
Molecular Cloning and Functional Analysis of Gene Clusters for the Biosynthesis of Indole-Diterpenes in Penicillium crustosum and P. janthinellum

Abstract
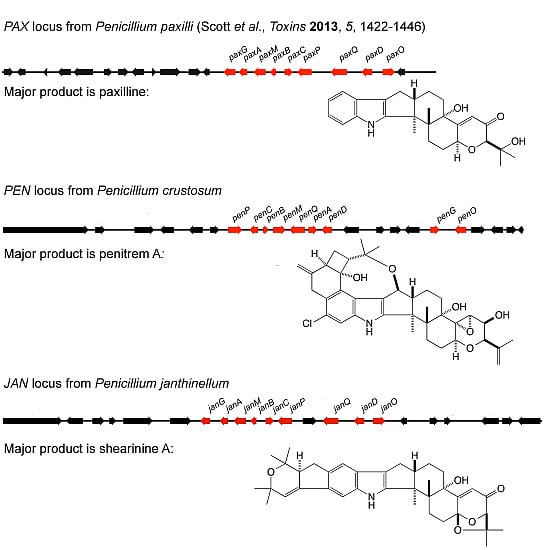
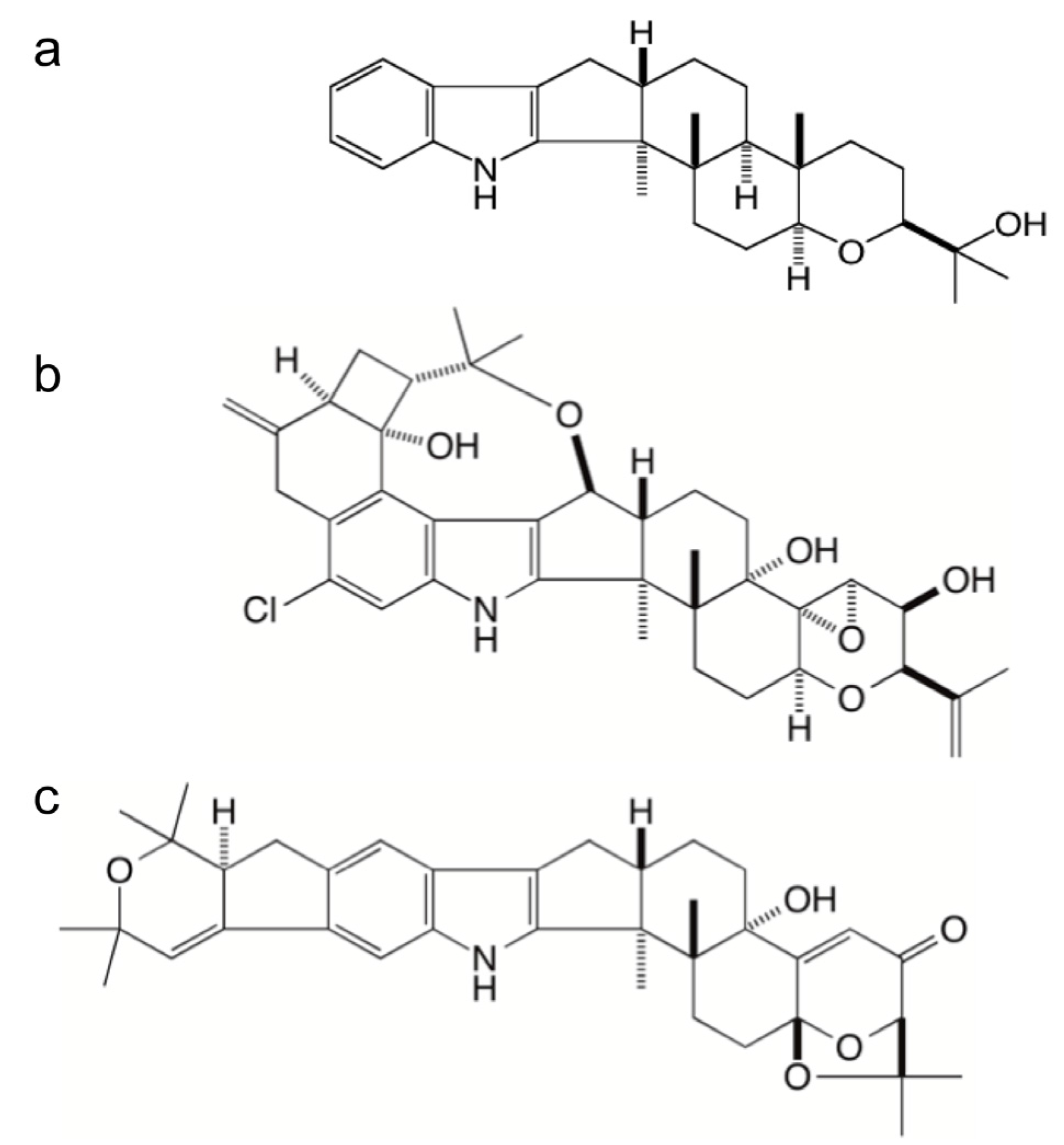
:
1. Introduction

2. Results
2.1. Identification of Gene Clusters for Indole-Diterpene Biosynthesis in P. crustosum and P. janthinellum
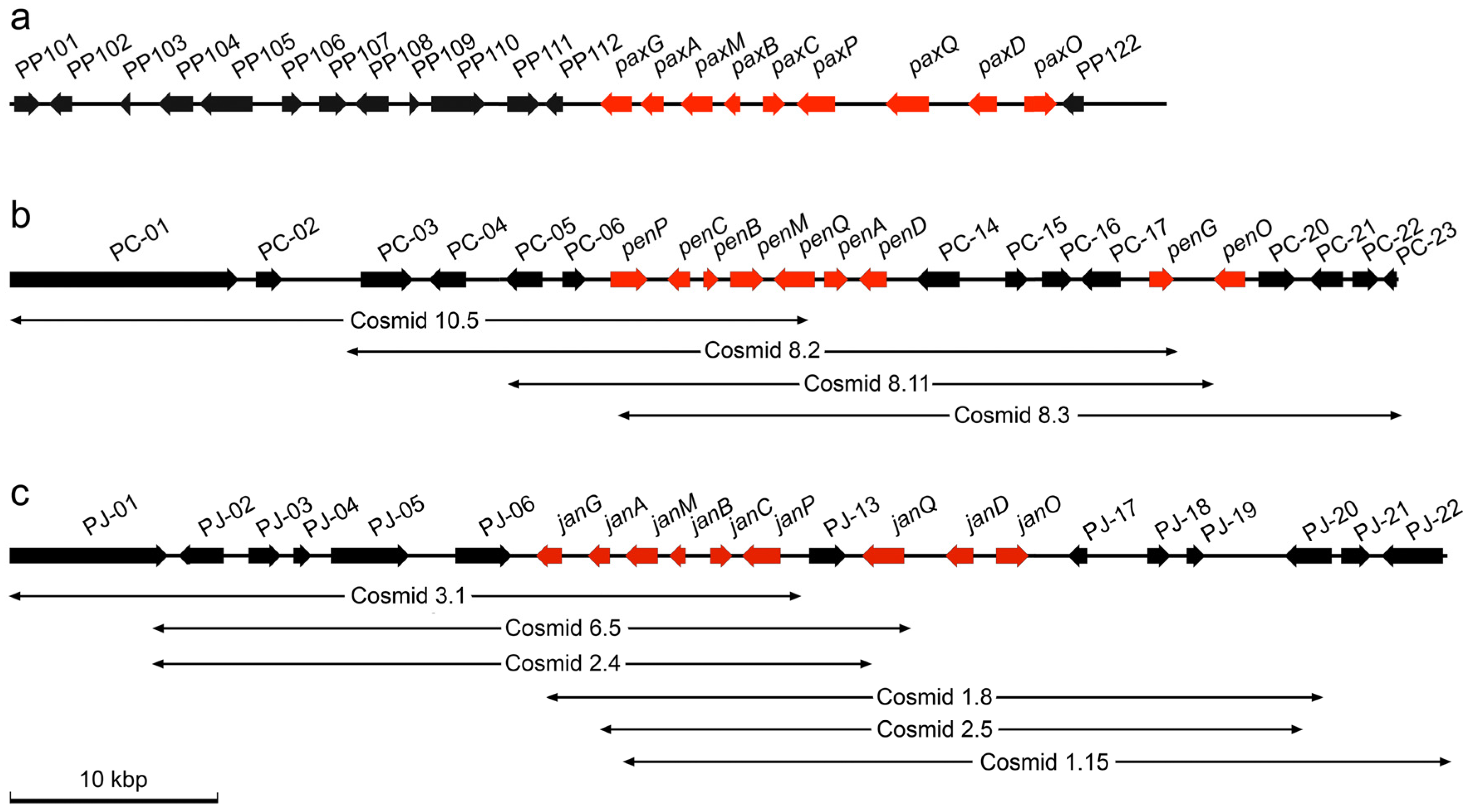

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/ORF | No. of Exons | Predicted Product Size (aa) | Predicted Function | Top BLASTp Match | ||
|---|---|---|---|---|---|---|
| Organism | E-Value | Accession No. | ||||
| PC-01 (partial) | ≥5 | ≥3555 | NRPS (HC-toxin synthase) | Penicillium digitatum | 0.0 | EKV04333 |
| PC-02 | 1 | 445 | Hypothetical | Aspergillus oryzae RIB40 | 1e−166 | XP_003189837 |
| PC-03 | 2 | 743 | Hypothetical cell wall protein | P. digitatum | 0.0 | EKV04334 |
| PC-04 | 5 | 492 | Conserved PWI domain | P. chrysogenum | 8e−132 | XP_002563178 |
| PC-05 | 6 | 335 | Cytochrome P450 monooxygenase (GA14 Synthase) | A. oryzae RIB40 | 2e−149 | XP_001827555 |
| PC-06 | 4 | 335 | NmrA-family transcriptional regulator | A. flavus NRRL3357 | 7e−120 | XP_002384043 |
| PC-07 (penP) | 6 | 520 | Cytochrome P450 monooxygenase | P. paxilli | 0.0 | AAK11528 |
| PC-08 (penC) | 3 | 342 | Prenyl transferase | P. chrysogenum | 2e−168 | XP_002562743 |
| PC-09 (penB) | 2 | 243 | Integral membrane protein | P. paxilli | 5e−130 | ADO29934 |
| PC-10 (penM) | 4 | 465 | FAD-dependent monooxygenase | P. paxilli | 0.0 | AAK11530 |
| PC-11 (penQ) | 9 | 510 | Cytochrome P450 monooxygenase | P. paxilli | 0.0 | AAK11527 |
| PC-12 (penA) | 2 | 368 | Integral membrane protein | P. paxilli | 2e−53 | ADO29933 |
| PC-13 (penD) | 2 | 427 | Aromatic prenyl transferase | P. paxilli | 3e−85 | AAAK11526 |
| PC-14 | 5 | 577 | Dimethylaniline monooxygenase | Metarhizium acridum | 2e−170 | EFY85025 |
| PC-15 | 3 | 271 | Oxidoreductase/short chain dehydrogenase | Glarea lozoyensis | 6e−77 | EPE26761 |
| PC-16 | 1 | 494 | Acetyltransferase | A. oryzae RIB40 | 5e−116 | XP_001822009 |
| PC-17 | 5 | 543 | MFS transporter | P. chrysogenum | 0.0 | XP_002562735 |
| PC-18 (penG) | 4 | 341 | Geranylgeranyl diphosphate synthase | A. niger ATCC1015 | 1e−133 | EHA20968 |
| PC-19 (penO) | 4 | 450 | FAD-binding oxidoreductase | A. oryzae RIB40 | 1e−108 | XP_001817261 |
| PC-20 | 4 | 453 | Cytochrome P450 monooxygenase | Pyrenophora tritici-repentis | 5e−49 | XP_001938007 |
| PC-21 | 3 | 510 | Cytochrome P450 monooxygenase | Exophiala dermatitidis | 1e−119 | EHY54727 |
| PC-22 | 2 | 425 | Aromatic prenyl transferase | Aspergillus sp. MF297 | 1 e−78 | ADM34132 |
