
Presence or Absence of mlr Genes and Nutrient Concentrations Co-Determine the Microcystin Biodegradation Efficiency of a Natural Bacterial Community

 and
and
Abstract
:1. Introduction
2. Results
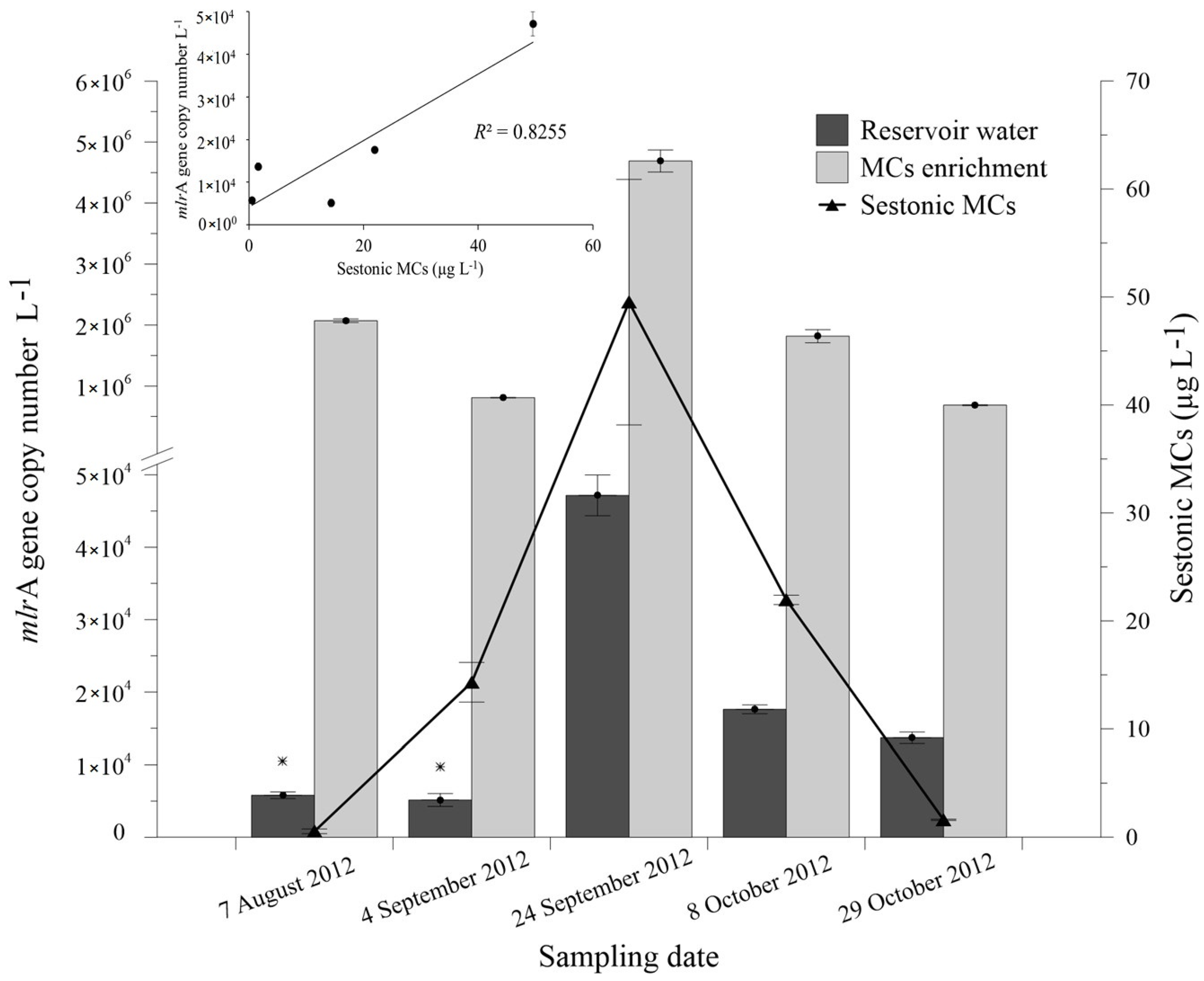
2.1. Cyanobacterial Bloom and Biodegradation Capacity of the Natural Bacterial Community
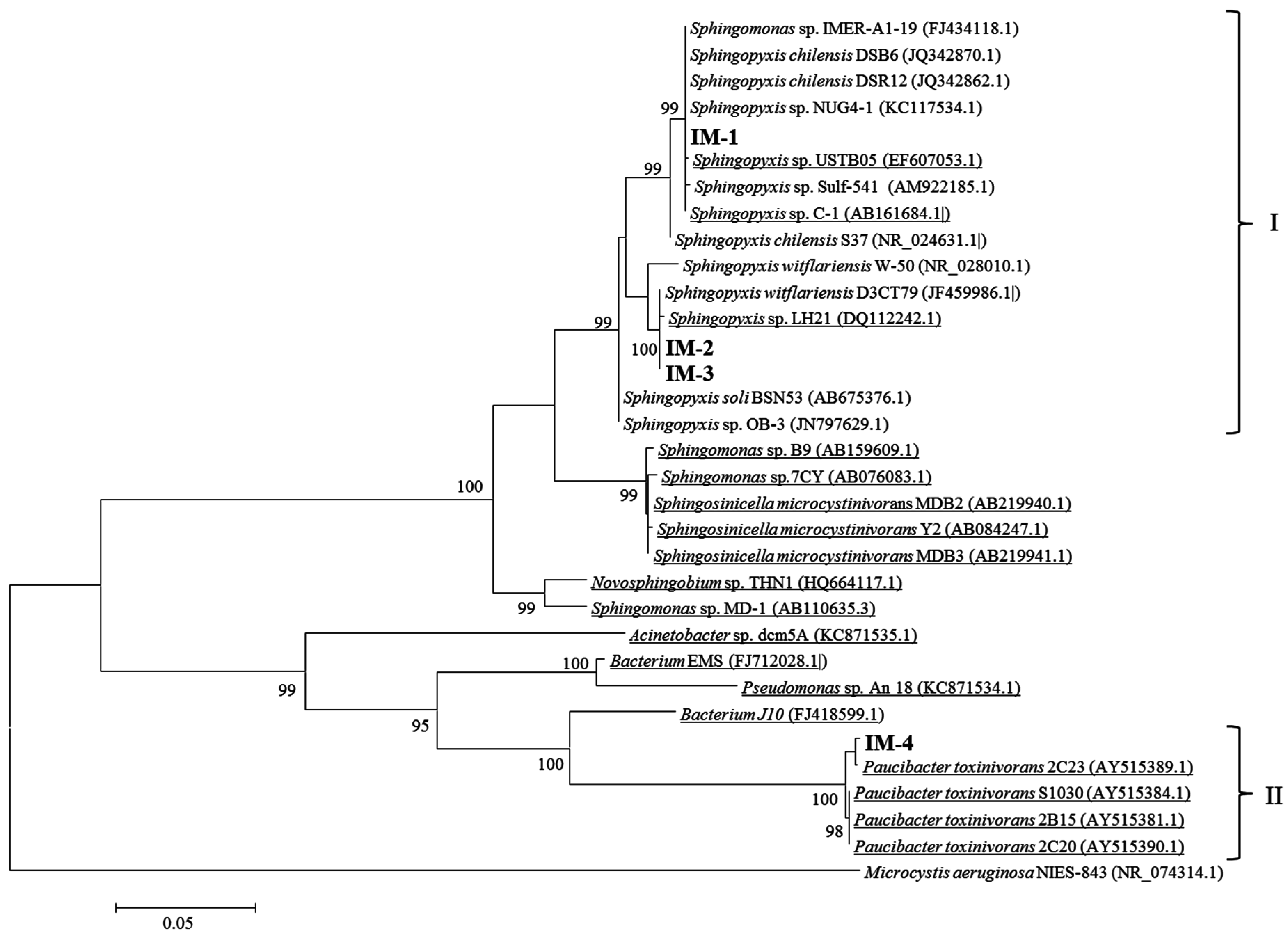
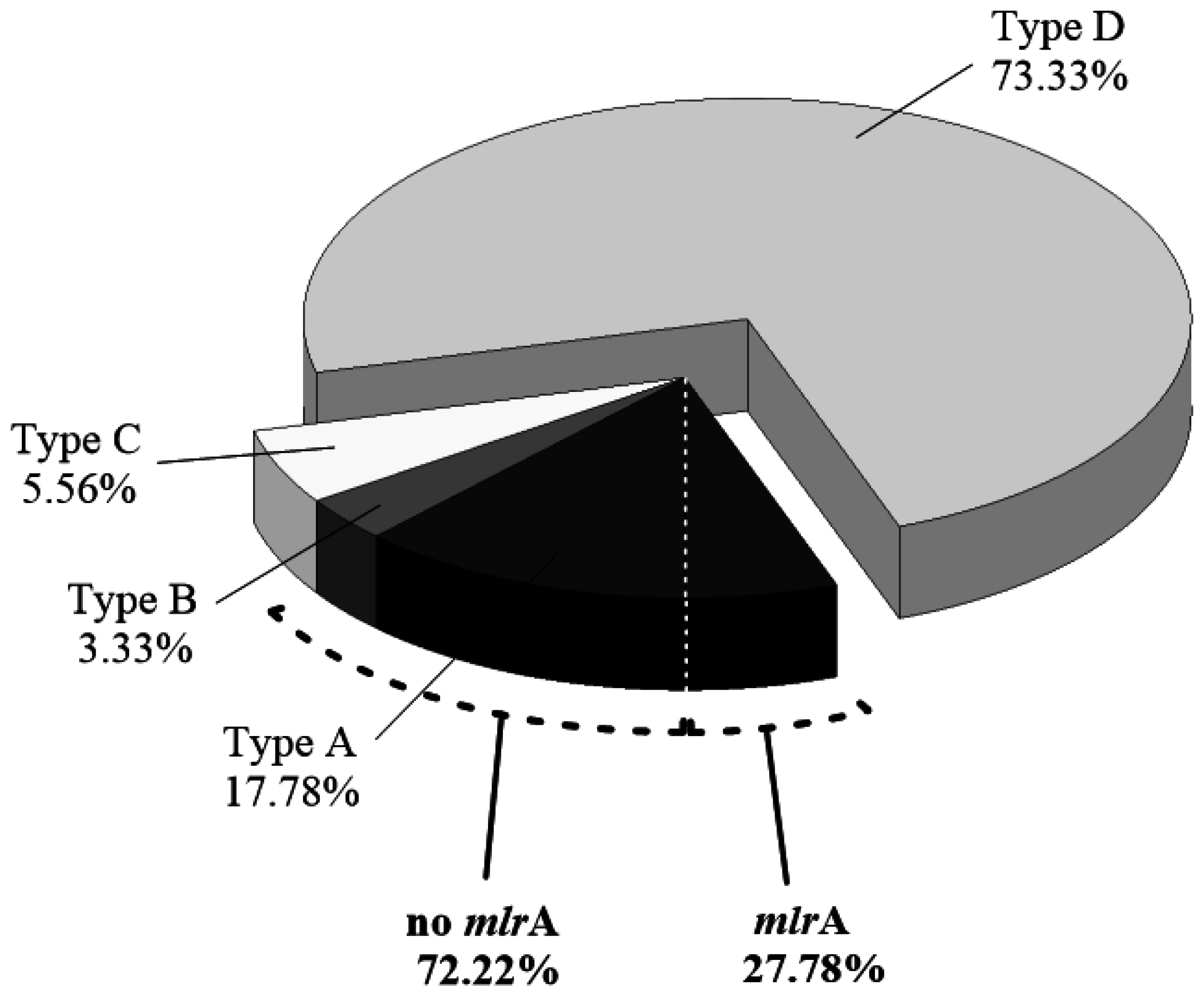
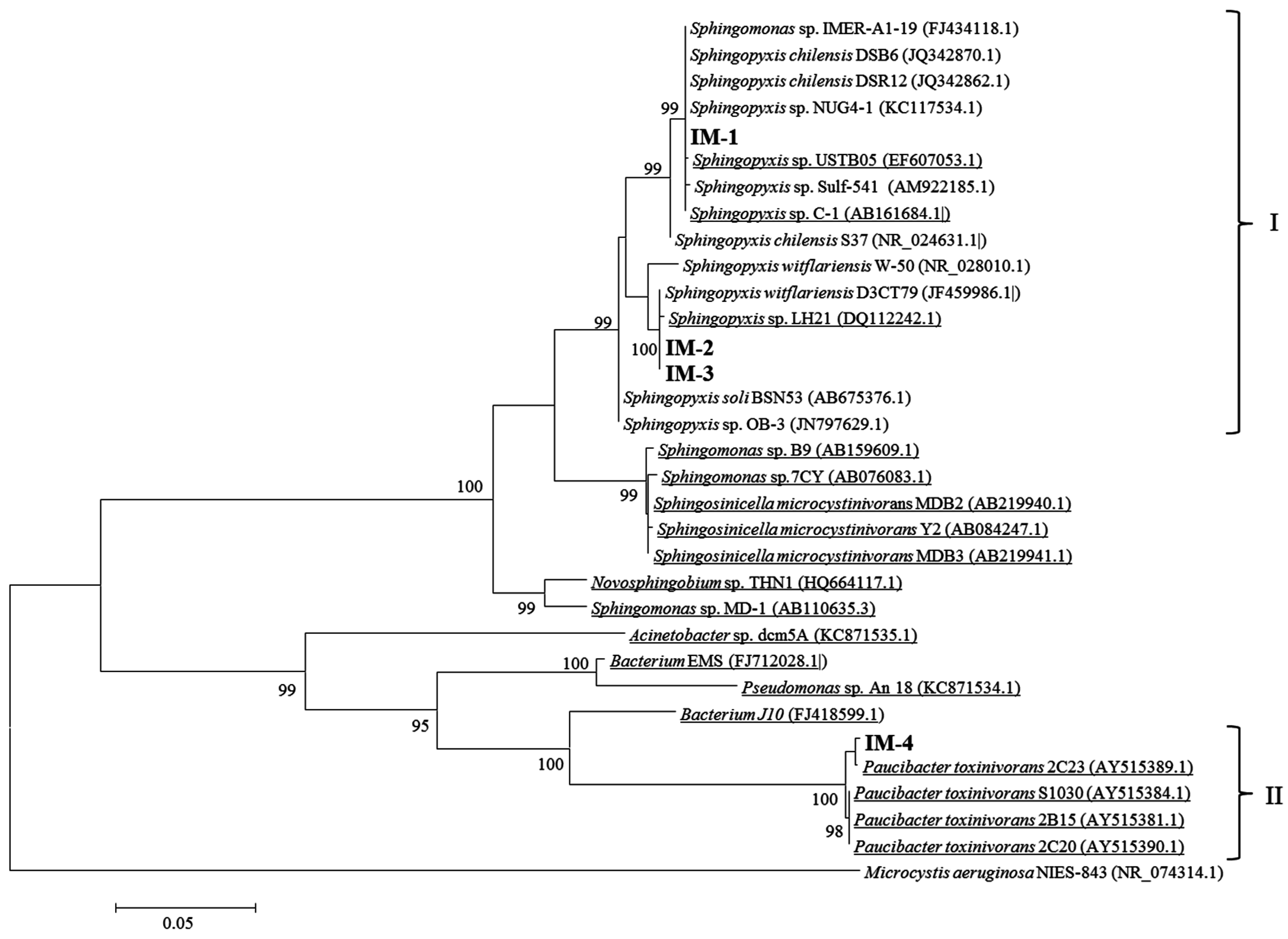
2.2. Isolation of MCs-Degrading Bacteria from the Bacterial Community
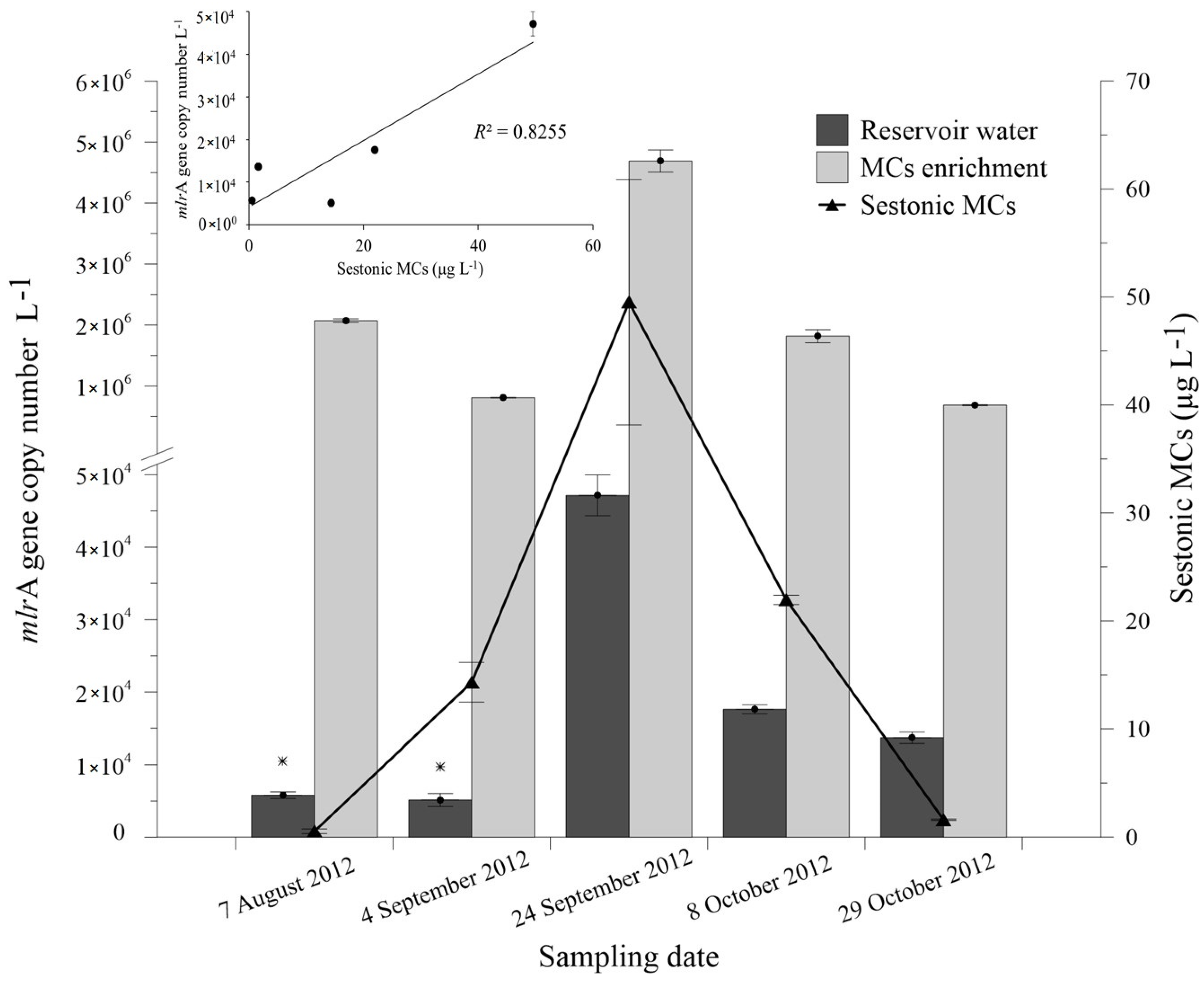
2.3. Abundance of mlrA Gene during the Bloom Episode
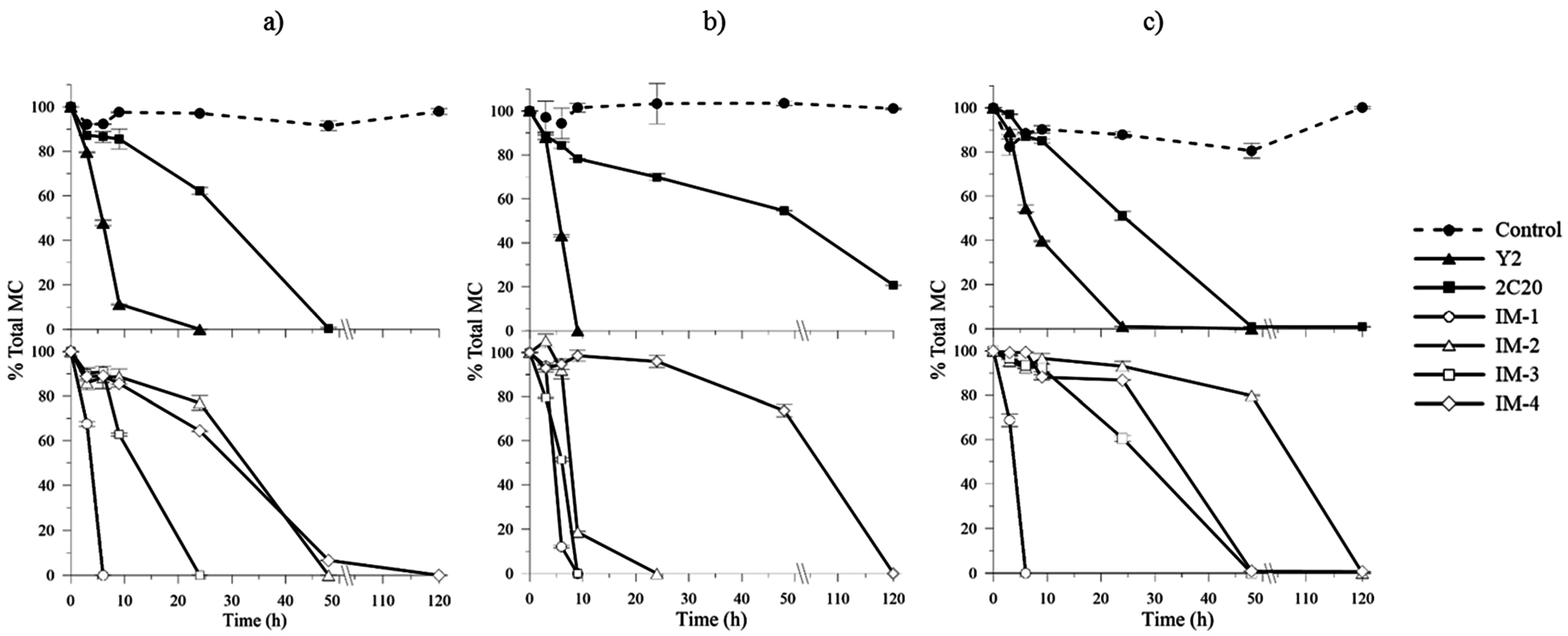
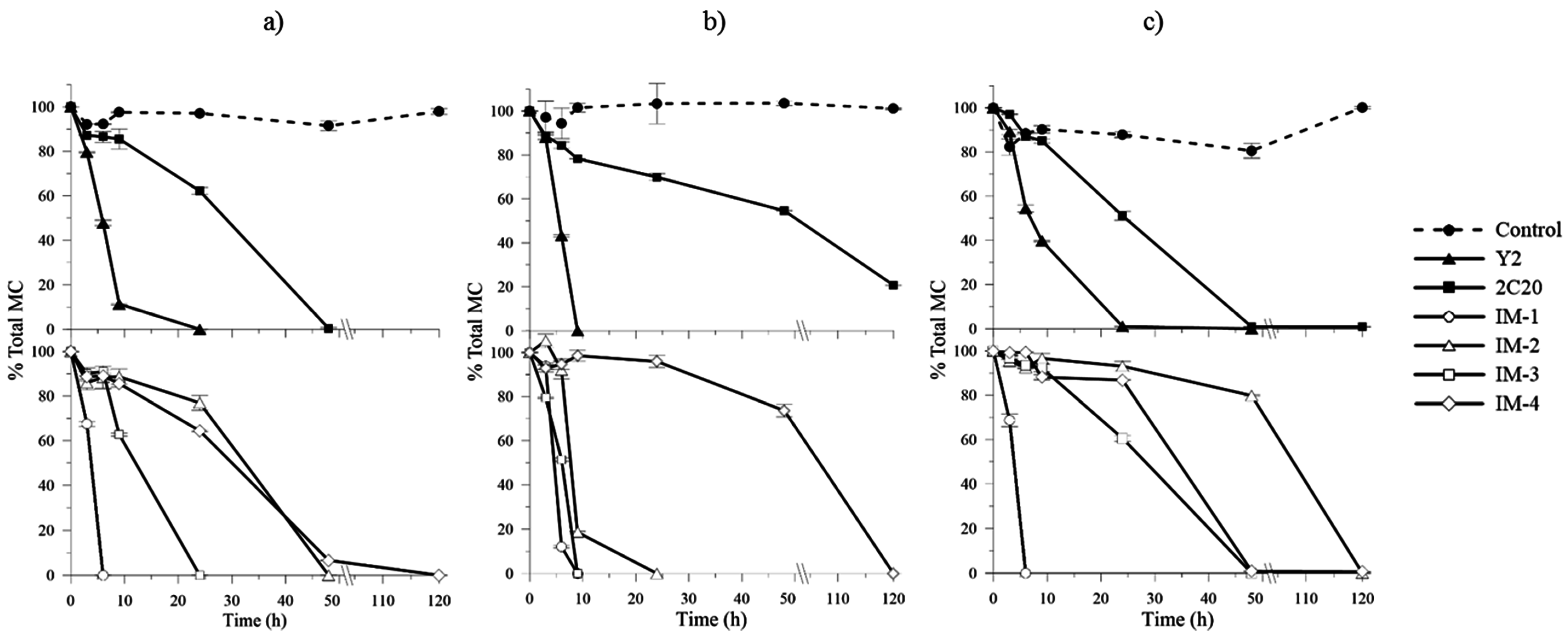
2.4. In Vitro MCs Biodegradation Assays and Kinetics
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Sampling
5.2. Microcystins Extraction
5.3. Reservoir’s MCs-Degradation Capacity
5.4. Screening and Isolation of MCs-Degrading Bacteria
5.5. MCs Degradation under Different TOC and TN Concentrations
5.6. Analysis of Microcystins
5.7. Genomic DNA Extraction
5.8. mlrA-D Genes Detection
5.9. mcyE Gene Detection
5.10. Quantification of mlrA Gene: Real-Time PCR
5.11. Identification of Bacterial Isolates Using 16S rRNA Gene Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflict of Interest
Abbreviations
| MCs | microcystins |
| mlrA+ | presence of mlrA gene |
| mlrA− | absence (or no PCR amplification) of mlrA gene |
| mlr+ | presence of the complete mlr gene cluster |
| mlr− | absence (or no PCR amplification) of the complete mlr gene cluster |
| TOC | Total Organic Carbon |
| TN | Total Nitrogen |
| PCR | Polymerase Chain Reaction |
| NCBI | National Center for Biotechnology Information |
| BLASTN | Basic Local Alignment Search Tool for Nucleotide |
| CyanoHAB | Cyanobacterial Harmful Algal Bloom |
References
