
Evolution of the Cytolytic Pore-Forming Proteins (Actinoporins) in Sea Anemones


Abstract
:1. Introduction
2. Results
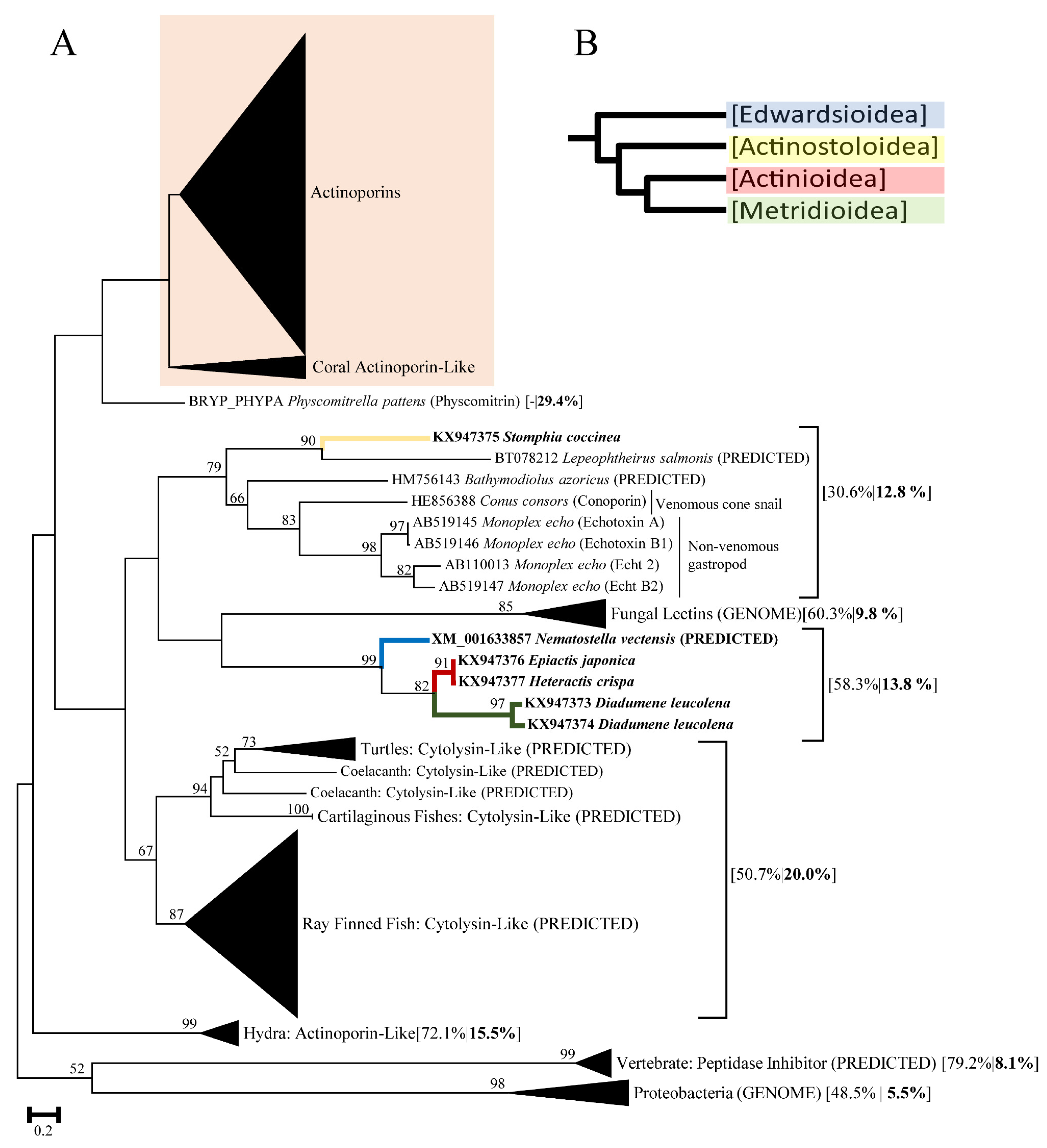
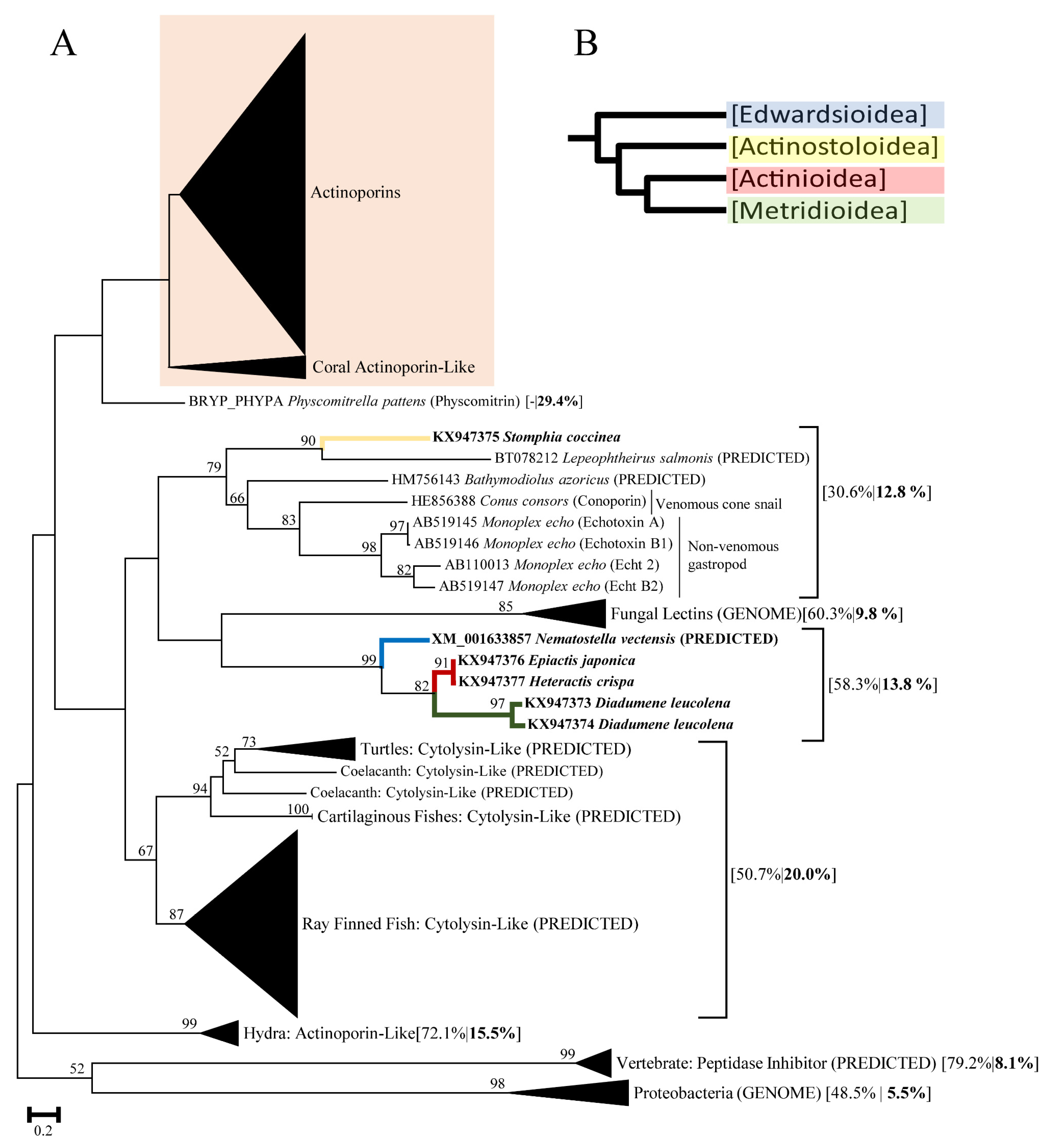
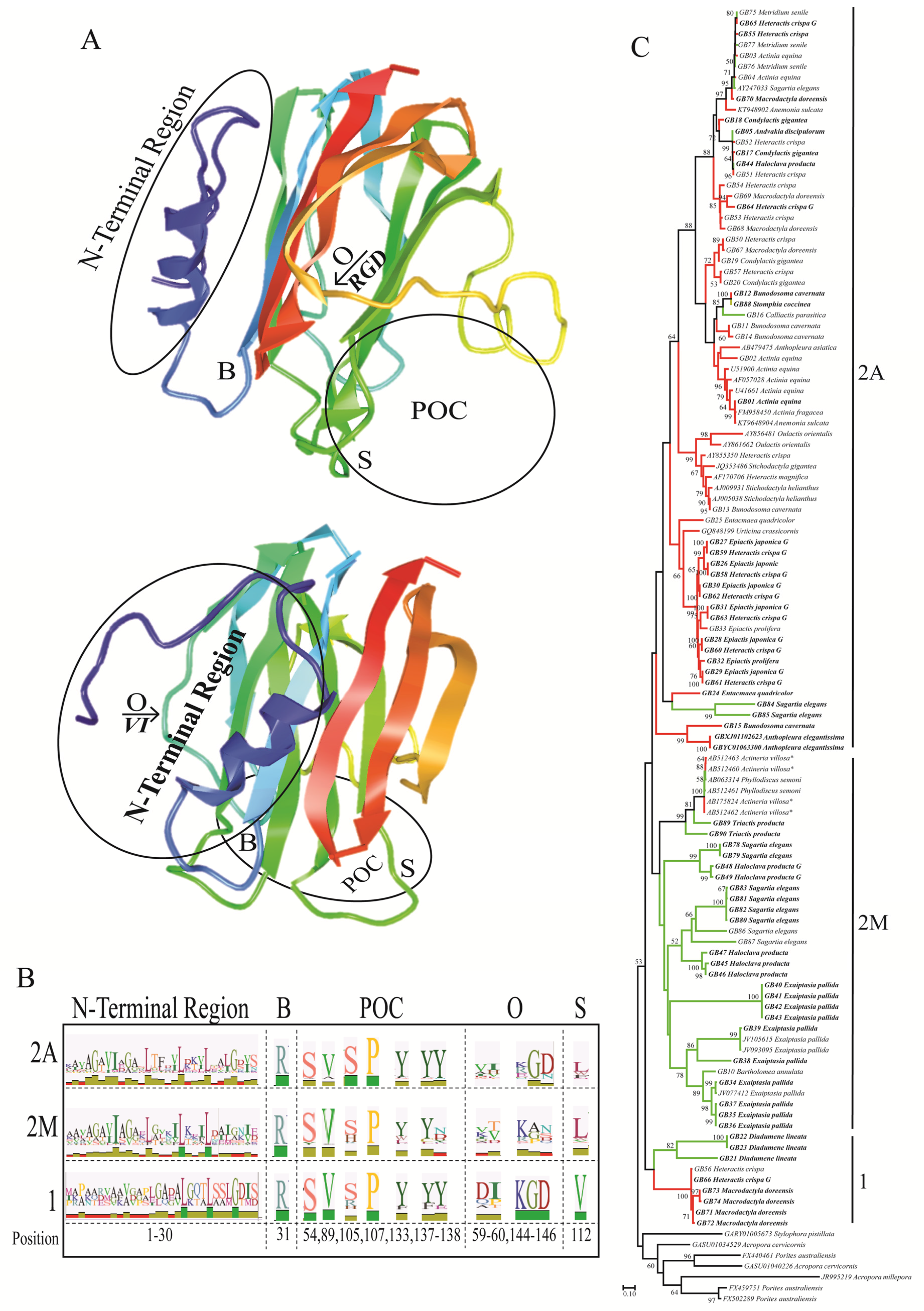
2.1. Actinoporin Alignment and Tree Reconstruction
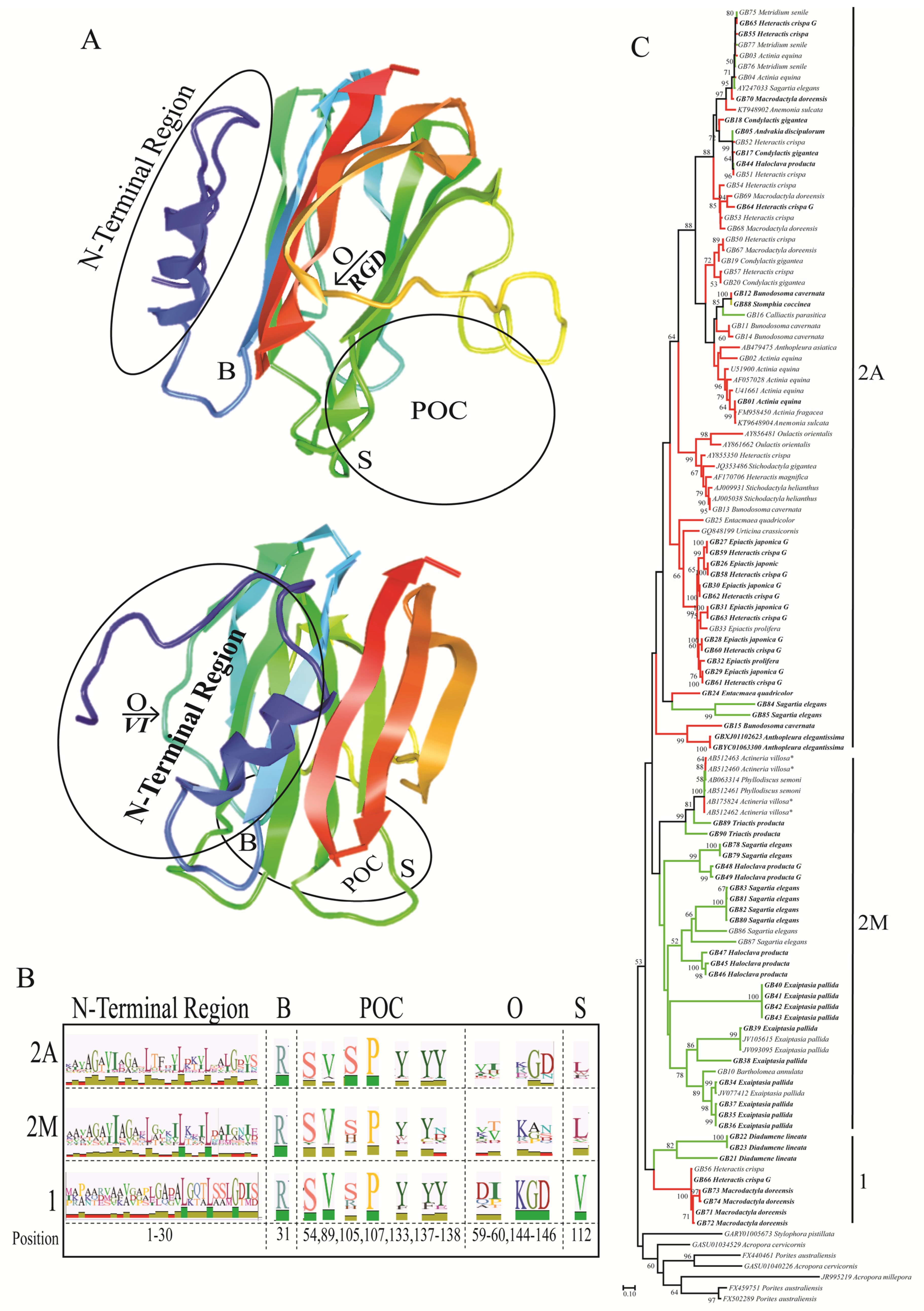
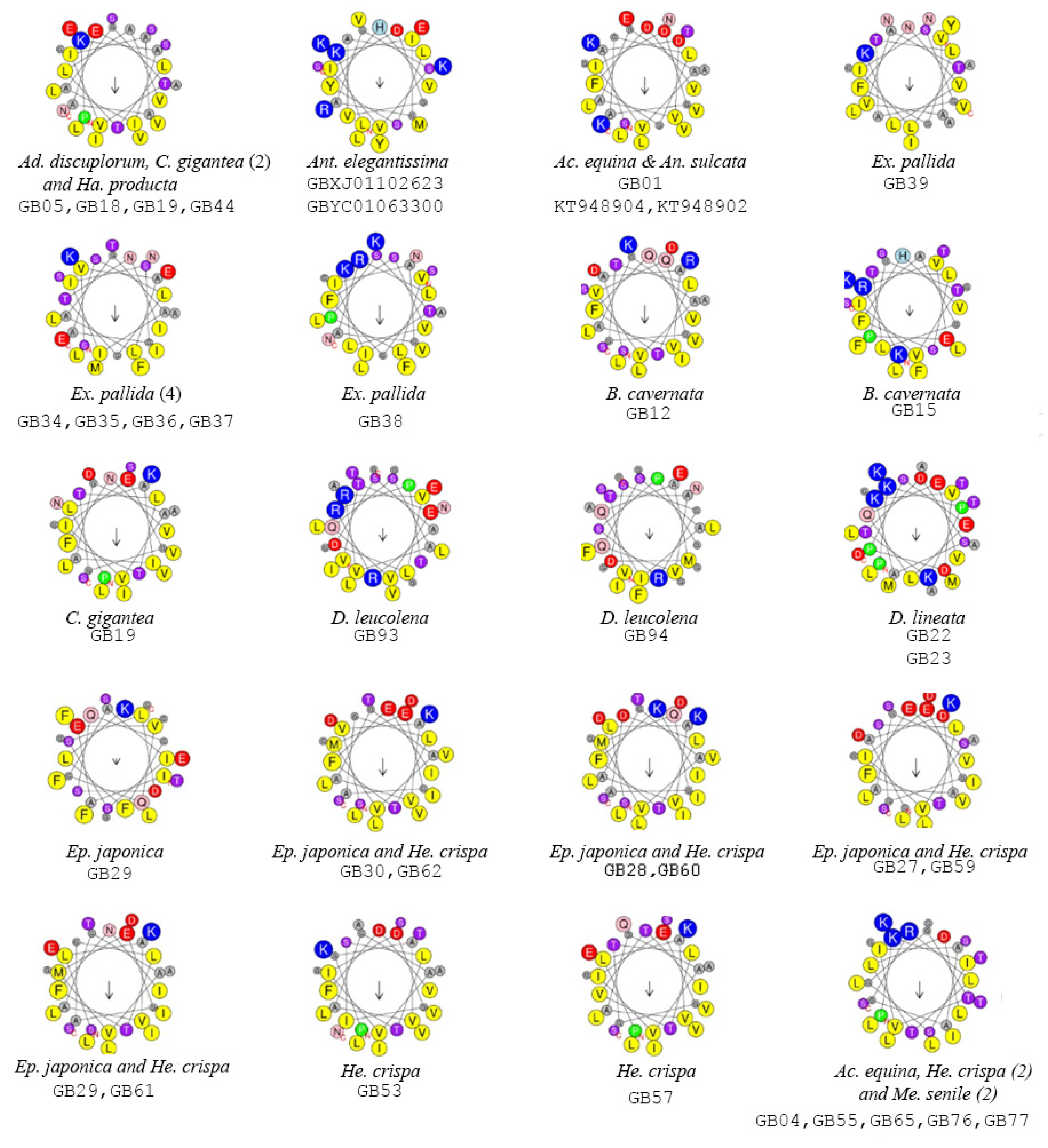
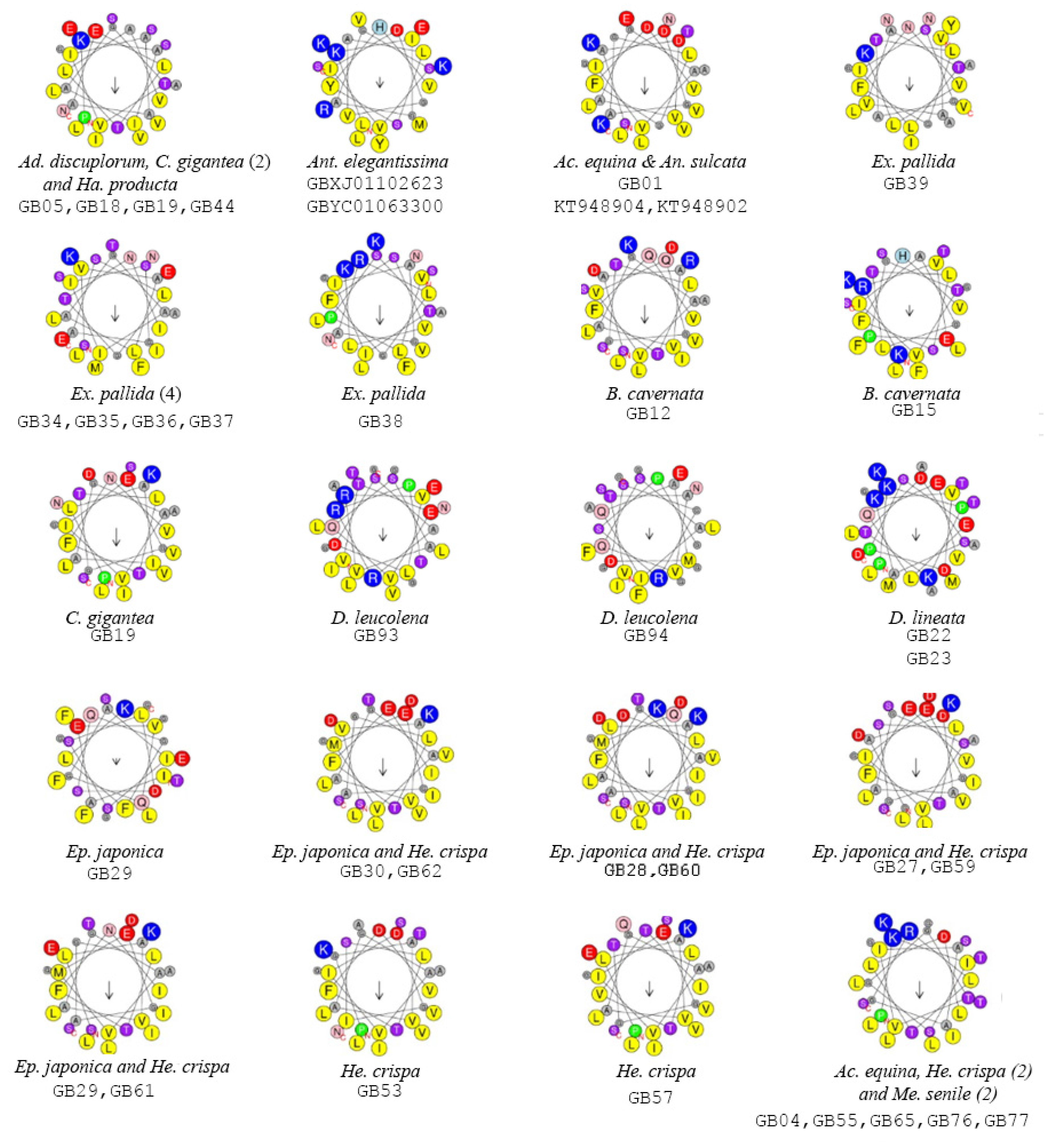
2.2. Functionally Important Residues
2.3. Selection Analyses
3. Discussion
4. Materials and Methods
4.1. Partial Transcriptome and Genome Sequencing and Assembly
4.2. Identification of Actinoporin and Actinoporin-Like Sequences
4.3. Evaluation of Functionally Important Residues
4.4. Selection Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frazão, B.; Vasconcelos, V.; Antunes, A. Sea anemone (Cnidaria, Anthozoa, Actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Oshiro, N.; Takuwa-Kuroda, K.; Iwanaga, S.; Nozaki, M.; Nakajima, T. Novel proteinaceous toxins from the nematocyst venom of the Okinawan sea anemone Phyllodiscus semoni Kwietniewski. Biochem. Biophys. Res. Commun. 2002, 294, 760–763. [Google Scholar] [CrossRef]
- Cline, E.I.; Wiebe, L.I.; Young, J.D.; Samuel, J. Toxic effects of the novel protein UPI from the sea anemone Urticina piscivora. Pharmacol. Res. 1995, 32, 309–314. [Google Scholar] [CrossRef]
- Radwan, F.F.Y. Comparative toxinological and immunological studies on the nematocyst venoms of the Red Sea fire corals Millepora dichotoma and M. platyphylla. Comp. Biochem. Physiol. Part C 2002, 3, 323–334. [Google Scholar] [CrossRef]
- Zhang, M.; Fishman, Y.; Sher, D.; Zlotkin, E. Hydralysin, a novel animal group-selective paralytic and cytolytic protein from a noncnidocystic origin in Hydra. Biochemistry (Mosc.) 2003, 42, 8939–8944. [Google Scholar] [CrossRef] [PubMed]
- Sher, D.; Fishman, Y.; Zhang, M.; Lebendiker, M.; Gaathon, A.; Mancheño, J.-M.; Zlotkin, E. Hydralysins, a new category of β-pore-forming toxins in Cnidaria. J. Biol. Chem. 2005, 280, 22847–22855. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, D.; Burnell, J. Identification, cloning and sequencing of two major venom proteins from the box jellyfish, Chironex fleckeri. Toxicon 2007, 50, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Maček, P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria). Toxicon 2002, 40, 111–124. [Google Scholar] [CrossRef]