| PC-23 | 3 | 204 | Cytochrome P450 monooxygenase (sterigmatocystin stcS) | A. flavus NRRL3357 | 1e−37 | XP_002384465 |
| Gene/ORF | No. of exons | Predicted product size (aa) | Predicted function | Top BLASTp match | ||
|---|---|---|---|---|---|---|
| Organism | E-value | Accession no. | ||||
| PJ-01 | 6 | 2361 | Pfs domain protein | Aspergillus fumigatus | 0.0 | XP_748404 |
| PJ-02 | 2 | 476 | Transcriptional regulator Ngg1 | Penicillium oxalicum | 0.0 | EPS27964 |
| PJ-03 | 2 | 500 | RING finger domain protein | P. chrysogenum | 5e−100 | XP_002561235 |
| PJ-04 | 3 | 203 | 60S ribosomal protein | P. oxalicum | 1e−146 | EPS27962 |
| PJ-05 | 3 | 1216 | Ubiquitin protein lyase | P. oxalicum | 0.0 | EPS27961 |
| PJ-06 | 7 | 800 | Beta-glucosidase | P. oxalicum | 0.0 | EPS27960 |
| PJ-07 (janG) | 4 | 312 | Geranylgeranyl diphosphate synthase | P. chrysogenum | 9e−171 | XP_002562745 |
| PJ-08 (janA) | 2 | 349 | Integral membrane protein | P. paxilli | 4e−104 | ADO29933 |
| PJ-09 (janM) | 3 | 463 | FAD-dependent monooxygenase | P. paxilli | 0.0 | AAK11530 |
| PJ-10 (janB) | 2 | 243 | Integral membrane protein | P. paxilli | 2e−134 | ADO229934 |
| PJ-11 (janC) | 3 | 327 | Prenyl transferase | P. chrysogenum | 0.0 | XP_002562743 |
| PJ-12 (janP) | 6 | 515 | Cytochrome P450 monooxygenase | P. paxilli | 0.0 | AAK11528 |
| PJ-13 | 3 | 566 | Cytochrome P450 monooxygenase | Ajellomyces dermatitidis | 7e−128 | EEQ86297 |
| PJ-14 (janQ) | 9 | 469 | Cytochrome P450 monooxygenase | P. paxilli | 0.0 | AAK11527 |
| PJ-15 (janD) | 2 | 438 | Aromatic prenyl transferase | P. paxilli | 0.0 | AAK11526 |
| PJ-16 (janO) | 4 | 448 | FAD-binding oxidoreductase | P. paxilli | 0.0 | ADO29935 |
| PJ-17 | 4 | 283 | Conserved hypothetical | Talaromyces marneffei | 6e−32 | XP_002147239 |
| PJ-18 | 2 | 348 | Alcohol dehydrogenase | A. terreus | 0.0 | XP_001212944 |
| PJ-19 | 2 | 272 | FRG1-like family protein | P. oxalicum | 0.0 | EPS27959 |
| PJ-20 | 3 | 720 | Conserved hypothetical | P. oxalicum | 0.0 | EPS27958 |
| PJ-21 | 2 | 514 | Glucoronyl hydrolase | P. oxalicum | 0.0 | EPS27957 |
| PJ-22 | 7 | 801 | Transcriptional regulator | P. oxalicum | 0.0 | EPS27956 |
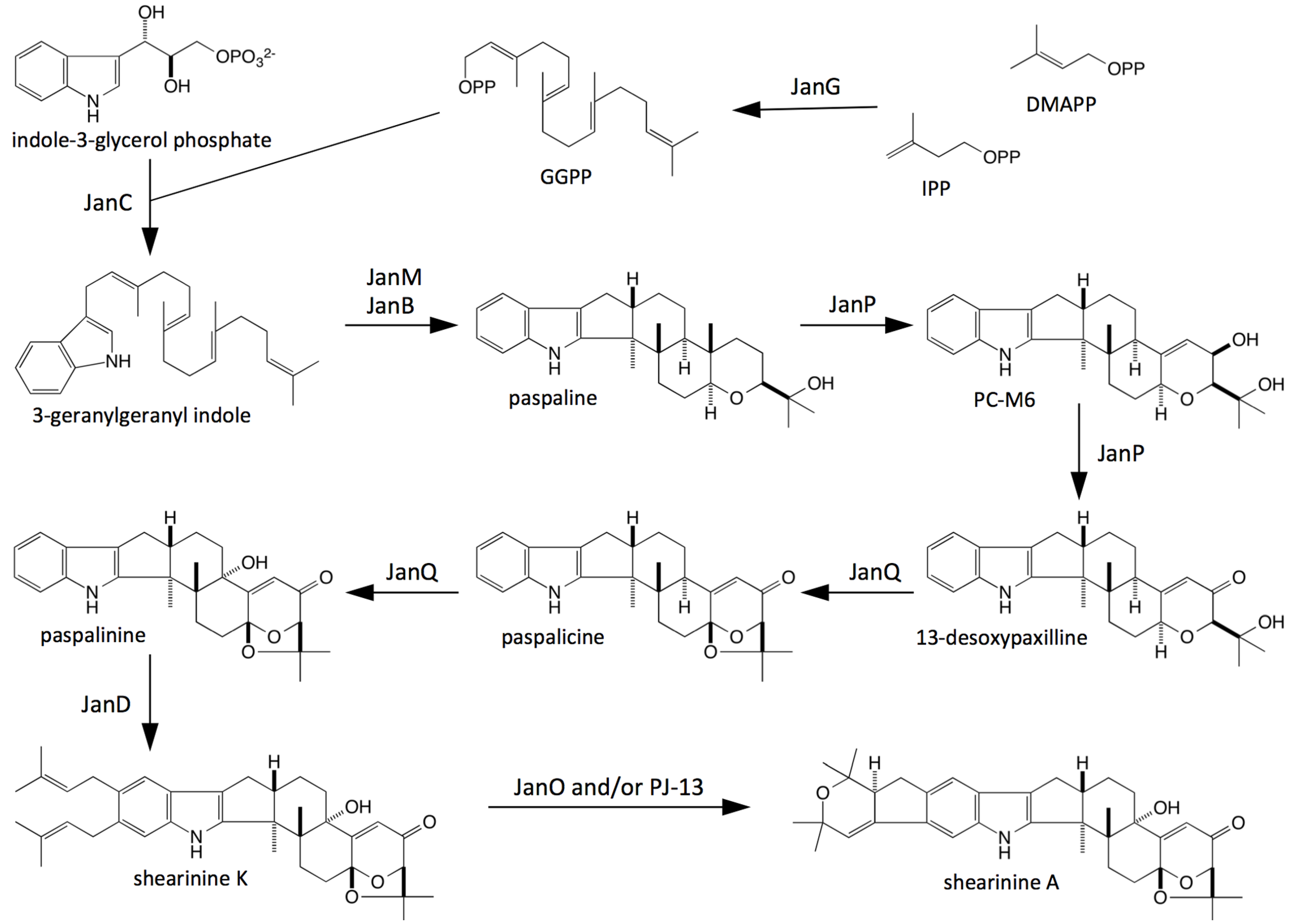
2.2. Bioinformatic Analysis of Indole-Diterpene Cluster Sequences
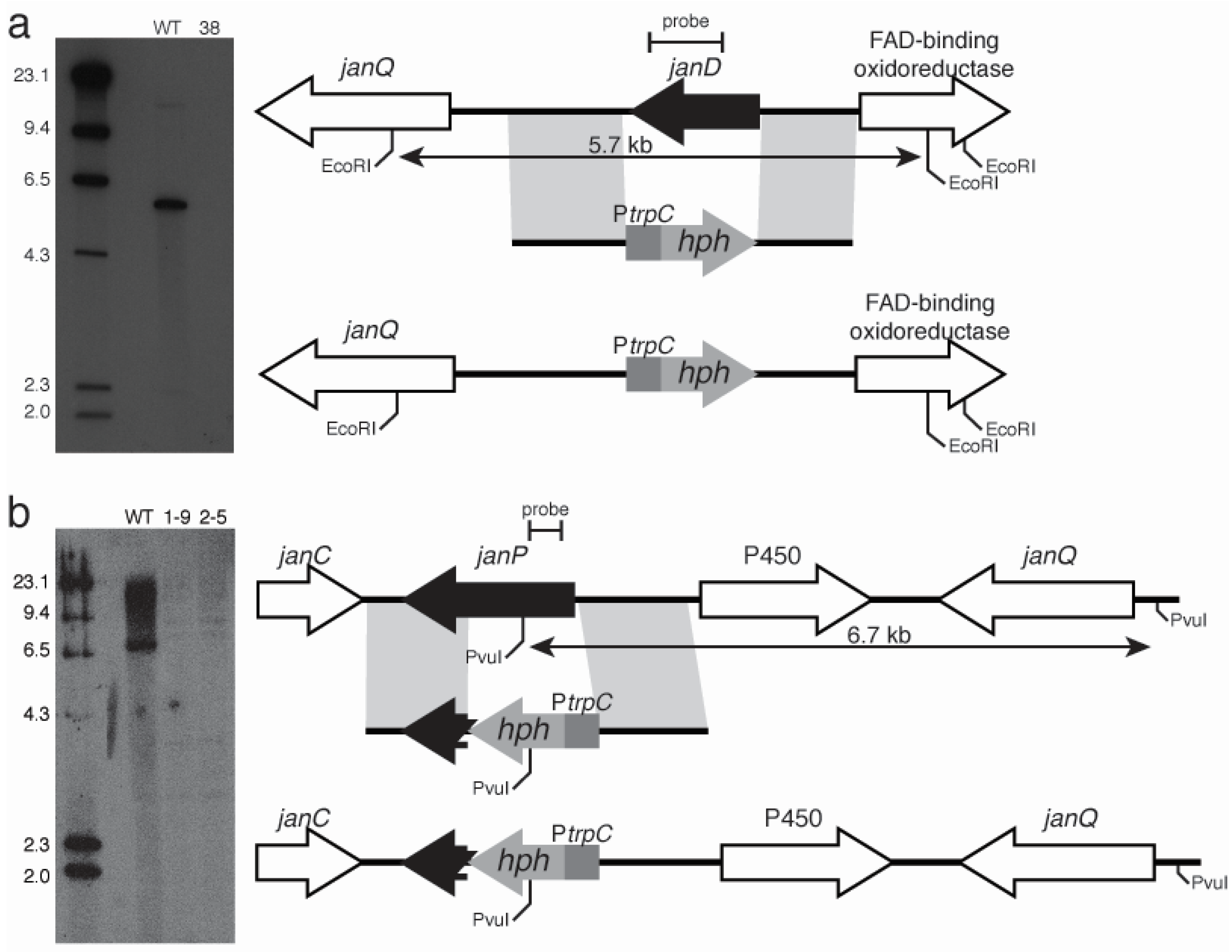
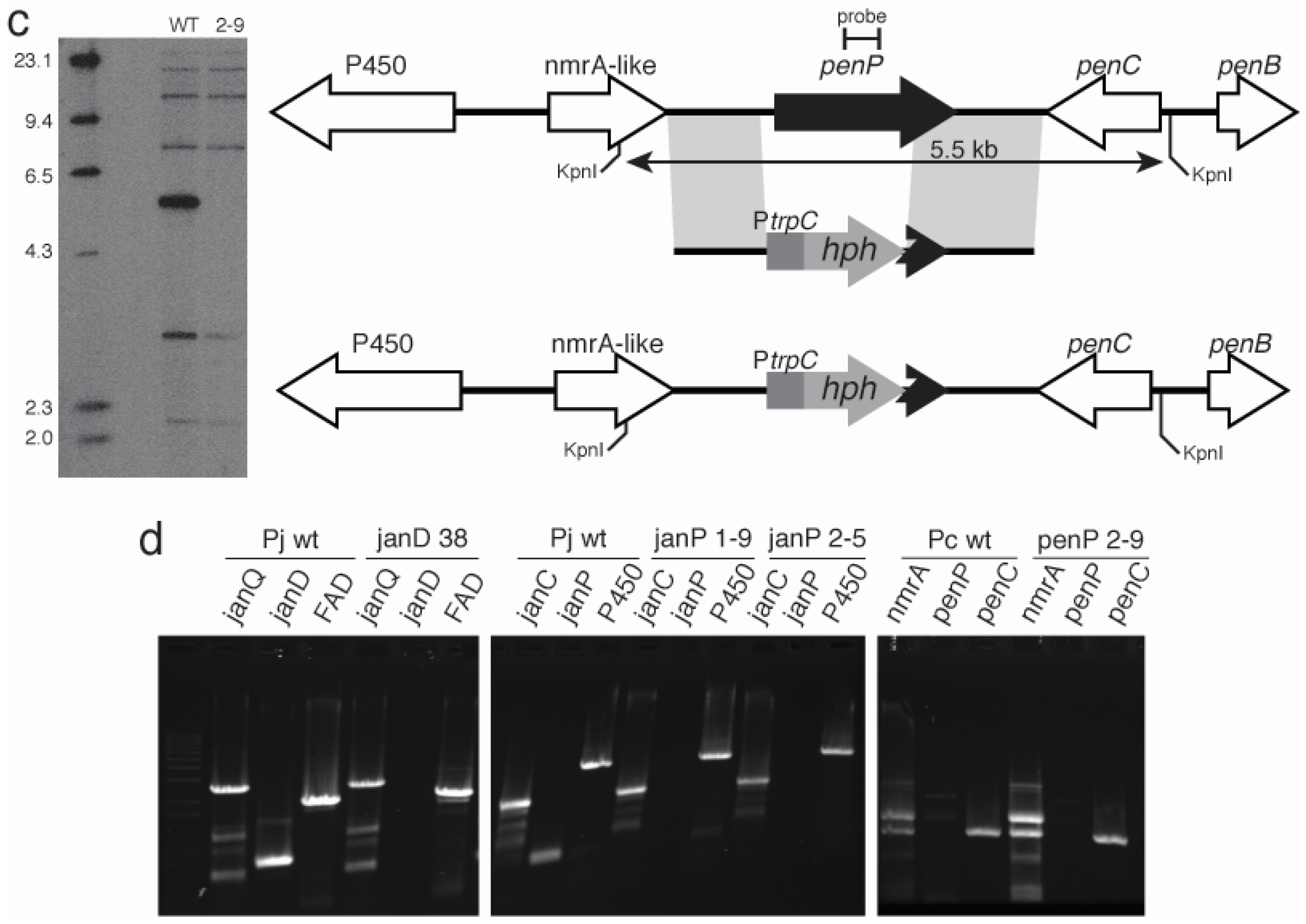
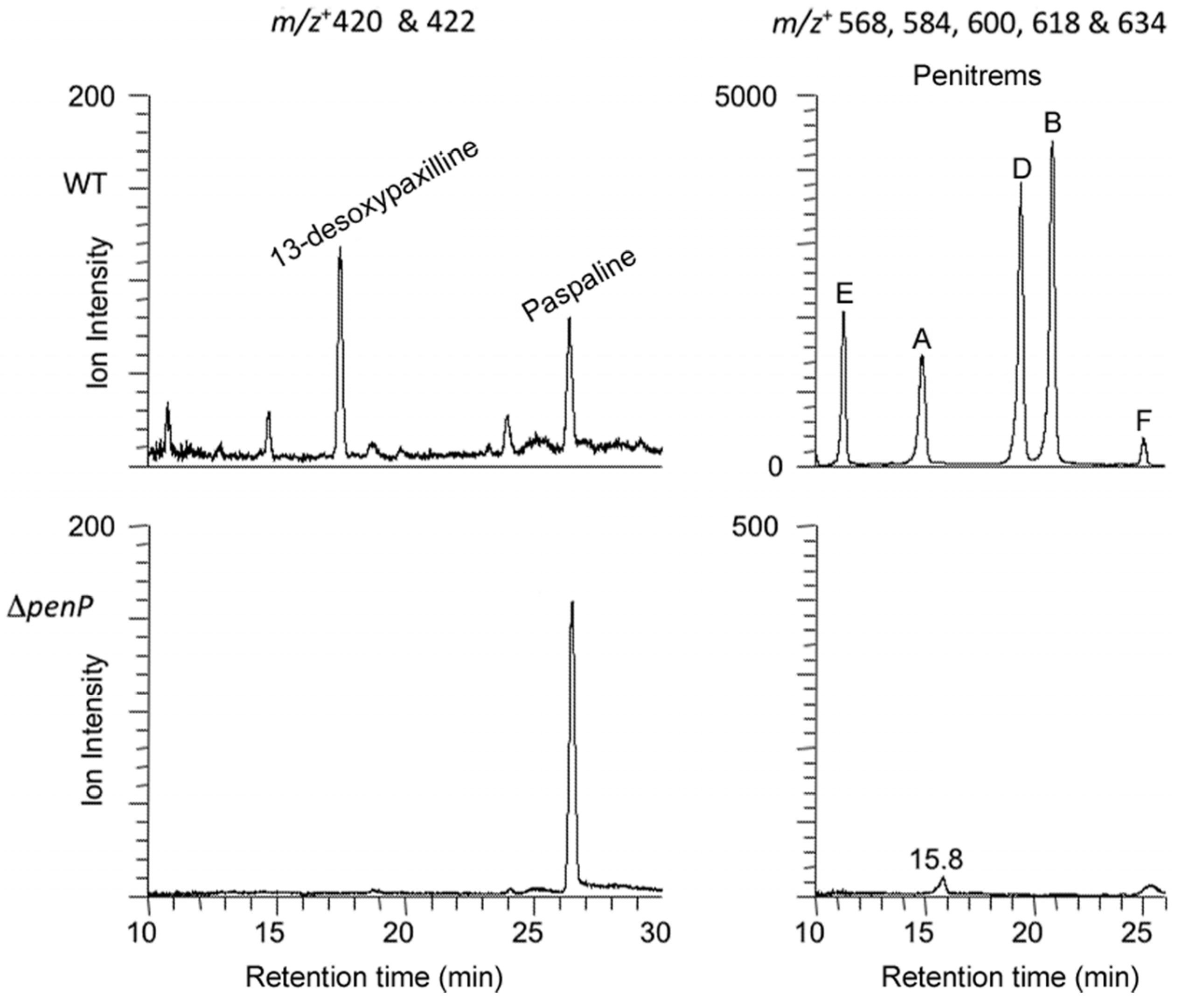
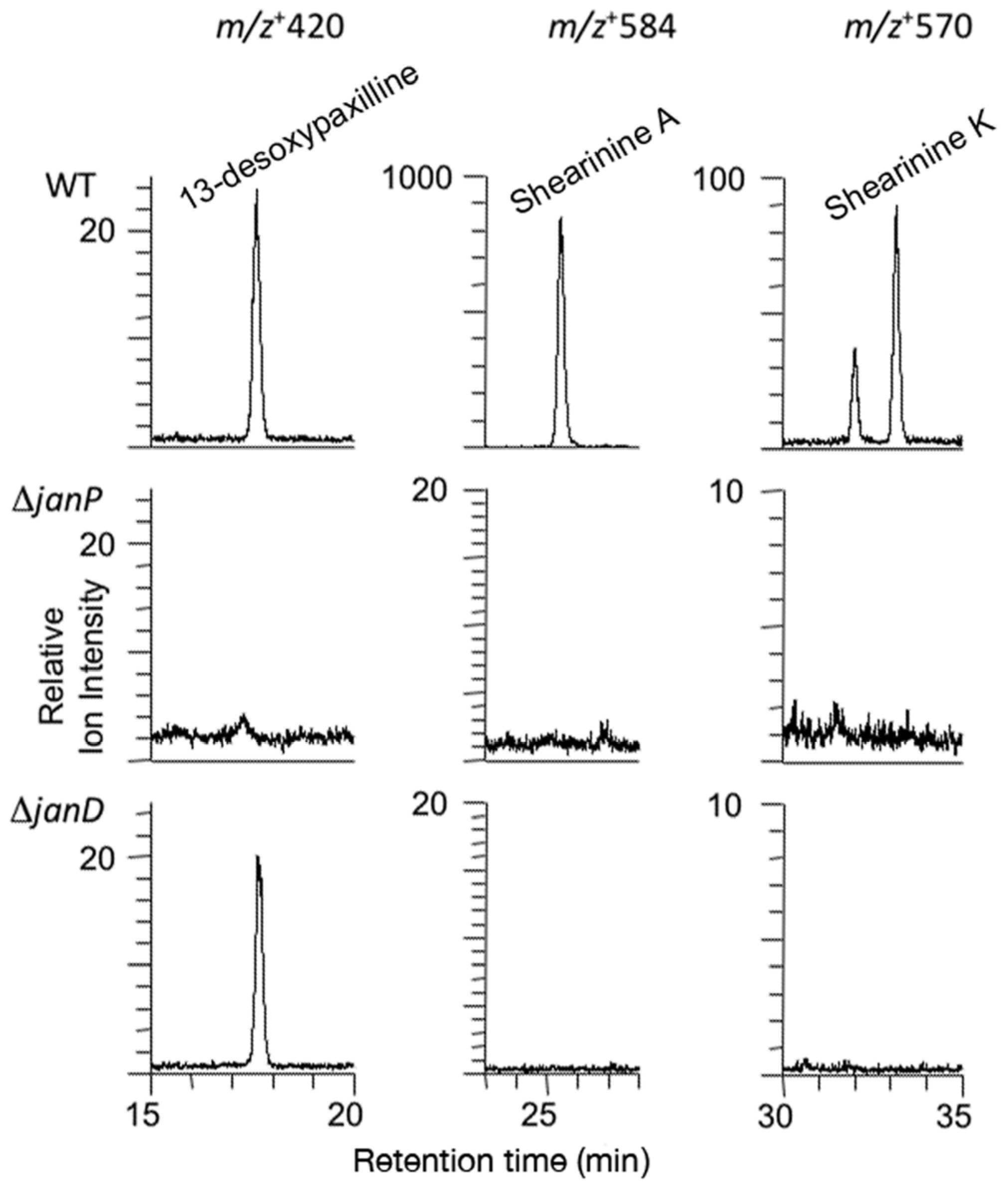
2.3. Disruption of Indole-Diterpene Biosynthesis by Deletion of Genes in the PEN and JAN Clusters of P. crustosum and P. janthinellum
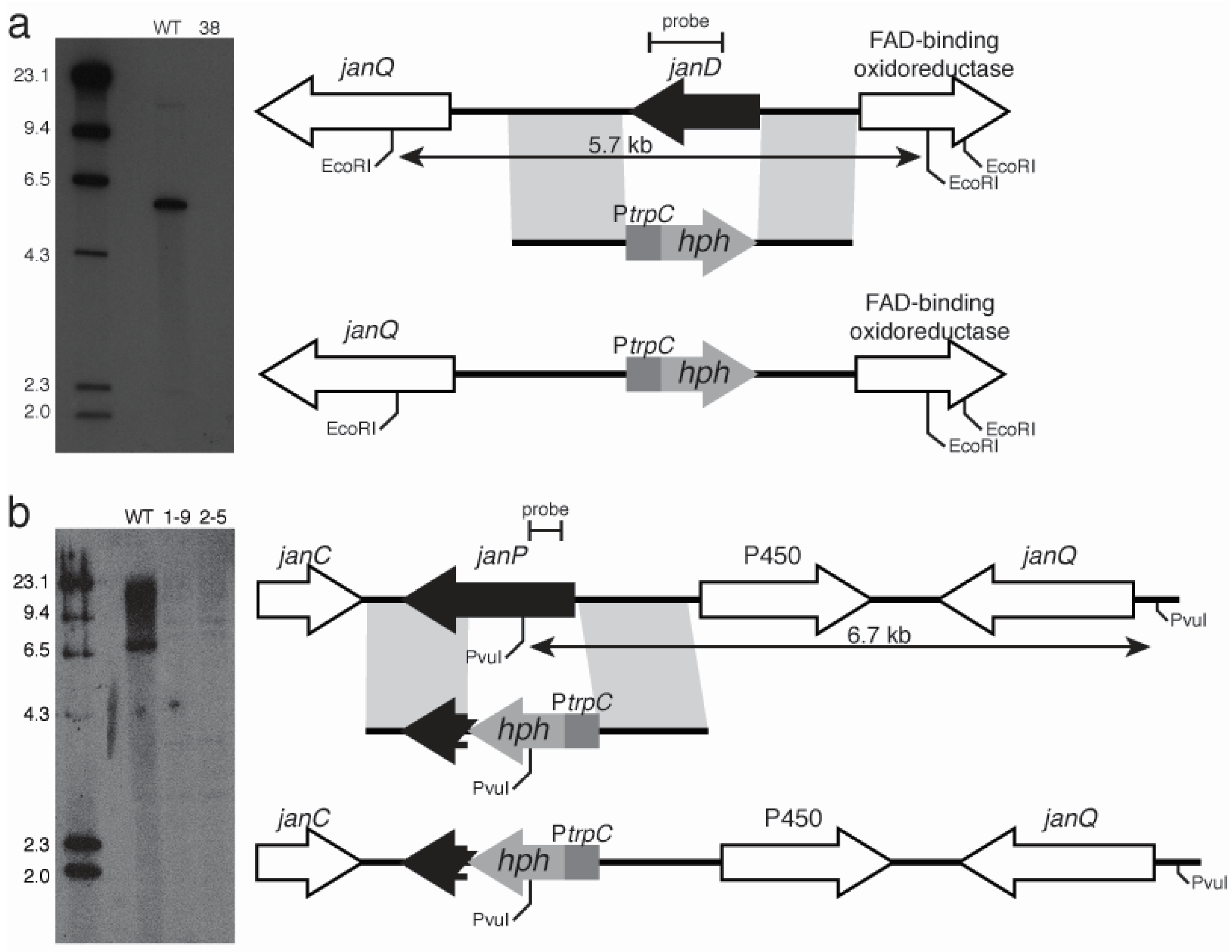
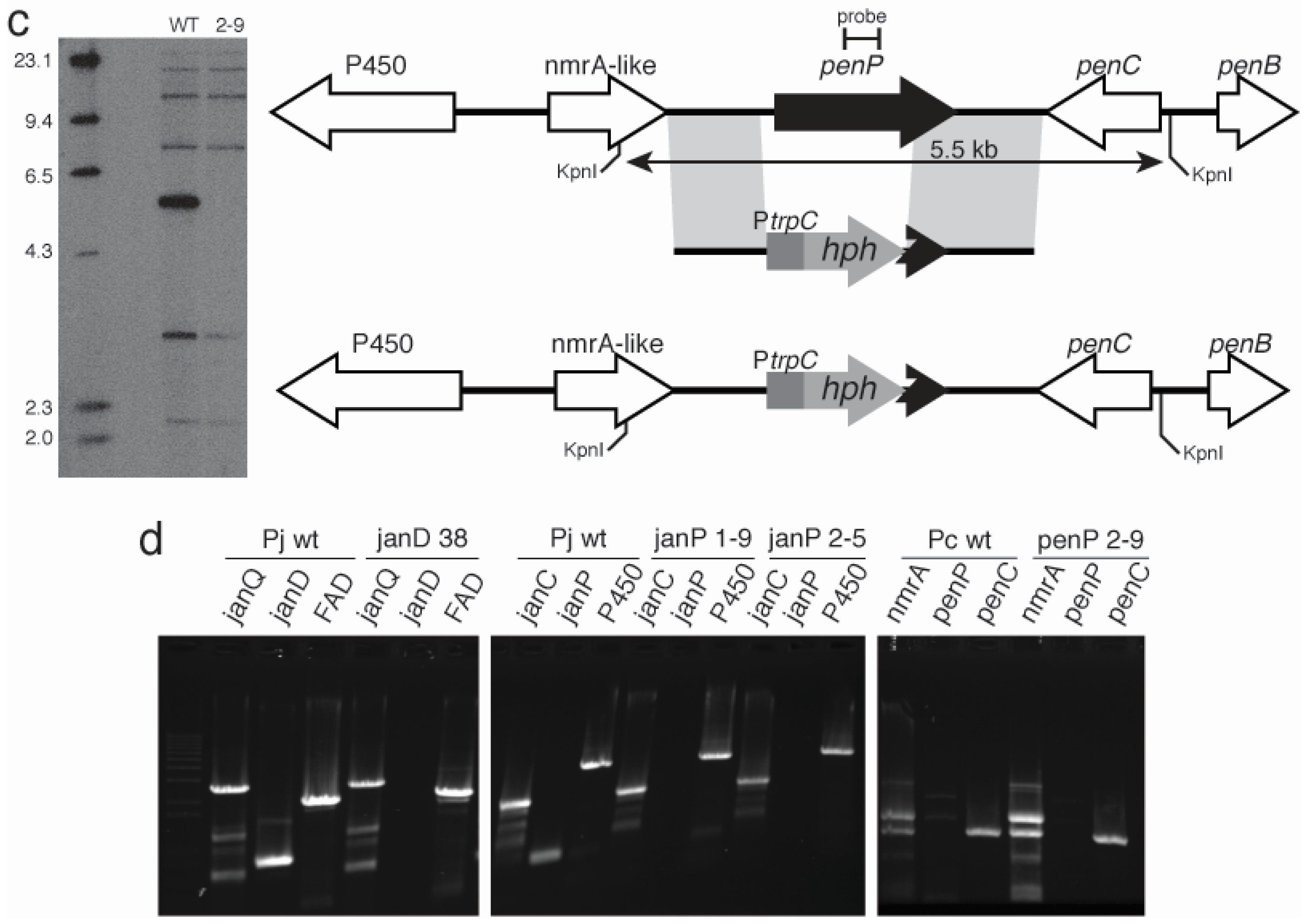