- El-Shehawy, R.; Gorokhova, E.; Fernández-Piñas, F.; del Campo, F.F. Global warming and hepatotoxin production by cyanobacteria: What can we learn from experiments? Water Res. 2012, 46, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Hall, N.S.; Calandrino, E.S. Controlling harmful cyanobacterial blooms in a world experiencing anthropogenic and climatic-induced change. Sci. Total Environ. 2011, 409, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Huisman, J. Blooms like it hot. Science 2008, 320, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Codd, G.A. Cyanobacterial toxins: Occurrence, properties and biological significance. Water Sci. Technol. 1995, 32, 149–156. [Google Scholar] [CrossRef]
- Codd, G.A.; Lindsay, J.; Young, F.M.; Morrison, L.F.; Metcalf, J.S. Harmful cyanobacteria. From mass mortalities to management measures. In Harmful Cyanobacteria; Huissman, J., Matthijs, H.C.P., Visser, P.M., Eds.; Aquatic Ecology Series; Springer: Dordrecht, The Netherlands, 2005; pp. 1–23. [Google Scholar]
- Sivonen, K.; Börner, T. Bioactive compounds produced by cyanobacteria. In The Cyanobacteria. Molecular Biology, Genetics and Evolution; Herrero, A., Flores, E., Eds.; Caister Academic Press: Norfolk, UK, 2008; pp. 159–197. [Google Scholar]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, K.; Jones, G. Cyanobacterial toxins. In Toxic Cyanobacteria in Water. A Guide to Their Public Health Consequences, Monitoring and Management; Chorus, I., Bartram, J., Eds.; F & FN Spon: London, UK, 1999; pp. 55–124. [Google Scholar]
- Cirés, S.; Quesada, A. Catálogo Cianobacterias Planctónicas Potencialmente Tóxicas de las Aguas Continentales Españolas; Ministerio de Medio Ambiente y Medio Rural y Marino: Madrid, Spain, 2011. [Google Scholar]
- MacKintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef]
- Spoof, L. Microcystins and nodularins. In TOXIC: Cyanobacterial Monitoring and Cyanotoxin Analysis; Meriluoto, J., Cood, G.A., Eds.; Åbo Akademi University Press: Åbo, Finland, 2005; pp. 15–39. [Google Scholar]
- Börner, T.; Dittmann, E. Molecular biology of cyanobacterial toxins. Genetic basis of microcystin production. In Harmful Cyanobacteria; Huisman, J., Matthijs, H.C.P., Visser, P.M., Eds.; Aquatic Ecology Series; Springer: Dortrecht, The Netherlands, 2005; pp. 25–40. [Google Scholar]