- Álvarez, C.; Mancheño, J.M.; Martínez, D.; Tejuca, M.; Pazos, F.; Lanio, M.E. Sticholysins, two pore-forming toxins produced by the Caribbean sea anemone Stichodactyla helianthus: Their interaction with membranes. Toxicon 2009, 54, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Šuput, D. In vivo effects of cnidarian toxins and venoms. Toxicon 2009, 54, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Bakrač, B.; Gutiérrez-Aguirre, I.; Podlesek, Z.; Sonnen, A.F.-P.; Gilbert, R.J.C.; Maček, P.; Lakey, J.H.; Anderluh, G. Molecular determinants of sphingomyelin specificity of a eukaryotic pore-forming toxin. J. Biol. Chem. 2008, 283, 18665–18677. [Google Scholar] [CrossRef] [PubMed]
- Črnigoj Kristan, K.; Viero, G.; Dalla Serra, M.; Maček, P.; Anderluh, G. Molecular mechanism of pore formation by actinoporins. Toxicon 2009, 54, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Sepčić, K.; Turk, T.; Maček, P. Cytolytic proteins from cnidarians—An overview. Acta Chim. Slov. 2011, 58, 724–729. [Google Scholar] [PubMed]
- Kawashima, Y.; Nagai, H.; Ishida, M.; Nagashima, Y.; Shiomi, K. Primary structure of echotoxin 2, an actinoporin-like hemolytic toxin from the salivary gland of the marine gastropod Monoplex echo. Toxicon 2003, 42, 491–497. [Google Scholar] [CrossRef]
- Valle, A.; Alvarado-Mesén, J.; Lanio, M.E.; Álvarez, C.; Barbosa, J.A.R.G.; Pazos, I.F. The multigene families of actinoporins (part I): Isoforms and genetic structure. Toxicon 2015, 103, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Hoang, Q.T.; Cho, S.H.; McDaniel, S.F.; Ok, S.H.; Quatrano, R.S.; Shin, J.S. An actinoporin plays a key role in water stress in the moss Physcomitrella patens. New Phytol. 2009, 184, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Aguirre, I.; Trontelj, P.; Maček, P.; Lakey, J.H.; Anderluh, G. Membrane binding of zebrafish actinoporin-like protein: AF domains, a novel superfamily of cell membrane binding domains. Biochem. J. 2006, 398, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Ohyanagi, H.; Hayakawa, S.; Osato, N.; Nishimiya-Fujisawa, C.; Ikeo, K.; David, C.N.; Fujisawa, T.; Gojobori, T. The evolutionary emergence of cell type-specific genes inferred from the gene expression analysis of Hydra. Proc. Natl. Acad. Sci. USA 2007, 104, 14735–14740. [Google Scholar] [CrossRef] [PubMed]
- Glasser, E.; Rachamim, T.; Aharonovich, D.; Sher, D. Hydra actinoporin-like toxin-1, an unusual hemolysin from the nematocyst venom of Hydra magnipapillata which belongs to an extended gene family. Toxicon 2014, 91, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Birck, C.; Damian, L.; Marty-Detraves, C.; Lougarre, A.; Schulze-Briese, C.; Koehl, P.; Fournier, D.; Paquereau, L.; Samama, J.