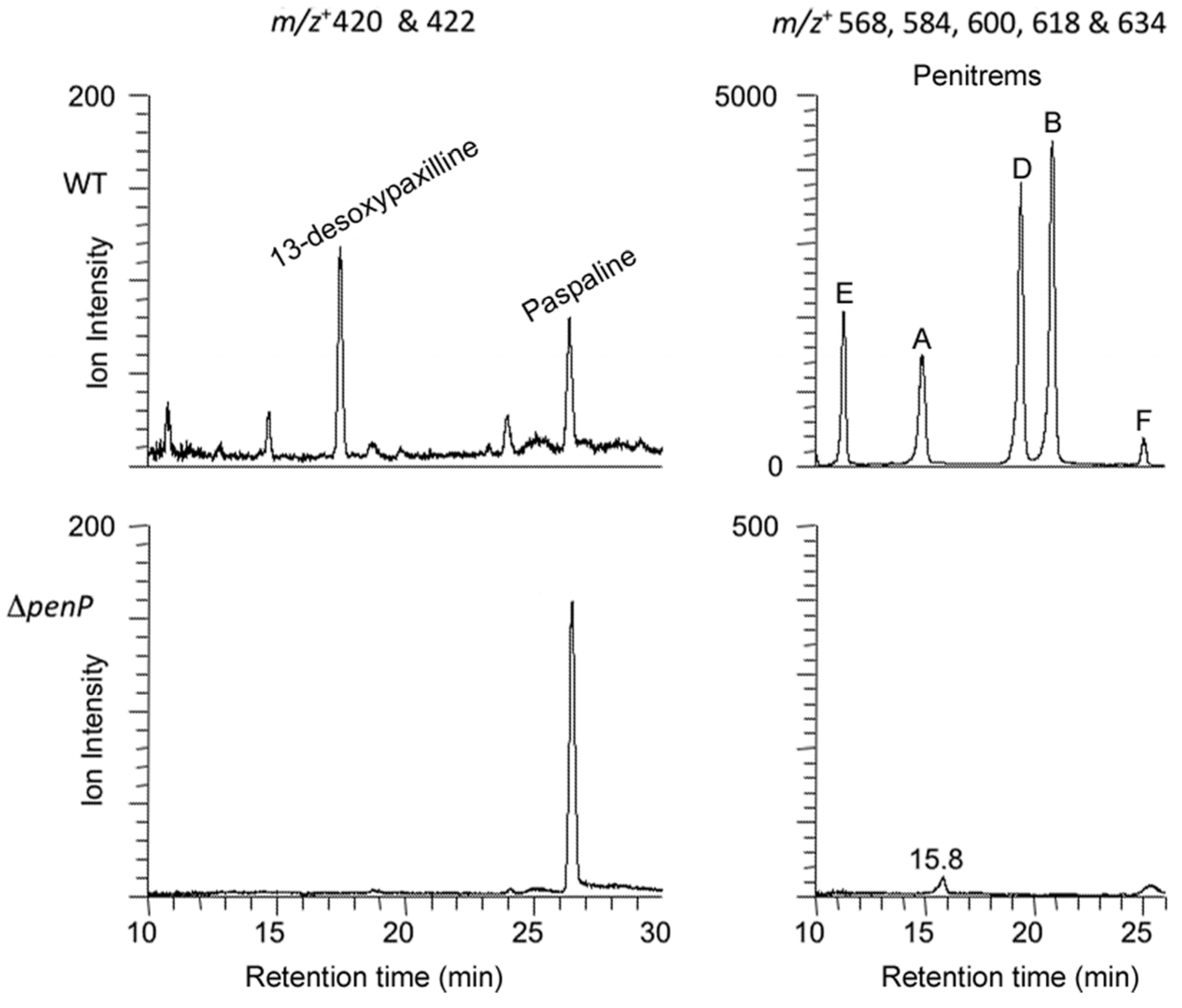
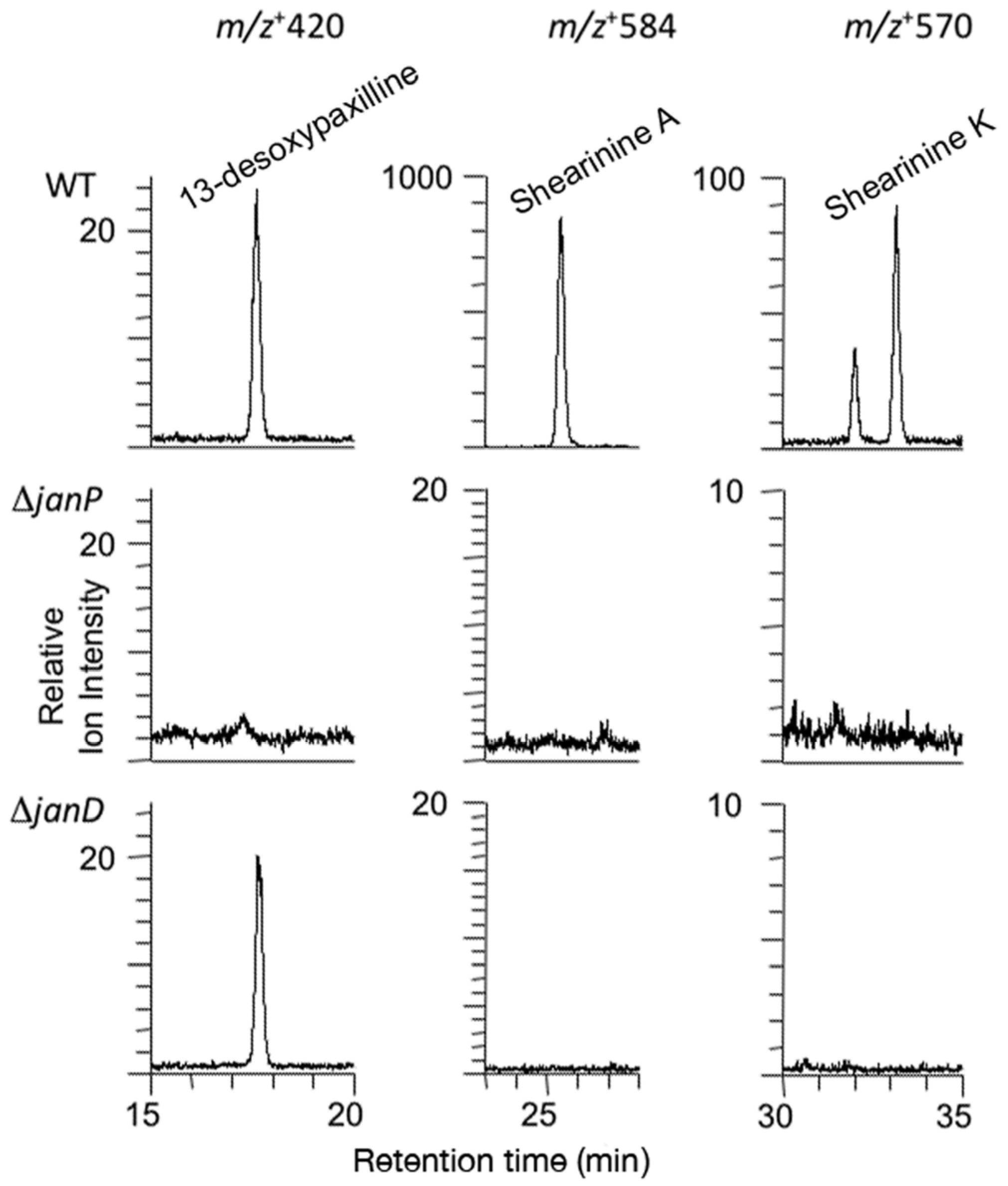
3. Discussion
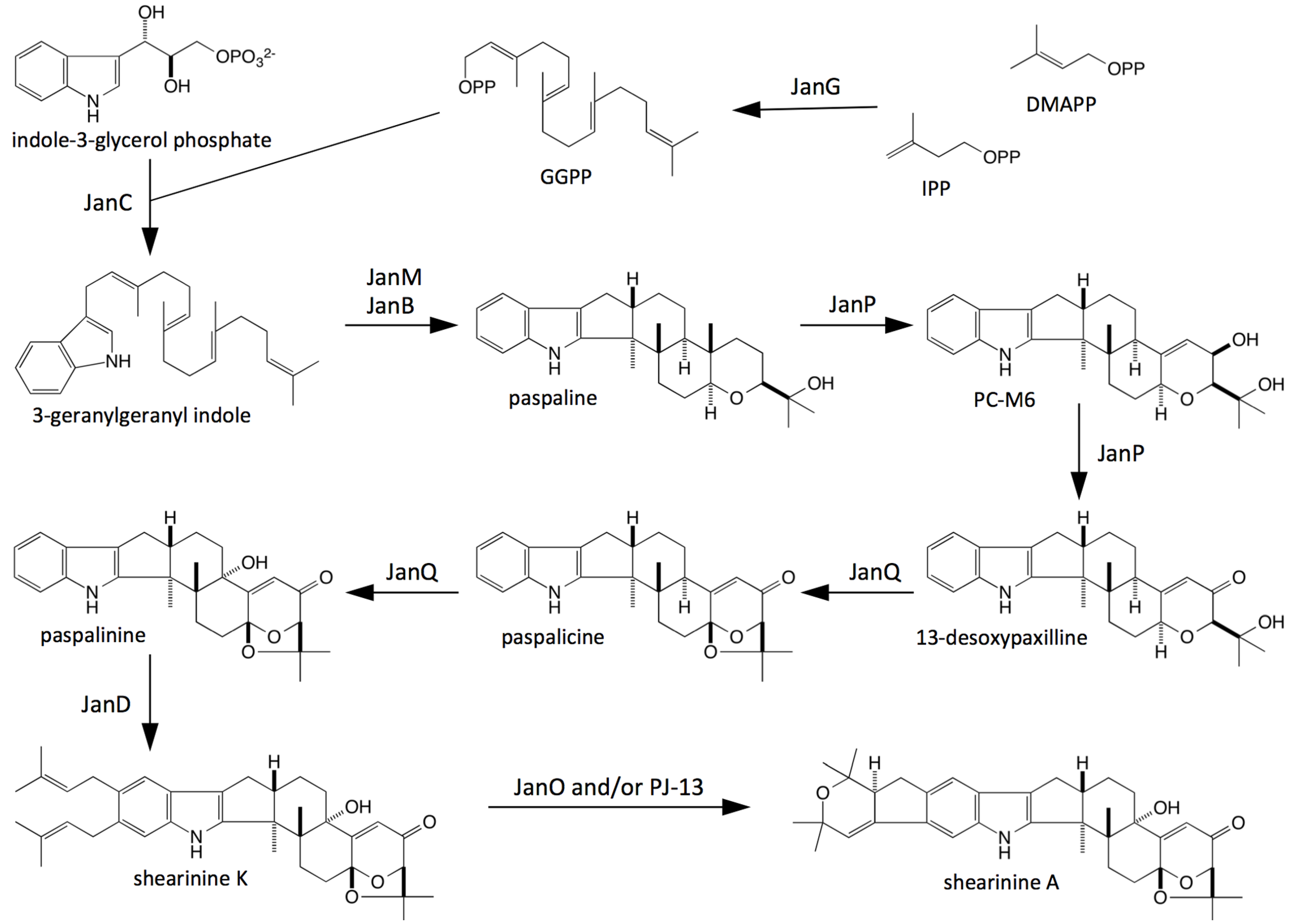
4. Experimental Section
4.1. Fungal Strains and Growth Conditions
4.2. Isolation, PCR-Amplification, and Sequencing of Genomic DNA
4.3. Cosmid Library Production and Screening
4.4. Cosmid Sequencing
4.5. Bioinformatics
4.6. Preparation of Gene Replacement Constructs and Southern Analysis
4.7. Fungal Transformation
4.8. Indole-Diterpene Chemical Analysis
4.9. Nucleotide Sequence Accession Numbers
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
| Primer 1 | Primer 2 | Target | Product (Species & Size) | ||
|---|---|---|---|---|---|
| Name | Sequence | Name | Sequence | ||
| ConC1 | TGCTTGATGATGGTCGAYGAT | ConC2 | GACATTCTTGCAGTCATTTTG | paxC homologs | P. crust. 529-bp P. janth. 53-bp |
| 2402F1 | GGATGATACCATGACCCTTGTCGCGTAT | PBR2 | TANGCNARNGCNGGNCCDATYTC | P. crust. C to P | P. crust. 1611-bp |
| 2402PF1 | CGGTTCTTCGCTGCCTTCGTCATGAAG | P. crust. C to P | P. crust. 1448-bp (probe) | ||
| PPF1 | AARCMNGAYGAYTTYYTNCARTGG | PPR2 | CANGCRTANCKNCCNARNCCRAARTG | paxP homologs | P. janth. 601-bp |
| 2408F1 | CTTTACCGTGATTGCGTAAGTCGCATT | 2408PF1 | GTGAGTACGGGTCCAAGTCATATGCAT | P. janth. C to P | 1045-bp (probe) |
| Primer Name | Sequence | Gene |
|---|---|---|
| CSnmrAF | AGTAGGGTAGGCAGCATCCA | Check for PC-06 (NmrA) |
| CSnmrAR | GCGCACTATTTCTGAGCAGC | Check for PC-06 |
| pen1 | ATGTCCAGATCCCTAACCCAC | Screen for penP deletion |
| pen2 | CATAATTGGGGTCACCTGATG | Screen for penP deletion |
| CSpenCF | CGTCGACAGAATCTCCAGCT | Check for penC |
| CSpenCR | TCATCCATCACACCGCGATT | Check for penC |
| CSjanCF | GGGGAAGGTAGCCATGCTTT | Check for janC |
| CSjanCR | TGCGGATATTACGAGGCGAC | Check for janC |
| jan1 | CCAATGTTAGAGTCCCAACG | Screen for jan P deletion |
| jan2 | GTCGTATACTCCCCGACATG | Screen for jan P deletion |
| CSP450F | ACGGTCACGGTCAAGTCTTC | Check for PJ-13 (P450) |
| CSP450R | CGTTACAGGGCCGGGTATTT | Check for PJ-13 (P450) |
| CSjanQF | TACAGGCCAGCTCTTCAACC | Check for janQ |
| CSjanQR | TCGGACATCTTTCGCACCAA | Check for janQ |
| CS janD intF | GGACTTACAAGGCTTTCGCA | Screen for janD deletion |
| CS janD intR | ACGCTTAAAGCCCAGAAACA | Screen for janD deletion |
| CSFADF | CTTCCTCGTGCCCTTGACAT | Check for janO (FAD) |
| CSFADR | GACCATGCTGTACCGAACGT | Check for janO (FAD) |
| Biological Material | Relevant Characteristics | Reference |
|---|---|---|
| Fungal strains | ||
| Penicillium crustosum | ||
| PN2402 (T2/3013) | Wild-type | Margaret di Menna |
| PN2897 (CE68/∆penP#2-9) | PN2402/∆penP::PtrpC-hph; HygR | This study |
| Penicillium janthinellum | ||