- Pearson, L.; Mihali, T.; Moffitt, M.; Kellmann, R.; Neilan, B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar. Drugs 2010, 8, 1650–1680. [Google Scholar] [CrossRef] [PubMed]
- Wörmer, L.; Huerta-Fontela, M.; Cirés, S.; Carrasco, D.; Quesada, A. Natural photodegradation of the cyanobacterial toxins microcystin and cylindrospermopsin. Environ. Sci. Technol. 2010, 44, 3002–3007. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Sinha, R.P.; Incharoensakdi, A. The cyanotoxin-microcystins: Current overview. Rev. Environ. Sci. Bio/Technol. 2014, 13, 215–249. [Google Scholar] [CrossRef]
- Dziga, D.; Wasylewski, M.; Wladyka, B.; Nybom, S.; Meriluoto, J. Microbial degradation of microcystins. Chem. Res. Toxicol. 2013, 26, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Graham, D.; Fowler, N.; Lawton, L.A. Biodegradation of microcystins and nodularin in freshwaters. Chemosphere 2008, 73, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Kormas, K.A.; Lymperopoulou, D.S. Cyanobacterial toxin degrading bacteria: Who are they? Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bourne, D.G.; Riddles, P.; Jones, G.J.; Smith, W.; Blakeley, R.L. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 2001, 16, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Bourne, D.G.; Jones, G.J.; Blakeley, R.L.; Jones, A.; Negri, A.P.; Riddles, P. Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl. Environ. Microbiol. 1996, 62, 4086–4094. [Google Scholar] [PubMed]
- Hoefel, D.; Adriansen, C.M.M.; Bouyssou, M.A.C.; Saint, C.P.; Newcombe, G.; Ho, L. Development of an mlrA gene-directed TaqMan PCR assay for quantitative assessment of microcystin-degrading bacteria within water treatment plant sand filter biofilms. Appl. Environ. Microbiol. 2009, 75, 5167–5169. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Okano, K.; Park, H.D.; Itayama, T.; Inamori, Y.; Neilan, B.A.; Burns, B.P.; Sugiura, N. Detection and sequencing of the microcystin LR-degrading gene, mlrA, from new bacteria isolated from Japanese lakes. FEMS Microbiol. Lett. 2003, 229, 271–276. [Google Scholar] [CrossRef]
