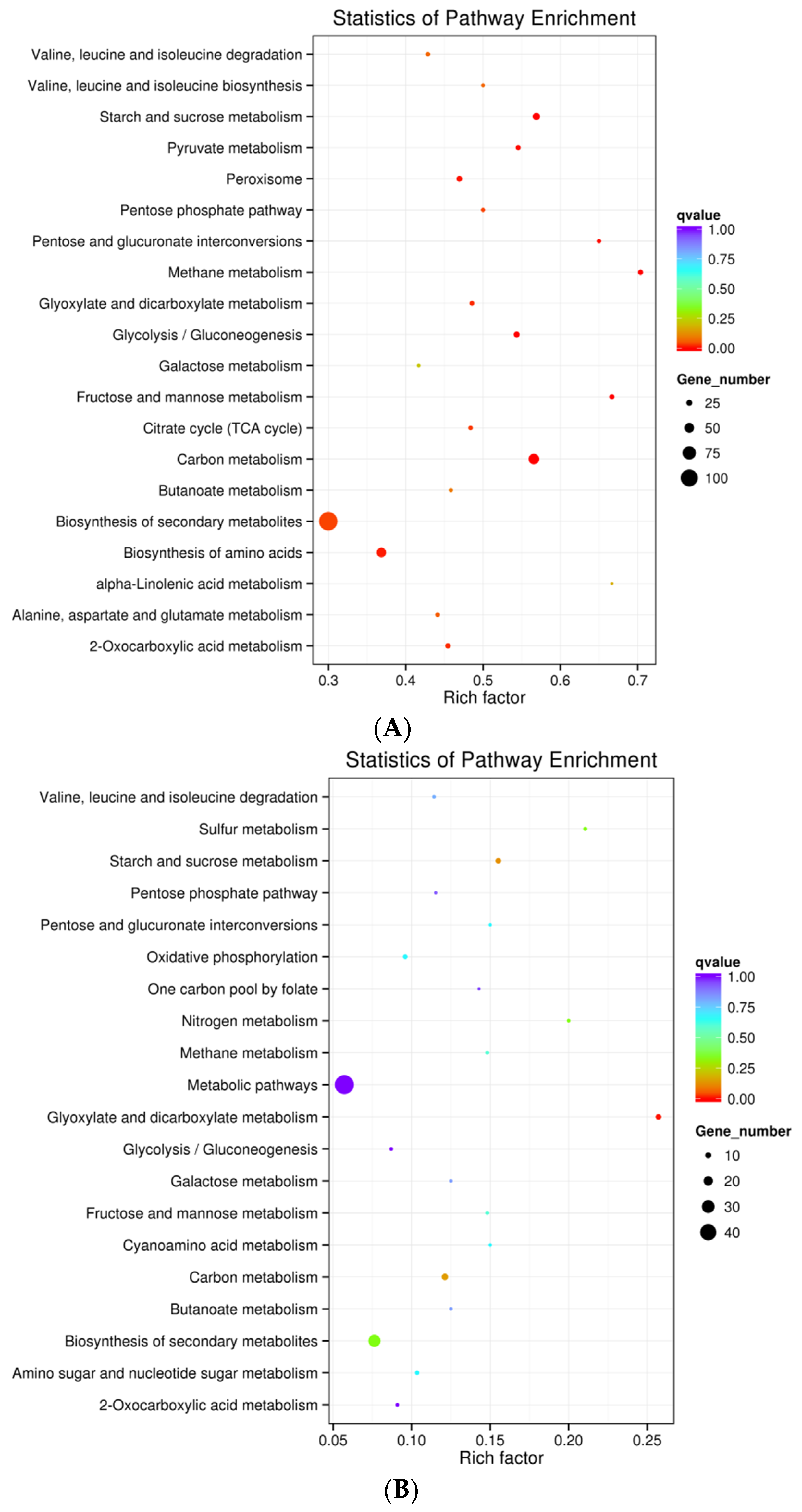
2.3. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Enrichment Analysis of DEGs
2.4. Expression Analysis of Development-Related Genes and Secondary Metabolism Gene Clusters in A. flavus
| Gene Name | Log2 (fold change) | Description | |||
|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | ||
| Mycelia | |||||
| AFLA_105900 | / | −2.6 | / | / | feruloyl esterase C |
| AFLA_110270 | / | −2.3 | / | / | feruloyl esterase B-1 |
| AFLA_128870 | / | −1.6 | / | / | feruloyl esterase B-2 |
| AFLA_115930 | / | −0.8 | / | / | terrelysin |
| AFLA_023340 | / | −2.3 | / | / | pectinesterase A |
| AFLA_020000 | / | −3.0 | / | / | pectinesterase |
| AFLA_104300 | / | −1.1 | / | / | alpha-N-arabinofuranosidase B |
| AFLA_062930 | / | −1.8 | / | 1.6 | alpha-N-arabinofuranosidase 2 |
| AFLA_063490 | / | −2.1 | / | / | alpha-L-arabinofuranosidaseaxhA |
| AFLA_070020 | / | −1.3 | / | / | alpha-N-arabinofuranosidase C |
| AFLA_089770 | / | −3.8 | / | / | alpha-N-arabinofuranosidase A |
| AFLA_038730 | / | −1.7 | / | / | mannan endo-1,4-beta-mannosidase E |
| AFLA_128610 | / | −1.2 | / | / | beta-mannosidase A |
| AFLA_117830 | / | −0.7 | / | / | beta-mannosidase B |
| AFLA_086360 | / | −2.8 | / | / | exopolygalacturonase C |
| AFLA_131770 | / | −1.2 | / | / | exopolygalacturonase X |
| AFLA_096690 | / | −3.3 | / | / | galacturan 1,4-alpha-galacturonidase A |
| Conidia | |||||
| AFLA_083110 | / | −2.0 | 4.2 | / | conidiation-specific protein Con-10 |
| AFLA_112100 | / | −0.4 | / | / | conidiation-specific protein Con-8 |
| AFLA_044790 | / | −3.3 | 3.7 | / | conidiation-specific family protein |
| AFLA_044800 | / | −1.8 | 6.5 | / | conidiation protein Con-6 |
| AFLA_098380 | / | −0.6 | 1.7 | / | conidial hydrophobinRodA/RolA |
| AFLA_031340 | / | −0.6 | / | / | transcription factor AtfA |
| AFLA_006170 | / | / | 2.2 | / | polyketidesynthetasePksP |
| Aflatoxin | |||||
| AFLA_139160 | / | −0.5 | / | / | aflX/ordB/monooxygenase/oxidase |
| AFLA_139210 | / | / | 1.2 | / | aflP/omtA/O-methyltransferase A |
| AFLA_139260 | / | / | 1.2 | / | aflG/avnA/P450 monooxygenase |
| AFLA_139270 | / | −0.6 | / | / | aflNa/hypD/hypothetical protein |
| AFLA_139280 | / | / | 1.2 | / | aflN/verA/monooxygenase |
| AFLA_139380 | / | / | 1.2 | / | aflA/fas-2/fatty acid synthase alpha subunit |
| AFLA_139390 | / | −1.4 | / | / | aflD/nor-1/reductase |
| AFLA_139400 | / | / | 1.1 | / | aflCa/hypC/hypothetical protein |
| AFLA_139410 | / | / | 1.2 | / | aflC/pksA/polyketide synthase |
| AFLA_112820 | / | −1.4 | / | / | toxin biosynthesis ketoreductase, putative |