-P. A new lectin family with structure similarity to actinoporins revealed by the crystal structure of Xerocomus chrysenteron lectin XCL. J. Mol. Biol. 2004, 344, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.D.; Mateo de Acosta, C.; Lage, A. A new immunotoxin built by linking a hemolytic toxin to a monoclonal antibody specific for immature T lymphocytes. Int. J. Cancer 1988, 42, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.D.; Mateo de Acosta, C.; Lage, A. A carcinoembryonic antigen-directed immunotoxin built by linking a monoclonal antibody to a hemolytic toxin. Int. J. Cancer 1989, 43, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Campos, A.M.; Pazos, F.; Alvarez, C.; Lanio, M.E.; Casallanovo, F.; Schreier, S.; Salinas, R.K.; Vergara, C.; Lissi, E. Properties of St I and St II, two isotoxins isolated from Stichodactyla helianthus: A comparison. Toxicon 2001, 39, 1547–1560. [Google Scholar] [CrossRef]
- Tejuca, M.; Anderluh, G.; Dalla Serra, M. Sea anemone cytolysins as toxic components of immunotoxins. Toxicon 2009, 54, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Morante, K.; Caaveiro, J.M.M.; Viguera, A.R.; Tsumoto, K.; González-Mañas, J.M. Functional characterization of Val60, a key residue involved in the membrane-oligomerization of fragaceatoxin C, an actinoporin from Actinia fragacea. FEBS Lett. 2015, 589, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Malovrh, P.; Barlic, A.; Podlesek, Z.; MaCek, P.; Menestrina, G.; Anderluh, G. Structure-function studies of tryptophan mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. J. 2000, 346, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Mancheño, J.M.; Martı́n-Benito, J.; Martı́nez-Ripoll, M.; Gavilanes, J.G.; Hermoso, J.A. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure 2003, 11, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- García-Linares, S.; Richmond, R.; García-Mayoral, M.F.; Bustamante, N.; Bruix, M.; Gavilanes, J.G.; Martínez-del-Pozo, Á. The sea anemone actinoporin (Arg-Gly-Asp) conserved motif is involved in maintaining the competent oligomerization state of these pore-forming toxins. FEBS J. 2014, 281, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, G.; Pederzolli, C.; Macek, P.; Menestrina, G. Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J. Membr. Biol. 1993, 131, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Malovrh, P.; Viero, G.; Serra, M.D.; Podlesek, Z.; Lakey, J.H.; Maček, P.; Menestrina, G.; Anderluh, G. A novel mechanism of pore formation membrane penetration by the N-terminal amphipathic region of equinatoxin. J. Biol. Chem. 2003, 278, 22678–22685. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, A.; Potrich, C.; Sabo, J.K.; Frisanco, M.; Guella, G.; Dalla Serra, M.; Anderluh, G.; Separovic, F.; Norton, R.S. Structure and activity of the N-terminal Region of the eukaryotic cytolysin equinatoxin II. Biochemistry (Mosc.) 2006, 45, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Monastyrnaya, M.; Leychenko, E.; Isaeva, M.; Likhatskaya, G.; Zelepuga, E.; Kostina, E.; Trifonov, E.; Nurminski, E.; Kozlovskaya, E. Actinoporins from the sea anemones, tropical Radianthus macrodactylus and northern Oulactis orientalis: Comparative analysis of structure–function relationships. Toxicon 2010, 56, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Uechi, G.; Toma, H.; Arakawa, T.; Sato, Y. Molecular characterization on the genome structure of hemolysin toxin isoforms isolated from sea anemone Actineria villosa and Phyllodiscus semoni. Toxicon 2010, 56, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Valcarcel, C.A.; Dalla Serra, M.; Potrich, C.; Bernhart, I.; Tejuca, M.; Martinez, D.; Pazos, F.; Lanio, M.E.; Menestrina, G. Effects of lipid composition on membrane permeabilization by sticholysin I and II, two cytolysins of the sea anemone Stichodactyla helianthus. Biophys. J. 2001, 80, 2761–2774. [Google Scholar] [CrossRef]
- Anderluh, G.; Serra, M.D.; Viero, G.; Guella, G.; Maček, P.; Menestrina, G. Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J. Biol. Chem. 2003, 278, 45216–45223. [Google Scholar] [CrossRef] [PubMed]
- Mechaly, A.E.; Bellomio, A.; Gil-Cartón, D.; Morante, K.; Valle, M.; González-Mañas, J.M.; Guérin, D.M.A. Structural insights into the oligomerization and architecture of eukaryotic membrane pore-forming toxins. Structure 2011, 19, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Caaveiro, J.M.M.; Morante, K.; González-Mañas, J.M.; Tsumoto, K. Structural basis for self-assembly of a cytolytic pore lined by protein and lipid. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Tejuca, M.; Serra, M.D.; Potrich, C.; Alvarez, C.; Menestrina, G. Sizing the radius of the pore formed in erythrocytes and lipid vesicles by the toxin sticholysin I from the sea anemone Stichodactyla helianthus. J. Membr. Biol. 2001, 183, 125–135. [Google Scholar] [CrossRef] [PubMed]