- Manage, P.M.; Edwards, C.; Singh, B.K.; Lawton, L.A. Isolation and identification of novel microcystin-degrading bacteria. Appl. Environ. Microbiol. 2009, 75, 6924–6928. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Lu, X.; Jacob, J.; Sun, S.; Heath, R. Metagenomic identification of bacterioplankton taxa and pathways involved in microcystin degradation in Lake Erie. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Hoefel, D.; Saint, C.P.; Newcombe, G. Isolation and identification of a novel microcystin-degrading bacterium from a biological sand filter. Water Res. 2007, 41, 4685–4695. [Google Scholar] [CrossRef] [PubMed]
- Valeria, A.M.; Ricardo, E.J.; Stephan, P.; Alberto, W.D. Degradation of microcystin-RR by Sphingomonas sp. CBA4 isolated from San Roque reservoir (Córdoba-Argentina). Biodegradation 2006, 17, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Yan, H.; Wang, J.; Wei, W.; Ning, J.; Pan, G. Microcystin-LR biodegradation by Sphingopyxis sp. USTB-05. Front. Environ. Sci. Eng. China 2010, 5, 526–532. [Google Scholar] [CrossRef]
- Nybom, S.M.K.; Salminen, S.J.; Meriluoto, J.A.O. Removal of microcystin-LR by strains of metabolically active probiotic bacteria. FEMS Microbiol. Lett. 2007, 270, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Maseda, H.; Lu, Z.; Utsumi, M.; Zhang, Z.; Sugiura, N. Investigations into the biodegradation of microcystin-LR mediated by the biofilm in wintertime from a biological treatment facility in a drinking-water treatment plant. Bioresour. Technol. 2012, 106, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Eleuterio, L.; Batista, J.R. Biodegradation studies and sequencing of microcystin-LR degrading bacteria isolated from a drinking water biofilter and a fresh water lake. Toxicon 2010, 55, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Sakharkar, M.K.; Utsumi, M.; Zhang, Z.; Sugiura, N. Comparative study for the effects of variable nutrient conditions on the biodegradation of microcystin-LR and concurrent dynamics in microcystin-degrading gene abundance. Bioresour. Technol. 