| PN2408 (C1P3) | Wild-type | Margaret di Menna |
| PN2898 (CE71/∆janP#1-9) | PN2408/∆janP::PtrpC-hph; HygR | This study |
| PN2899 (CE70/∆janP#2-5) | PN2408/∆janP::PtrpC-hph; HygR | This study |
| PN2883 (∆janD#38) | PN2408/∆janD::PtrpC-hph; HygR | This study |
| Yeast strains | ||
| Saccharomyces cerevisiae | ||
| FY834 (PN2806) | MATa his3 200ð ura3-52 leu2 1 lys2 202 trp1 63 | [43] |
| Bacterial strains | ||
| Escherichia coli | ||
| DH5α | F−, ϕ80lacZ, DM15, ∆(lacZYA-argF), U169, recA1, endA1, hsdR17 (rk−, mk−), phoA, supE44, λ−, thi-1, gyrA96, relA1 | Invitrogen |
| VCS257 | supE44 supF58 hsdS3(rB− mB−) dapD8 lacY1 glnV44 Δ(gal-uvrB)47 tyrT58 gyrA29 tonA53 Δ(thyA57) | Stratagene |
| PN4138 | DH5α/pRS426 | |
| PN4139 | DH5α/pCE50 penP1 | This study |
| PN4140 | DH5α/pCE51 janP1 | This study |
| PN4142 | DH5α/pCE52 janP2 | This study |
| PN4172 | DH5α/pCS5 janD | This study |
| Plasmids | ||
| pII99 (PN1687) | PtrpC-nptII-TtrpC, AmpR/GenR | [44] |
| pMOcosX | AmpR | [36] |
| pSF15.15 | pSP72 containing 1.4-kb HindIII PtrpC-hph from pCB1004 cloned into SmaI site | [45] |
| pRS426 | ori(f1)-lacZ-T7 promoter-MCS (KpnI-SacI)-T3 promoter-lacI-ori(pMB1)-ampR-ori (2 micron), URA3, AmpR | Fungal Genetics Stock Center |
| pCE50 | pRS426 containing 5′penP-PtrpC-hph-3′penP; AmpR/HygR | This study |
| pCE51 | pRS426 containing 5′japP-PtrpC-hph-3′janP; AmpR/HygR | This study |
| pCS5 | pRS426 containing 5′janD-PtrpC-hph-3′janD; AmpR/HygR | This study |
| Primer 1 | Primer 2 | Target | ||
|---|---|---|---|---|
| Name | Sequence | Name | Sequence | |
| pRS426-penP-F | GTAACGCCAGGGTTTTCCCAGTCACGACAAGCTTTCTGCCCATGTAGCTGCTCAG | penP-hph-R | ATGCTCCTTCAATATCAGTTCCAAGCTGCAAGGCGTTGAATCACCCTG | penP 5' flank |
| hph-penP-F | CCAGCACTCGTCCGAGGGCAAAGGAATAGACGCAGAACCTGGCAGTCTCG | penP-pRS426-R | GCGGATAACAATTTCACACAGGAAACAGCCTCGAGGGAGATTCTGTCGACGGAATG | penP 3' flank |
| pRS426-janP-F | GTAACGCCAGGGTTTTCCCAGTCACGACGAATTCGCTCGTATCACTTCATAGCAG | janP-hph-R | AAATGCTCCTTCAATATCAGTTCCAAGCTGAAGTGGATGGTGTAGGAAGC | janP 5' flank |
| hph-janP-F | CCAGCACTCGTCCGAGGGCAAAGGAATAGACCTGGCTCAACGACTGCTTG | janP-pRS426-R | GCGGATAACAATTTCACACAGGAAACAGCCTCGAGTTGCTGGATGGATAGACTTCG | janP 3' flank |
| CSPjantD1F | GTAACGCCAGGGTTTTCCCAGTCACGACATGCCTTTGTATTAACCGCT | CSPjantD2R | ATGCTCCTTCAATATCAGTTCCAAGCTAATCTTCTAGAGACTTTGAGGG | janD 5' flank |
| CSPjantD3F | CCAGCACTCGTCCGAGGGCAAAGGAATAGGAAGTGGAAAGATTAGGTTTGGTC | CSPjantD4R | GCGGATAACAATTTCACACAGGAAACAGCTCGGGCTTAAATAGATGTCAAGG | janD 3' flank |
| hph-F | AGCTTGGAACTGATATTGAAGG | hph-R | CTATTCCTTTGCCCTCGGACG | PtrpC-hph (hygromycin resistance) |
References
- Steyn, P.S.; Vleggaar, R. Tremorgenic mycotoxins. Prog. Chem. Org. Nat. Prod. 1985, 48, 1–80. [Google Scholar]
