
Cell-to-Cell Propagation of the Bacterial Toxin CNF1 via Extracellular Vesicles: Potential Impact on the Therapeutic Use of the Toxin

 ,
, 

Abstract
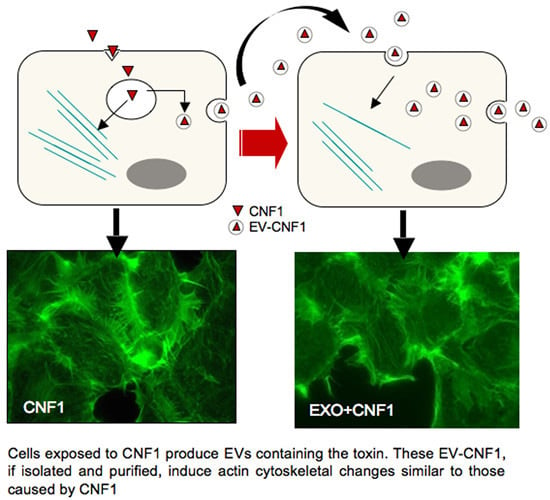
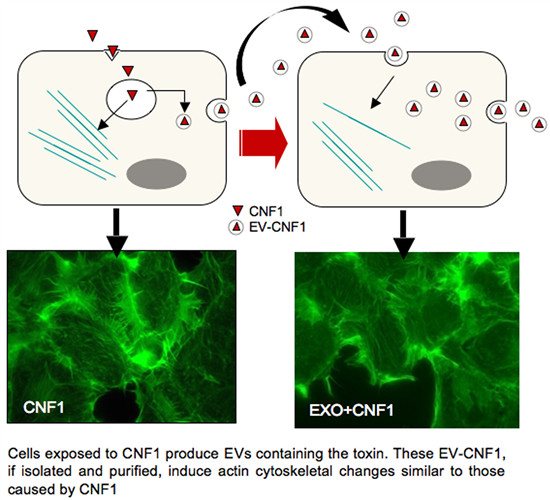