2011, 102, 9509–9517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, H.; Liu, A.; Cao, Z.; Hao, J.; Gong, R. Identification of a new microcystin-degrading bacterium isolated from Lake Chaohu, China. Bull. Environ. Contam. Toxicol. 2015, 94, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Surono, I.S.; Collado, M.C.; Salminen, S.; Meriluoto, J. Effect of glucose and incubation temperature on metabolically active Lactobacillus plantarum from dadih in removing microcystin-LR. Food Chem. Toxicol. 2008, 46, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Kómarek, J.; Anagnostidis, K. Cyanoprokaryota 1. Teil Chroococales. In Süsswasserflora von Mitteleuropa; Ettl, H., Gärtner, G., Heynig, H., Mollenhauer, D., Eds.; Gustav Fisher: Jena, Germany, 1999; p. 548. [Google Scholar]
- World Health Organization (WHO). Guidelines for Drinking-Water Quality, 2nd ed.; WHO: Geneva, Switzerland, 1998; Volume 2. [Google Scholar]
- World Health Organization (WHO). Guidelines for Safe Recreational Water Environments. Coastal and Freshwaters; WHO: Geneva, Switzerland, 2003; Volume 1. [Google Scholar]
- Agha, R.; Cirés, S.; Wörmer, L.; Domínguez, J.A.; Quesada, A. Multi-scale strategies for the monitoring of freshwater cyanobacteria: Reducing the sources of uncertainty. Water Res. 2012, 46, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.; Moreno, E.; Sanchis, D.; Wörmer, L.; Paniagua, T.; Del Cueto, A.; Quesada, A. Cyanobacterial abundance and microcystin occurrence in Mediterranean water reservoirs in Central Spain: Microcystins in the Madrid area. Eur. J. Phycol. 2006, 41, 281–291. [Google Scholar] [CrossRef]
- Moreno, I.; Repetto, G.; Carballal, E.; Gago, A.; Cameán, A.M. Cyanobacteria and microcystins occurrence in the Guadiana River (SW Spain). Int. J. Environ. Anal. Chem. 2005, 85, 461–474. [Google Scholar] [CrossRef]
- Gkelis, S.; Papadimitriou, T.; Zaoutsos, N.; Leonardos, I. Anthropogenic and climate-induced change favors toxic cyanobacteria blooms: Evidence from monitoring a highly eutrophic, urban Mediterranean lake. Harmful Algae 2014, 39, 322–333. [Google Scholar] [CrossRef]