- García-Linares, S.; Castrillo, I.; Bruix, M.; Menéndez, M.; Alegre-Cebollada, J.; Martínez-del-Pozo, Á.; Gavilanes, J.G. Three-dimensional structure of the actinoporin sticholysin I. Influence of long-distance effects on protein function. Arch. Biochem. Biophys. 2013, 532, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yap, L.L.; Chua, K.L.; Khoo, H.E. A multigene family of Heteractis magnificalysins (HMgs). Toxicon 2008, 51, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.; Barbeitos, M.S.; Brugler, M.R.; Crowley, L.M.; Grajales, A.; Gusmão, L.; Häussermann, V.; Reft, A.; Daly, M. Hidden among sea anemones: The first comprehensive phylogenetic reconstruction of the order Actiniaria (Cnidaria, Anthozoa, Hexacorallia) reveals a novel group of hexacorals. PLOS ONE 2014, 9, e96998. [Google Scholar] [CrossRef] [PubMed]
- Hinds, M.G.; Zhang, W.; Anderluh, G.; Hansen, P.E.; Norton, R.S. Solution structure of the eukaryotic pore-forming cytolysin equinatoxin II: Implications for pore formation. J. Mol. Biol. 2002, 315, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Broe, M.; Daly, M. Tissue-specific venom composition and differential gene expression in sea anemones. Genome Biol. Evol. 2016, 8, 2358–2375. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Broe, M.; Daly, M. Multi-copy venom genes hidden in de novo transcriptome assemblies, a cautionary tale with the snakelocks sea anemone Anemonia sulcata (Pennant, 1977). Toxicon 2015, 108, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Brugler, M.R.; Daly, M. A RNA-seq approach to identify putative toxins from acrorhagi in aggressive and non-aggressive Anthopleura elegantissima polyps. BMC Genom. 2015, 16, 221. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.; Simakov, O.; Esherick, L.Y.; Liew, Y.J.; Lehnert, E.M.; Michell, C.T.; Li, Y.; Hambleton, E.A.; Guse, A.; Oates, M.E.; et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. USA 2015, 112, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.V.; et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 2007, 317, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Stefanik, D.J.; Lubinski, T.J.; Granger, B.R.; Byrd, A.L.; Reitzel, A.M.; DeFilippo, L.; Lorenc, A.; Finnerty, J.R. Production of a reference transcriptome and transcriptomic database (EdwardsiellaBase) for the lined sea anemone, Edwardsiella lineata, a parasitic cnidarian. BMC Genom. 2014, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Daly, M. Functional and genetic diversity of toxins in sea anemones. In Evolution of Venomous Animals and Their Toxins; Gopalakrishnakone, P., Malhotra, A., Eds.; Toxinology; Springer: Dordrecht, The Netherlands, 2016; pp. 1–18. [Google Scholar]