- Gallagher, R.T.; White, E.P.; Mortimer, P.H. Ryegrass staggers: Isolation of potent neurotoxins lolitrem A and lolitrem B from staggers-producing pastures. N. Z. Vet. J. 1981, 29, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.J.; Dorner, J.W.; Lansden, J.A.; Cox, R.H.; Pape, C.; Cunfer, B.M.; Nicholson, S.S.; Bendell, D.M. Paspalum staggers: Isolation and identification of tremorgenic metabolites from sclerotia of Claviceps paspali. J. Agric. Food Chem. 1977, 25, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.J.; Scott, D.B. Indole-diterpene biosynthesis in ascomycetous fungi. In Handbook of Industrial Mycology; An, Z., Ed.; Marcel Dekker: New York, NY, USA, 2004; Chapter 14; Volume 70, pp. 405–426. [Google Scholar]
- Gloer, J.B. Antiinsectan natural products from fungal sclerotia. Acc. Chem. Res. 1995, 28, 343–350. [Google Scholar] [CrossRef]
- Ball, O.J.-P.; Prestidge, R.A. Endophyte associated alkaloids, insect resistance and animal disorders: An interrelated complex. N. Z. Vet. J. 1993, 41, 216. [Google Scholar]
- Knaus, H.-G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garcia-Calvo, M.; Helms, L.M.H.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P.; Smith, A.B.; et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M.; Warren, V.A.; Thomas, B.S.; Brochu, R.M.; Ertel, E.A.; Rohrer, S.; Schaeffer, J.; Schmatz, D.; Petuch, B.R.; Tang, Y.S.; et al. Nodulisporic acid opens insect glutamate-gated chloride channels: Identification of a new high-affinity modulator. Biochemistry 2000, 39, 5543–5554. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-H.; Tomoda, H.; Nishida, H.; Masuma, R.; Omura, S. Terpendoles, novel acat inhibitors produced by Albophoma yamanashiensis. I. Production, isolation and biological properties. J. Antibiot. 1995, 48, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; McMillan, L.; Telfer, E.; Scott, B. Molecular cloning and genetic analysis of an indole-diterpene gene cluster from Penicillium paxilli. Mol. Microbiol. 2001, 39, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.; Young, C.A.; Saikia, S.; McMillan, L.K.; Monahan, B.J.; Koulman, A.; Astin, J.; Eaton, C.J.; Bryant, A.; Wrenn, R.E.; et al. Deletion and gene expression analyses define the paxilline biosynthetic gene cluster in Penicillium paxilli. Toxins 2013, 5, 1422–1446. [Google Scholar] [CrossRef] [PubMed]