- Jančula, D.; Straková, L.; Sadílek, J.; Maršálek, B.; Babica, P. Survey of cyanobacterial toxins in Czech water reservoirs-the first observation of neurotoxic saxitoxins. Environ. Sci. Pollut. Res. 2014, 21, 8006–8015. [Google Scholar] [CrossRef] [PubMed]
- Messineo, V.; Bogialli, S.; Melchiorre, S.; Sechi, N.; Lugliè, A.; Casiddu, P.; Mariani, M.A.; Padedda, B.M.; Corcia, A.D.; Mazza, R.; et al. Cyanobacterial toxins in Italian freshwaters. Limnol. Ecol. Manag. Inland Waters 2009, 39, 95–106. [Google Scholar] [CrossRef]
- Orihel, D.M.; Bird, D.F.; Brylinsky, M.; Chen, H.; Donald, D.B.; Huang, D.Y.; Giani, A.; Kinniburgh, D.; Kling, H.; Kotak, B.G.; et al. High microcystin concentrations occur only at low nitrogen-to-phosphorus ratios in nutrient-rich Canadian lakes. Can. J. Fish. Aquat. Sci. 2012, 69, 1457–1462. [Google Scholar] [CrossRef]
- Su, X.; Xue, Q.; Steinman, A.; Zhao, Y.; Xie, L. Spatiotemporal dynamics of microcystin variants and relationships with environmental parameters in Lake Taihu, China. Toxins (Basel) 2015, 7, 3224–3244. [Google Scholar] [CrossRef] [PubMed]
- De Senerpont Domis, L.N.; Elser, J.J.; Gsell, A.S.; Huszar, V.L.M.; Ibelings, B.W.; Jeppesen, E.; Kosten, S.; Mooij, W.M.; Roland, F.; Sommer, U.; et al. Plankton dynamics under different climatic conditions in space and time. Freshw. Biol. 2013, 58, 463–482. [Google Scholar] [CrossRef]
- Romo, S.; Soria, J.; Fernández, F.; Ouahid, Y.; Barón-Solá, Á. Water residence time and the dynamics of toxic cyanobacteria. Freshw. Biol. 2013, 58, 513–522. [Google Scholar] [CrossRef]
- Berg, K.A.; Lyra, C.; Sivonen, K.; Paulin, L.; Suomalainen, S.; Tuomi, P.; Rapala, J. High diversity of cultivable heterotrophic bacteria in association with cyanobacterial water blooms. ISME J. 2009, 3, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xing, P.; Wu, Q.L. The high resilience of the bacterioplankton community in the face of a catastrophic disturbance by a heavy Microcystis bloom. FEMS Microbiol. Ecol. 2012, 82, 192–201. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, L.; Wu, Y.; Song, L.; Gan, N. Ecological dynamics of toxic Microcystis spp. and microcystin-degrading bacteria in Dianchi Lake, China. Appl. Environ. Microbiol. 2014, 80, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Akasako, H.; Lu, Z.; Akiyama, S.; Goto, M.; Utsumi, M.; Sugiura, N. Assessment of the factors contributing to the variations in microcystins biodegradability of the biofilms on a practical biological treatment facility. Bioresour. Technol. 2015, 175, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Hoefel, D.; Palazot, S.; Sawade, E.; Newcombe, G.; Saint, C.P.; Brookes, J.D. Investigations into the biodegradation of microcystin-LR in wastewaters. J. Hazard. Mater. 2010, 180, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Peng, L.; Li, J.; Qiao, Y. Divergent responses of functional gene expression to various nutrient conditions during microcystin-LR biodegradation by Novosphingobium sp. THN1 strain. Bioresour. Technol. 2014, 156, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Tang, T.; Monis, P.T.; Hoefel, D. Biodegradation of multiple cyanobacterial metabolites in drinking water supplies. Chemosphere 2012, 87, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Park, H.D.; Sasaki, Y.; Maruyama, T.; Yanagisawa, E.; Hiraishi, A.; Kato, K. Degradation of the cyanobacterial hepatotoxin microcystin by a new bacterium isolated from a hypertrophic lake. Environ. Toxicol. 2001, 16, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rapala, J.; Berg, K.A.; Lyra, C.; Niemi, R.M.; Manz, W.; Suomalainen, S.; Paulin, L.; Lahti, K. Paucibacter toxinivorans gen. nov., sp. nov., a bacterium that degrades cyclic cyanobacterial hepatotoxins microcystins and nodularin. Int. J. Syst. Evol. Microbiol. 2005, 55, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Addinsoft XLstat 2016: Leading data analysis and statistical solution for microsoft excel. Avaliable online: www.xlstat.com (accessed on 2 November 2016).