- Uechi, G.; Toma, H.; Arakawa, T.; Sato, Y. Biochemical and physiological analyses of a hemolytic toxin isolated from a sea anemone Actineria villosa. Toxicon 2005, 45, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Maula, T.; Isaksson, Y.J.E.; García-Linares, S.; Niinivehmas, S.; Pentikäinen, O.T.; Kurita, M.; Yamaguchi, S.; Yamamoto, T.; Katsumura, S.; Gavilanes, J.G.; et al. 2NH and 3OH are crucial structural requirements in sphingomyelin for sticholysin II binding and pore formation in bilayer membranes. Biochim. Biophys. Acta 2013, 1828, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.K.; Yao, S.; Rojko, N.; Anderluh, G.; Lybrand, T.P.; Downton, M.T.; Wagner, J.; Separovic, F. Characterization of the lipid-binding site of equinatoxin II by NMR and molecular dynamics simulation. Biophys. J. 2015, 108, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Cea, M.A.; Castrillo, I.; Alegre-Cebollada, J.; Martínez-del-Pozo, Á.; Gavilanes, J.G.; Bruix, M. Intrinsic local disorder and a network of charge–charge interactions are key to actinoporin membrane disruption and cytotoxicity. FEBS J. 2011, 278, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Moran, Y.; Praher, D.; Schlesinger, A.; Ayalon, A.; Tal, Y.; Technau, U. Analysis of soluble protein contents from the nematocysts of a model sea anemone sheds light on venom evolution. Mar. Biotechnol. N. Y. N 2013, 15, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Moran, Y.; Genikhovich, G.; Gordon, D.; Wienkoop, S.; Zenkert, C.; Özbek, S.; Technau, U.; Gurevitz, M. Neurotoxin localization to ectodermal gland cells uncovers an alternative mechanism of venom delivery in sea anemones. Proc. R. Soc. B Biol. Sci. 2012, 279, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez de la Vega, R.C.; Giraud, T. Intragenome diversity of gene families encoding toxin-like proteins in venomous animals. Integr. Comp. Biol. 2016, 56, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Barlow, D.J.; Long, P.F. Machine learning can differentiate venom toxins from other proteins having non-toxic physiological functions. PeerJ Comput. Sci. 2016, 2, e90. [Google Scholar] [CrossRef]
- Jouiaei, M.; Sunagar, K.; Gross, A.F.; Scheib, H.; Alewood, P.F.; Moran, Y.; Fry, B.G. Evolution of an ancient venom: Recognition of a novel family of cnidarian toxins and the common evolutionary origin of sodium and potassium neurotoxins in sea anemone. Mol. Biol. Evol. 2015, 32, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Chung, R.; Dunn, S.R.; Weston, A.J.; Jaimes-Becerra, A.; Marques, A.C.; Morandini, A.C.; Hranueli, D.; Starcevic, A.; Ward, M.; et al. Gene duplications are extensive and contribute significantly to the toxic proteome of nematocysts isolated from Acropora digitifera (Cnidaria: Anthozoa: Scleractinia). BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-Y.; Yang, W.; Chen, H.