- McMillan, L.K.; Carr, R.L.; Young, C.A.; Astin, J.W.; Lowe, R.G.T.; Parker, E.J.; Jameson, G.B.; Finch, S.C.; Miles, C.O.; McManus, O.B.; et al. Molecular analysis of two cytochrome P450 monooxygenase genes required for paxilline biosynthesis in Penicillium paxilli, and effects of paxilline intermediates on mammalian maxi-K ion channels. Mol. Gen. Genomics 2003, 270, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Saikia, S.; Parker, E.J.; Koulman, A.; Scott, B. Four gene products are required for the fungal synthesis of the indole diterpene paspaline. FEBS Lett. 2006, 580, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Liu, C.; Minami, A.; Noike, M.; Isaka, T.; Fueki, S.; Shichijo, Y.; Toshima, H.; Gomi, K.; Dairi, T.; et al. Reconstitution of biosynthetic machinery for indole-diterpene paxilline in Aspergillus oryzae. J. Am. Chem. Soc. 2013, 135, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Saikia, S.; Parker, E.J.; Koulman, A.; Scott, B. Defining paxilline biosynthesis in Penicillium paxilli: Functional characterization of two cytochrome P450 monooxygenases. J. Biol. Chem. 2007, 282, 16829–16837. [Google Scholar] [CrossRef] [PubMed]
- Young, C.A.; Bryant, M.K.; Christensen, M.J.; Tapper, B.A.; Bryan, G.T.; Scott, B. Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol. Gen. Genomics 2005, 274, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Young, C.A.; Felitti, S.; Shields, K.; Spangenberg, G.; Johnson, R.D.; Bryan, G.T.; Saikia, S.; Scott, B. A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet. Biol. 2006, 43, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Monahan, B.J.; Tkacz, J.S.; Scott, B. An indole-diterpene gene cluster from Aspergillus flavus. Appl. Environ. Microbiol. 2004, 70, 6875–6883. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.J.; Koulman, A.; Monahan, B.J.; Pritchard, B.L.; Payne, G.A.; Scott, B. Identification of two aflatrem biosynthesis gene loci in Aspergillus flavus and metabolic engineering of Penicillium paxilli to elucidate their function. Appl. Environ. Microbiol. 2009, 75, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Sings, H.; Singh, S.B. Tremorgenic and nontremorgenic 2,3-fused indole diterpenoids. Alkaloids 2004. [Google Scholar] [CrossRef]
- Smith, B.L.; McLeay, L.M.; Embling, P.P. Effects of the mycotoxins penitrem, paxilline and lolitrem B on the electromyographic activity of skeletal and gastrointestinal smooth muscle of sheep. Res. Vet. Sci. 1997, 62, 111–116. [Google Scholar] [CrossRef]
- de Jesus, A.E.; Gorst-Allman, C.P.; Steyn, P.S.; van Heerden, F.R.; Vleggar, R.; Wessels, P.L.; Hull, W.E. Tremorgenic mycotoxins from Penicillium crustosum. Biosynthesis of penitrem A. J. Chem. Soc. Perkin Trans. 1983, 1, 1863–1868. [Google Scholar] [CrossRef]
- Gallagher, R.T.; Latch, G.C.M.; Keogh, R.G. The janthitrems: Fluorescent tremorgenic toxins produced by Penicillium janthinellum isolates from ryegrass pastures. Appl. Environ. Microbiol. 1980, 39, 272–273. [Google Scholar] [PubMed]