| AFLA_050450 | / | −1.2 | / | / | toxin biosynthesis protein |
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Aspergillus flavus Strain and Culture Conditions
5.2. RNA Isolation and cDNA Library Construction
5.3. Mapping of Reads to the Reference Genome and Quantification of Gene Expression
5.4. Identification and Enrichment Analysis of Differentially Expressed Genes
Supplementary Materials
Acknowledgments
Author Contributions
Conflict of Interest
References
- Duran, R.M.; Gregersen, S.; Smith, T.D.; Bhetariya, P.J.; Cary, J.W.; Harris-Coward, P.Y.; Mattison, C.P.; Grimm, C.; Calvo, A.M. The role of Aspergillus flavus vea in the production of extracellular proteins during growth on starch substrates. Appl. Microbiol. Biot. 2014, 98, 5081–5094. [Google Scholar] [CrossRef] [PubMed]
- Amaike, S.; Keller, N.P. Aspergillus flavus. Annu. Rev. Phytopathol. 2011, 49, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Anjum, F.M.; Butt, M.S.; Imran, P.; Randhawa, M.A.; Farhan, S.; Khalid, W. Aflatoxins: Biosynthesis, occurrence, toxicity, and remedies. Crit Rev. Food Sci. 2013, 53, 862–874. [Google Scholar]
- Kobuchi Nakai, V.; de Oliveira Rocha, L.; Goncalez, E.; Fonseca, H.; Moises Marcos Ortega, E.; Correa, B. Distribution of fungi and aflatoxins in a stored peanut variety. Food Chem. 2008, 106, 285–290. [Google Scholar] [CrossRef]
- Wang, H.; Lei, Y.; Yan, L.; Cheng, K.; Dai, X.; Wan, L.; Guo, W.; Cheng, L.; Liao, B. Deep sequencing analysis of transcriptomes in Aspergillus flavus in response to resveratrol. BMC Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cary, J.; Han, Z.; Yin, Y.; Lohmar, J.; Shantappa, S.; Harris-Coward, P.; Mack, B.; Ehrlich, K.; Wei, Q.; Arroyo-Manzanares, N. Transcriptome analysis of Aspergillus flavus reveals vea-dependent regulation of secondary metabolite gene clusters, including the novel aflavarin cluster. Eukaryot. Cell. 2015, 14, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.M.; Barros, G.G.; Palacios, S.A.; Chulze, S.N.; Battilani, P. Review on pre- and post-harvest management of peanuts to minimize aflatoxin contamination. Food Res. Int. 2014, 62, 11–19. [Google Scholar] [CrossRef]
- Bhatnagar-Mathur, P.; Sunkara, S.; Bhatnagar-Panwar, M.; Waliyar, F.; Sharma, K.K. Biotechnological advances for combating Aspergillus flavus and aflatoxin contamination in crops. Plant Sci. 2015, 234, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Liang, Y.; Zhang, J.; Chen, Z.; Liu, C.