-P.; Tu, H.-B.; Wu, W.-Y.; Wei, J.-W.; Wang, J.; Liu, W.-H.; Xu, A.-L. Cloning and characterization of an acidic cytolysin cDNA from sea anemone Sagartia rosea. Toxicon 2002, 40, 1563–1569. [Google Scholar] [CrossRef]
- Rachamim, T.; Morgenstern, D.; Aharonovich, D.; Brekhman, V.; Lotan, T.; Sher, D. The dynamically evolving nematocyst content of an anthozoan, a scyphozoan, and a hydrozoan. Mol. Biol. Evol. 2015, 32, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, A.; Özbek, S. The Nematocyst: A molecular map of the cnidarian stinging organelle. Int. J. Dev. Biol. 2012, 56, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Basulto, A.; Pérez, V.M.; Noa, Y.; Varela, C.; Otero, A.J.; Pico, M.C. Immunohistochemical targeting of sea anemone cytolysins on tentacles, mesenteric filaments and isolated nematocysts of Stichodactyla helianthus. J. Exp. Zoolog. A Comp. Exp. Biol. 2006, 305A, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Clementi, G.; Cunietti, M.; Porres, C.; Oñaderra, M.; Gavilanes, J.G.; Pozo, A.M. D. Silent mutations at the 5’-end of the cDNA of actinoporins from the sea anemone Stichodactyla helianthus allow their heterologous overproduction in Escherichia coli. J. Biotechnol. 2007, 127, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Weisenfeld, N.I.; Yin, S.; Sharpe, T.; Lau, B.; Hegarty, R.; Holmes, L.; Sogoloff, B.; Tabbaa, D.; Williams, L.; Russ, C.; et al. Comprehensive variation discovery in single human genomes. Nat. Genet. 2014, 46, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package) version 3.6 Distributed by the author. 2005; Department of Genome Sciences, University of Washington: Seattle, WC, USA, 2005. [Google Scholar]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J.; Osguthorpe, D.J.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978, 120, 97–120. [Google Scholar] [CrossRef]
- Ros, U.; Rodríguez-Vera, W.; Pedrera, L.; Valiente, P.A.; Cabezas, S.; Lanio, M.E.; García-Sáez, A.J.; Alvarez, C. Differences in activity of actinoporins are related with the hydrophobicity of their N-terminus. Biochimie 2015, 116, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily/Species | N | pl | MW (kDa) | (<H>) * | (<μH>) * | Net Charge |
|---|---|---|---|---|---|---|
| Actinioidea | 52 | |||||
| Actinia equina | 4 | 9.47 | 21.6 | 0.424 | 0.337 | −2 |