- Xu, M.J.; Gessner, G.; Groth, I.; Lange, C.; Christner, A.; Bruhn, T.; Deng, Z.W.; Li, X.; Heinemann, S.H.; Grabley, S.; et al. Shearinines D-K, new indole triterpenoids from an endophytic Penicillium sp. (strain HKI0459) with blocking activity on large-conductance calcium-activated potassium channels. Tetrahedron 2007, 63, 435–444. [Google Scholar] [CrossRef]
- di Menna, M.E.; Lauren, D.R.; Wyatt, P.A. Effect of culture conditions on tremorgen production by some Penicillium species. Appl. Environ. Microbiol. 1986, 51, 821–824. [Google Scholar] [PubMed]
- Leuchtmann, A.; Bacon, C.W.; Schardl, C.L.; White, J.F., Jr.; Tadych, M. Nomenclatural realignment of Neotyphodium species with genus Epichloë. Mycologia 2014, 106, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Tudzynski, B.; Rojas, M.C.; Gaskin, P.; Hedden, P. The gibberellin 20-oxidase of Gibberella fujikuroi is a multifunctional monooxygenase. J. Biol. Chem. 2002, 277, 21246–21253. [Google Scholar] [CrossRef] [PubMed]
- Graham-Lorence, S.E.; Peterson, J.A. Structural alignments of P450s and extrapolations to the unknown. Meth. Enzymol. 1996, 272, 315–326. [Google Scholar] [PubMed]
- Belofsky, G.N.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Antiinsectan alkaloids: Shearinines A–C and a new paxilline derivative from the ascostromata of Eupenicillium shearii. Tetrahedron 1995, 51, 3959–3968. [Google Scholar] [CrossRef]
- Smetanina, O.F.; Kalinovsky, A.I.; Khudyakova, Y.V.; Pivkin, M.V.; Dmitrenok, P.S.; Fedorov, S.N.; Ji, H.; Kwak, J.Y.; Kuznetsova, T.A. Indole alkaloids produced by a marine fungus isolate of Penicillium janthinellum biourge. J. Nat. Prod. 2007, 70, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Minami, A.; Fujii, R.; Liu, C.; Tanaka, M.; Gomi, K.; Dairi, T.; Oikawa, H. Rapid reconstitution of biosynthetic machinery for fungal metabolites in Aspergillus oryzae: Total biosynthesis of aflatrem. Chembiochem 2014, 15, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Noike, M.; Minami, A.; Oikawa, H.; Dairi, T. Functional analysis of a prenyltransferase gene (paxD) in the paxilline biosynthetic gene cluster. Appl. Microbiol. Biotechnol. 2014, 98, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tagami, K.; Minami, A.; Matsumoto, T.; Frisvad, J.C.; Suzuki, H.; Ishikawa, J.; Gomi, K.; Oikawa, H. Reconstitution of biosynthetic machinery for the synthesis of the highly elaborated indole diterpene penitrem. Angew. Chem. 2015, 54, 5748–5752. [Google Scholar] [CrossRef] [PubMed]