-M. Autoxidated linolenic acid inhibits aflatoxin biosynthesis in Aspergillus flavus via oxylipin species. Fungal Genet. Biol. 2014, 81, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, J.; Lei, Y.; Yan, L.; Wang, S.; Jiang, H.; Ren, X.; Lou, Q.; Liao, B. Relationship of resveratrol content and resistance to aflatoxin accumulation caused by Aspergillus flavus in peanut seeds. Acta Agron. Sin. 2012, 38, 1875–1883. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, S.; Liu, C.; Chen, F.; Wang, T. Proteomic analysis reveals an aflatoxin-triggered immune response in cotyledons of Arachis hypogaea infected with Aspergillus flavus. J. Proteome Res. 2012, 11, 2739–2753. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, E.H.; Chen, X.P.; Li, L.; Liang, X.Q. Identification of seed proteins associated with resistance to pre-harvested aflatoxin contamination in peanut (Arachis hypogaea L.). BMC Plant Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhuang, W.; Tang, R.; Zhang, X.; Shan, S.; Jiang, H.; Huang, J. Peanut aflatoxin and genomics research in China: Progress and perspectives. Peanut Sci. 2009, 36, 21–28. [Google Scholar] [CrossRef]
- Guo, B.; Chen, X.; Dang, P.; Scully, B.T.; Liang, X.; Holbrook, C.C.; Yu, J.; Culbreath, A.K. Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection. BMC Dev. Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, M.; Guo, B. Resistance mechanisms to Aspergillus flavus infection and aflatoxin contamination in peanut (Arachis hypogaea). Plant Pathol. J. 2006, 5, 115–124. [Google Scholar]
- Liang, X.; Holbrook, C.; Lynch, R.; Guo, B. Β-1, 3-glucanase activity in peanut seed (Arachis hypogaea) is induced by inoculation with Aspergillus flavus and copurifies with a conglutin-like protein. Phytopathology 2005, 95, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Chen, Z.-Y.; Lee, R.D.; Scully, B.T. Drought stress and preharvest aflatoxin contamination in agricultural commodity: Genetics, genomics and proteomics. J. Integr. Plant Biol. 2008, 50, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- DeLucca, A.J., II; Palmgren, M.S.; Daigle, D.J. Depression of aflatoxin production by flavonoid-type compounds from peanut shells. Phytopathology 1987, 77, 1560–1563. [Google Scholar] [CrossRef]
- Burow, G.B.; Nesbitt, T.C.; Dunlap, J.; Keller, N.P. Seed lipoxygenase products modulate Aspergillus mycotoxin biosynthesis. Mol. Plant Microb. Interact. 1997, 10, 380–387. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, U97–U99. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Q.; Zhao, X.-X.; Zhi, Q.-Q.; Zhao, M.; He, Z.-M. Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet. Biol. 2013, 56, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Guo, G.; Wang, C.; Lin, Y.; Wang, X.; Zhao, M.; Guo, Y.; He, M.; Zhang, Y.; Pan, L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38, 5075–5087. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.J.; Rogers, J.; Bahler, J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Linz, J.E.; Wee, J.; Roze, L.V. Aspergillus parasiticus su-1 genome sequence, predicted chromosome structure, and comparative gene expression under aflatoxin-inducing conditions: Evidence that differential expression contributes to species phenotype. Eukaryot. Cell. 2014, 13, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Mack, B.; Yu, J.; Ehrlich, K.C. Transcriptomic profiles of Aspergillus flavus CA42, a strain that produces small sclerotia, by decanal treatment and after recovery. Fungal Genet. Biol. 2014, 68, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Khaldi, N.; Seifuddin, F.T.; Turner, G.; Haft, D.; Nierman, W.C.; Wolfe, K.H.; Fedorova, N.D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010, 47, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.R.; Kale, S.P.; Bhatnagar, D.; Yu, J.; Ehrlich, K.C. Expression profiling of non-aflatoxigenic Aspergillus parasiticus mutants obtained by 5-azacytosine treatment or serial mycelial transfer. Toxins 2011, 3, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, M.; Ludovici, M.; Sanzani, S.M.; Ippolito, A.; Cigliano, R.A.; Sanseverino, W.; Scarpari, M.; Scala, V.; Fanelli, C.; Reverberi, M. Menadione-induced oxidative stress re-shapes the oxylipin profile of Aspergillus flavus and its lifestyle. Toxins 2015, 7, 4315–4329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, Z.; Zhong, H.; Wang, S.; Yang, W.; Liu, Y.; Wang, S. RNA-seq based transcriptome analysis of aflatoxigenic Aspergillus flavus in response to water activity. Toxins 2014, 6, 3187–3207. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fedorova, N.D.; Montalbano, B.G.; Bhatnagar, D.; Cleveland, T.E.; Bennett, J.W.; Nierman, W.C. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-seq. Fems Microbiol. Lett. 2011, 322, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Aspergillus flavus genome database. Available online: http://fungi.ensembl.org/Aspergillus_flavus/Info/Annotation/#assembly (accessed on 10 September 2015).
- National Center for Biotechnology Information (NCBI) non-redundant (Nr) protein database. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 14 September 2015).
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Secondary Metabolite Unique Regions Finder. Available online: http://www.jcvi.org/smurf/index.php (accessed on 30 September 2015).