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
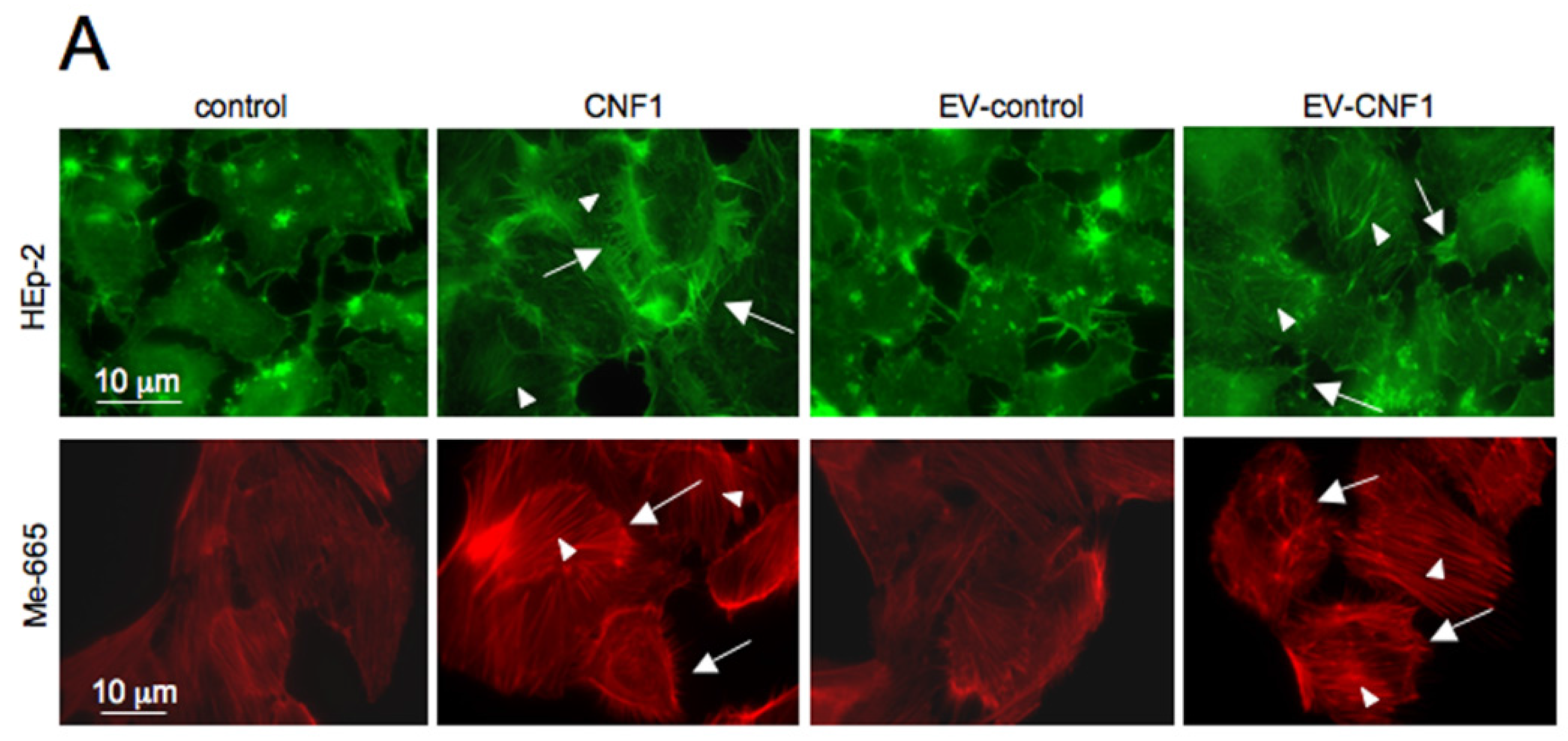
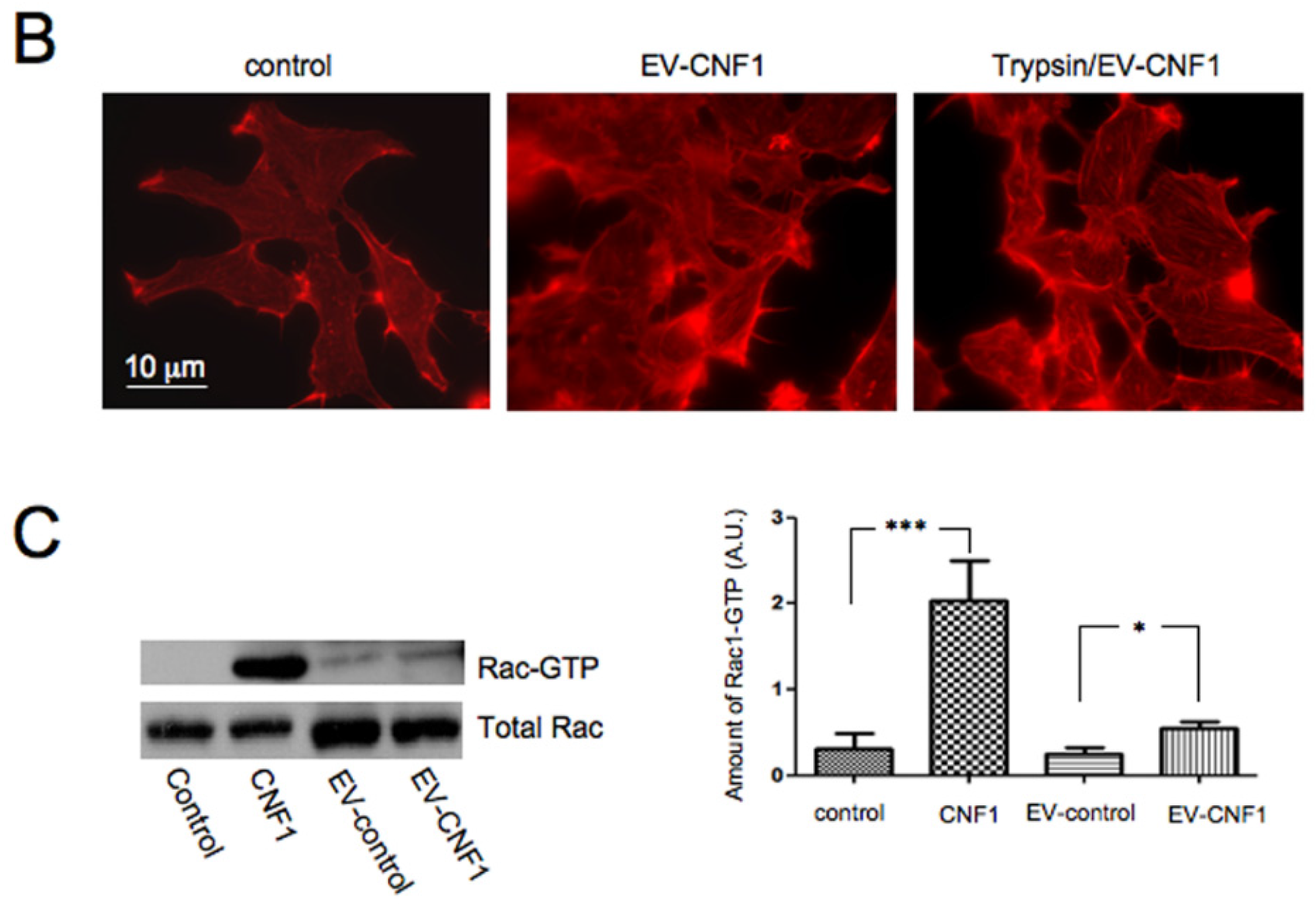