- Jungblut, A.-D.; Neilan, B.A. Molecular identification and evolution of the cyclic peptide hepatotoxins, microcystin and nodularin, synthetase genes in three orders of cyanobacteria. Arch. Microbiol. 2006, 185, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [PubMed]
- Turner, S.; Pryer, K.M.; Miao, V.P.W.; Palmer, J.D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999, 46, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Sestonic MCs (µg·L−1) | mcyE | |||
|---|---|---|---|---|---|
| MC-LR | MC-RR | MC-YR | Total MCs | ||
| 7 August 2012 | 0.31 ± 0.20 | 0.16 ± 0.02 | 0.07 ± 0.00 | 0.54 ± 0.22 | + |
| 4 September 2012 | 10.17 ± 1.59 | 3.14 ± 0.18 | 0.99 ± 0.07 | 14.31 ±1.84 | + |
| 24 September 2012 | 25.75 ± 6.10 | 16.82 ± 3.82 | 6.95 ± 1.45 | 49.52 ± 11.37 | + |
| 8 October 2012 | 13.64 ± 1.43 | 5.89 ± 0.74 | 2.42 ± 0.26 | 21.95 ± 0.43 | + |
| 29 October 2012 | 0.85 ± 0.26 | 0.52 ± 0.14 | 0.23 ± 0.06 | 1.60 ± 0.06 | + |
| Date | Control (µg·L−1) | Reservoir Water (µg·L−1) | ||
|---|---|---|---|---|
| Day 0 | Day 15 | Day 0 | Day 15 | |
| 7 August 2012 | 1315 ± 32 | 1239 ± 26 | 1196 ± 80 | n.d. |
| 4 September 2012 | 1238 ± 35 | 1219 ± 95 | 1206 ± 22 | n.d. |
| 24 September 2012 | 1303 ± 15 | 1321 ± 52 | 1200 ± 30 | n.d. |
| 8 October 2012 | 1145 ± 69 | 1179 ± 125 | 1173 ± 65 | n.d. |
| 29 October 2012 | 1183 ± 45 | 1284 ± 22 | 1211 ± 44 | n.d. |
| Bacterial Strains | mlrA-D Genes | Degradation Rates (µg MC L−1 h−1) | ||
|---|---|---|---|---|
| MSM | ¼ R2A Medium | Reservoir Water | ||
| Y2 | + | 73.24 ± 6.09 | 113.36 ± 22.33 | 48.2 ± 16.80 |
| 2C20 | − | 12.75 ± 2.93 | 8.04 ± 4.94 | 20.62 ± 4.00 |
| IM-1 | + | 171.15 ± 15.34 | 146.92 ± 23.79 | 144.9 ± 54.02 |
| IM-2 | + | 25.35 ± 6.04 | 97.03 ± 40.47 | 11.39 ± 5.28 |
| IM-3 | + | 37.10 ± 7.20 | 112.14 ± 21.36 | 16.30 ± 0.71 |
| IM-4 | − | 19.42 ± 1.12 | 4.12 ± 1.37 | 21.26 ± 3.36 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lezcano, M.Á.; Morón-López, J.; Agha, R.; López-Heras, I.; Nozal, L.; Quesada, A.; El-Shehawy, R. Presence or Absence of mlr Genes and Nutrient Concentrations Co-Determine the Microcystin Biodegradation Efficiency of a Natural Bacterial Community. Toxins 2016, 8, 318. https://doi.org/10.3390/toxins8110318
Lezcano MÁ, Morón-López J, Agha R, López-Heras I, Nozal L, Quesada A, El-Shehawy R. Presence or Absence of mlr Genes and Nutrient Concentrations Co-Determine the Microcystin Biodegradation Efficiency of a Natural Bacterial Community. Toxins. 2016; 8(11):318. https://doi.org/10.3390/toxins8110318
Chicago/Turabian StyleLezcano, María Ángeles, Jesús Morón-López, Ramsy Agha, Isabel López-Heras, Leonor Nozal, Antonio Quesada, and Rehab El-Shehawy. 2016. "Presence or Absence of mlr Genes and Nutrient Concentrations Co-Determine the Microcystin Biodegradation Efficiency of a Natural Bacterial Community" Toxins 8, no. 11: 318. https://doi.org/10.3390/toxins8110318
APA StyleLezcano, M. Á., Morón-López, J., Agha, R., López-Heras, I., Nozal, L., Quesada, A., & El-Shehawy, R. (2016). Presence or Absence of mlr Genes and Nutrient Concentrations Co-Determine the Microcystin Biodegradation Efficiency of a Natural Bacterial Community. Toxins, 8(11), 318. https://doi.org/10.3390/toxins8110318