| Anemonia sulcata [44,45] | 2 | 9.50 | 21.7 | 0.424 | 0.337 | −2 |
| Anthopleura elegantissima [46] | 2 | 8.63–8.64 | 21.2–21.3 | 0.443 | 0.207 | 2 |
| Bunodosoma cavernata | 5 | 9.23–10.65 | 20.4–21.0 | 0.482 | 0.356 | 0 |
| 0.586 | 0.21 | 2 | ||||
| Condylactis gigantea | 4 | 4.56–6.71 | 21.57–21.58 | 0.589 | 0.35 | −1 |
| Entacmaea quadricolor | 2 | 5.87 | 21.8 | 0.472 | 0.309 | −3 |
| Epiactis japonica | 6 | 4.86–9.38 | 21.4–25.2 | 0.494 | 0.122 | −2 |
| 0.58 | 0.336 | −3 | ||||
| 0.592 | 0.363 | −1 | ||||
| 0.42 | 0.361 | −4 | ||||
| 0.59 | 0.342 | −2 | ||||
| 0.474 | 0.396 | 1 | ||||
| Epiactis prolifera | 2 | 8.00 | 21.5 | 0.614 | 0.366 | −2 |
| Heteractis crispa [44] | 17 | 4.71–9.38 | 19.5–21.2 | 0.599 | 0.351 | −1 |
| 0.616 | 0.288 | −1 | ||||
| 0.567 | 0.327 | 2 | ||||
| 0.442 | 0.288 | −1 | ||||
| Macrodactyla doreensis | 8 | 5.72–7.75 | 19.1–21.6 | 0.629 | 0.287 | 2 |
| 0.523 | 0.202 | −2 | ||||
| 0.398 | 0.249 | −1 | ||||
| 0.615 | 0.328 | −1 | ||||
| Metridioidea | 37 | |||||
| Andvakia discipulorum | 1 | 4.63 | 21.6 | 0.583 | 0.322 | −1 |
| Bartholomea annulata | 1 | - | - | - | - | - |
| Calliactis parasitica | 1 | - | - | - | - | - |
| Diadumene lineata | 3 | 8.72 | 23.4 | 0.211 | 0.245 | −1 |
| Exaiptasia pallida [47] | 10 | 9.45–10.26 | 21.7–23.0 | 0.64 | 0.268 | 1 |
| 0.605 | 0.383 | 3 | ||||
| 0.556 | 0.319 | −1 | ||||
| Haloclava producta | 6 | 4.51–9.91 | 21.6–27.2 | 0.474 | 0.317 | −1 |
| 0.682 | 0.37 | 1 | ||||
| Metridium senile | 3 | - | - | - | - | - |
| Sagartia elegans | 10 | 8.33–9.66 | 21.6–25.3 | 0.606 | 0.2 | −1 |
| 0.514 | 0.279 | 3 | ||||
| 0.533 | 0.28 | 2 | ||||
| 0.631 | 0.325 | 0 | ||||
| Triactis producta | 2 | 8.56–9.7 | 21.1 | 0.424 | 0.386 | −1 |
| Actinostoloidea | 1 | |||||
| Stomphia coccinea | 1 | 9.12 | 18.4 | 0.481 | 0.312 | −1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macrander, J.; Daly, M. Evolution of the Cytolytic Pore-Forming Proteins (Actinoporins) in Sea Anemones. Toxins 2016, 8, 368. https://doi.org/10.3390/toxins8120368
Macrander J, Daly M. Evolution of the Cytolytic Pore-Forming Proteins (Actinoporins) in Sea Anemones. Toxins. 2016; 8(12):368. https://doi.org/10.3390/toxins8120368
Chicago/Turabian StyleMacrander, Jason, and Marymegan Daly. 2016. "Evolution of the Cytolytic Pore-Forming Proteins (Actinoporins) in Sea Anemones" Toxins 8, no. 12: 368. https://doi.org/10.3390/toxins8120368
APA StyleMacrander, J., & Daly, M. (2016). Evolution of the Cytolytic Pore-Forming Proteins (Actinoporins) in Sea Anemones. Toxins, 8(12), 368. https://doi.org/10.3390/toxins8120368