- Amare, M.G.; Keller, N.P. Molecular mechanisms of Aspergillus flavus secondary metabolism and development. Fungal Genet. Biol. 2014, 66, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Manavathu, E.K.; Chandrasekar, P.H. Aspergillus flavus: An emerging non-fumigatus Aspergillus species of significance. Mycoses 2009, 52, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, R.A.; Hawkins, L.K.; Mylroie, J.E.; Oliveira, D.A.; Smith, J.S.; Ozkan, S.; Windham, G.L.; Williams, W.P.; Warburton, M.L. Characterization of the maize chitinase genes and their effect on Aspergillus flavus and aflatoxin accumulation resistance. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Panahirad, S.; Zaare-Nahandi, F.; Mohammadi, N.; Alizadeh-Salteh, S.; Safaie, N. Effects of salicylic acid on Aspergillus flavus infection and aflatoxin B-1 accumulation in pistachio (Pistacia vera L.) fruit. J. Sci. Food Agric. 2014, 94, 1758–1763. [Google Scholar] [CrossRef] [PubMed]
- Astoreca, A.; Vaamonde, G.; Dalcero, A.; Marin, S.; Ramos, A. Abiotic factors and their interactions influence on the co-production of aflatoxin B-1 and cyclopiazonic acid by Aspergillus flavus isolated from corn. Food Microbiol. 2014, 38, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Warburton, M.L.; Williams, W.P.; Windham, G.L.; Murray, S.C.; Xu, W.W.; Hawkins, L.K.; Duran, J.F. Phenotypic and genetic characterization of a maize association mapping panel developed for the identification of new sources of resistance to Aspergillus flavus and aflatoxin accumulation. Crop. Sci. 2013, 53, 2374–2383. [Google Scholar] [CrossRef]
- Nierman, W.C.; Yu, J.J.; Fedorova-Abrams, N.D.; Losada, L.; Cleveland, T.E.; Bhatnagar, D.; Bennett, J.W.; Dean, R.; Payne, G.A. Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.J.; Chang, P.K.; Ehrlich, K.C.; Cary, J.W.; Bhatnagar, D.; Cleveland, T.E.; Payne, G.A.; Linz, J.E.; Woloshuk, C.P.; Bennett, J.W. Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2004, 70, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Roze, L.V.; Hong, S.-Y.; Linz, J.E. Aflatoxin biosynthesis: Current frontiers. Annu. Rev. Food Sci. Technol. 2013, 4, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, M. The information highways of a biotechnological workhorse-signal transduction in Hypocrea jecorina. BMC Genomics 2008. [Google Scholar] [CrossRef] [PubMed]
- Pullan, S.T.; Daly, P.; Delmas, S.; Ibbett, R.; Kokolski, M.; Neiteler, A.; van Munster, J.M.; Wilson, R.; Blythe, M.J.; Gaddipati, S. RNA-sequencing reveals the complexities of the transcriptional response to lignocellulosic biofuel substrates in Aspergillus niger. Fungal Biol. Biotechnol. 2014, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fatima, U.; Senthil-Kumar, M. Plant and pathogen nutrient acquisition strategies. Front. Plant Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jashni, M.K.; Mehrabi, R.; Collemare, J.; Mesarich, C.H.; de Wit, P. The battle in the apoplast: Further insights into the roles of proteases and their inhibitors in plant-pathogen interactions. Front. Plant Sci. 2015. [Google Scholar] [CrossRef]
- Carpita, N.C.; Gibeaut, D.M. Structural models of primary cell walls in flowering plants: Consistency of molecular structure with the physical properties of the walls during growth. Plant J. 1993, 3, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Jia, Q.; Chen, L.; Chen, Q.; Yang, Q. Recombinant expression and inhibition mechanism analysis of pectin methylesterase from Aspergillus flavus. Fems Microbiol. Lett. 2014, 355, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Lebeda, A.; Luhova, L.; Sedlarova, M.; Jancova, D. The role of enzymes in plant-fungal pathogens interactions-review. Z. Pflanz. Pflanz. 2001, 108, 89–111. [Google Scholar]