- Yoder, O.C. Cochliobolus heterostrophus, cause of southern corn leaf blight. Adv. Plant Pathol. 1988, 6, 93–112. [Google Scholar]
- Byrd, A.D.; Schardl, C.L.; Songlin, P.J.; Mogen, K.L.; Siegel, M.R. The β-tubulin gene of Epichloë typhina from perennial ryegrass (Lolium perenne). Curr. Genet. 1990, 18, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Orbach, M.J. A cosmid with a Hyr marker for fungal library construction and screening. Gene 1994, 150, 159–162. [Google Scholar] [CrossRef]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. Solexaqa: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7 (Suppl. 1). [Google Scholar] [CrossRef] [PubMed]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.M.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Meth. Enzymol. 2002, 350, 87–96. [Google Scholar] [PubMed]
- Winston, F.; Dollard, C.; Ricupero-Hovasse, S.L. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 1995, 11, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Namiki, F.; Matsunaga, M.; Okuda, M.; Inoue, I.; Nishi, K.; Fujita, Y.; Tsuge, T. Mutation of an arginine biosynthesis gene causes reduced pathogenicity in Fusarium oxysporum f. sp. melonis. Mol. Plant-Microbe Interact. 2001, 14, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, D.; Tanaka, A.; Scott, B. A p67phox-like regulator is recruited to control hyphal branching in a fungal-grass mutualistic symbiosis. Plant Cell 2006, 18, 2807–2821. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholson, M.J.; Eaton, C.J.; Stärkel, C.; Tapper, B.A.; Cox, M.P.; Scott, B. Molecular Cloning and Functional Analysis of Gene Clusters for the Biosynthesis of Indole-Diterpenes in Penicillium crustosum and P. janthinellum. Toxins 2015, 7, 2701-2722. https://doi.org/10.3390/toxins7082701
Nicholson MJ, Eaton CJ, Stärkel C, Tapper BA, Cox MP, Scott B. Molecular Cloning and Functional Analysis of Gene Clusters for the Biosynthesis of Indole-Diterpenes in Penicillium crustosum and P. janthinellum. Toxins. 2015; 7(8):2701-2722. https://doi.org/10.3390/toxins7082701
Chicago/Turabian StyleNicholson, Matthew J., Carla J. Eaton, Cornelia Stärkel, Brian A. Tapper, Murray P. Cox, and Barry Scott. 2015. "Molecular Cloning and Functional Analysis of Gene Clusters for the Biosynthesis of Indole-Diterpenes in Penicillium crustosum and P. janthinellum" Toxins 7, no. 8: 2701-2722. https://doi.org/10.3390/toxins7082701
APA StyleNicholson, M. J., Eaton, C. J., Stärkel, C., Tapper, B. A., Cox, M. P., & Scott, B. (2015). Molecular Cloning and Functional Analysis of Gene Clusters for the Biosynthesis of Indole-Diterpenes in Penicillium crustosum and P. janthinellum. Toxins, 7(8), 2701-2722. https://doi.org/10.3390/toxins7082701