- Hong, S.-Y.; Linz, J.E. Functional expression and subcellular localization of the aflatoxin pathway enzyme Ver-1 fused to enhanced green fluorescent protein. Appl. Environ. Microb. 2008, 74, 6385–6396. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Linz, J.E. Functional expression and sub-cellular localization of the early aflatoxin pathway enzyme Nor-1 in Aspergillus parasiticus. Fungal Biol. 2009, 113, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Kang, S.; Artymovich, K.A.; Hicks, G.R.; Raikhel, N.V.; Calvo, A.M.; Linz, J.E. A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 19533–19538. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Roze, L.V.; Linz, J.E. A possible role for exocytosis in aflatoxin export in Aspergillus parasiticus. Eukaryot. Cell. 2010, 9, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Yabe, K.; Nakajima, H. Enzyme reactions and genes in aflatoxin biosynthesis. Appl. Microbiol. Biotechnol. 2004, 64, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Minto, R.E.; Townsend, C.A. Enzymology and molecular biology of aflatoxin biosynthesis. Chem. Rev. 1997, 97, 2537–2555. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C.; Li, P.; Scharfenstein, L.; Chang, P.-K. Hypc, the anthrone oxidase involved in aflatoxin biosynthesis. Appl. Environ. Microbiol. 2010, 76, 3374–3377. [Google Scholar] [CrossRef] [PubMed]
- Wicklow, D.T.; Wilson, D.M.; Nelsen, T.C. Survival of Aspergillus flavus sclerotia and conidia buried in soil in Illinois or Georgia. Phytopathology 1993, 83, 1141–1147. [Google Scholar] [CrossRef]
- Lara-Rojas, F.; Sanchez, O.; Kawasaki, L.; Aguirre, J. Aspergillus nidulans transcription factor atfa interacts with the MAPK SakA to regulate general stress responses, development and spore functions. Mol. Microbiol. 2011, 80, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, D.; Asano, Y.; Yamashino, T.; Mizuno, T. Characterization of bZIP-type transcription factor AtfA with reference to stress responses of conidia of Aspergillus nidulans. Biosci. Biotechnol. Biochem. 2008, 72, 2756–2760. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, K.; Jahn, B.; Gehringer, H.; Schmidt, A.; Wanner, G.; Brakhage, A.A. Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med. Microbiol. Immun. 1998, 187, 79–89. [Google Scholar] [CrossRef]
- Bayry, J.; Beaussart, A.; Dufrene, Y.F.; Sharma, M.; Bansal, K.; Kniemeyer, O.; Aimanianda, V.; Brakhage, A.A.; Kaveri, S.V.; Kwon-Chung, K.J.; et al. Surface structure characterization of Aspergillus fumigatusconidia mutated in the melanin synthesis pathway and their human cellular immune response. Infect. Immun. 2014, 82, 3141–3153. [Google Scholar] [CrossRef] [PubMed]
- Aspergillus flavus genome and gene annotation files. Available online: ftp://ftp.ensemblgenomes.org/pub/release-17/fungi/fasta/Aspergillus_flavus/dna/ (accessed on 7 September 2015).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Lei, Y.; Yan, L.; Wan, L.; Ren, X.; Chen, S.; Dai, X.; Guo, W.; Jiang, H.; Liao, B. Functional Genomic Analysis of Aspergillus flavus Interacting with Resistant and Susceptible Peanut. Toxins 2016, 8, 46. https://doi.org/10.3390/toxins8020046
Wang H, Lei Y, Yan L, Wan L, Ren X, Chen S, Dai X, Guo W, Jiang H, Liao B. Functional Genomic Analysis of Aspergillus flavus Interacting with Resistant and Susceptible Peanut. Toxins. 2016; 8(2):46. https://doi.org/10.3390/toxins8020046
Chicago/Turabian StyleWang, Houmiao, Yong Lei, Liying Yan, Liyun Wan, Xiaoping Ren, Silong Chen, Xiaofeng Dai, Wei Guo, Huifang Jiang, and Boshou Liao. 2016. "Functional Genomic Analysis of Aspergillus flavus Interacting with Resistant and Susceptible Peanut" Toxins 8, no. 2: 46. https://doi.org/10.3390/toxins8020046
APA StyleWang, H., Lei, Y., Yan, L., Wan, L., Ren, X., Chen, S., Dai, X., Guo, W., Jiang, H., & Liao, B. (2016). Functional Genomic Analysis of Aspergillus flavus Interacting with Resistant and Susceptible Peanut. Toxins, 8(2), 46. https://doi.org/10.3390/toxins8020046