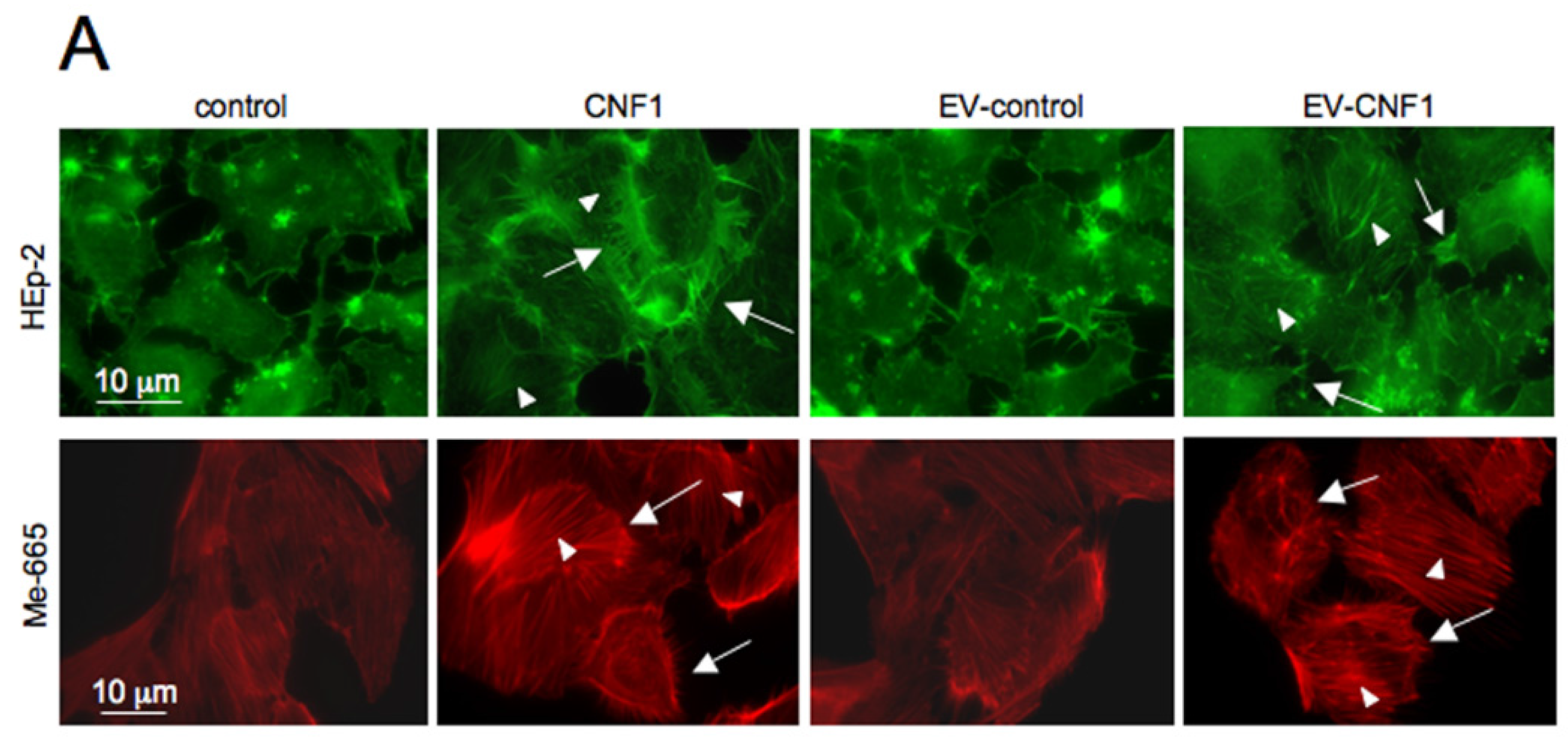
2.1. EV-CNF1 Induces Cytoskeletal Changes and Rac1 Activation
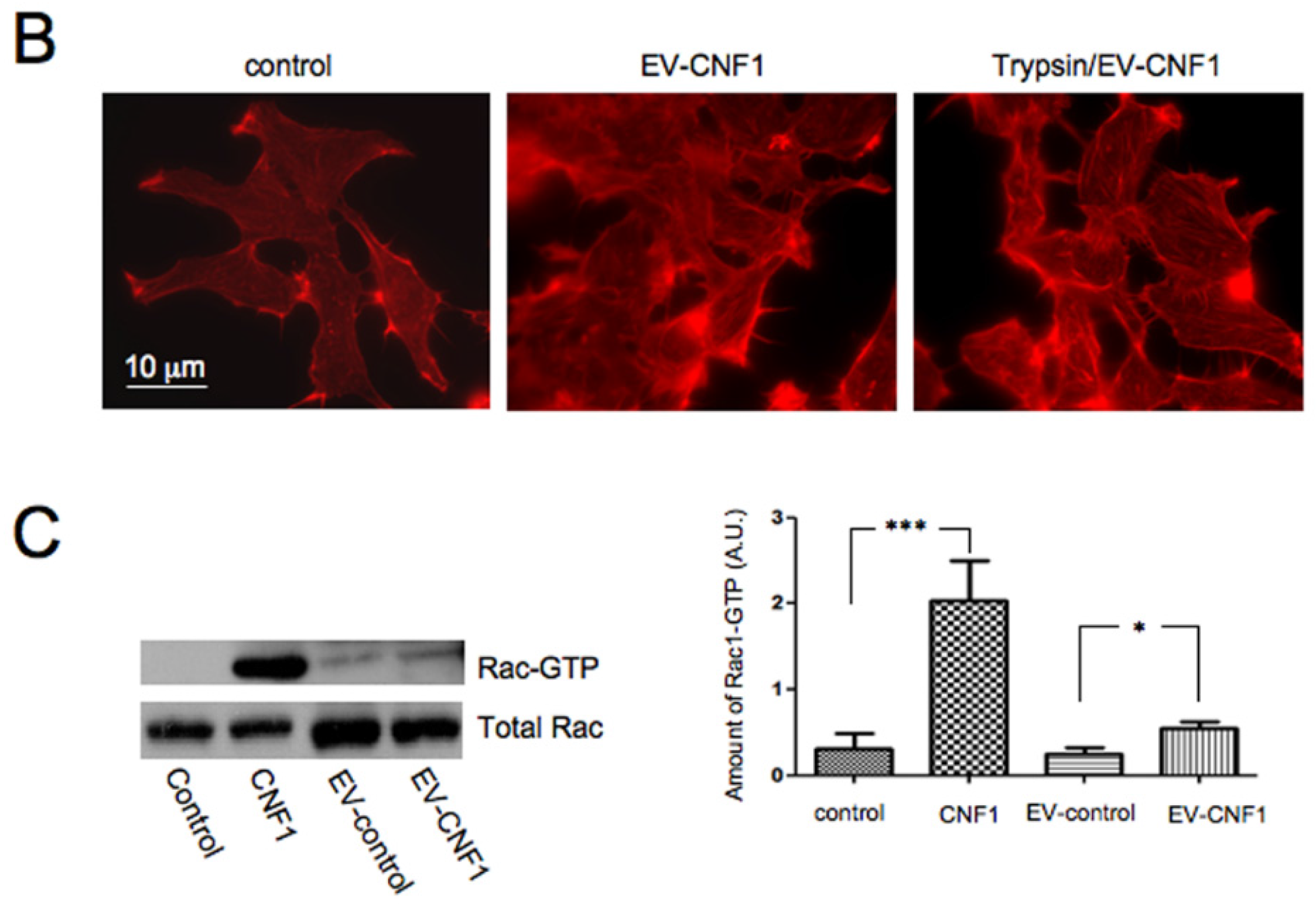
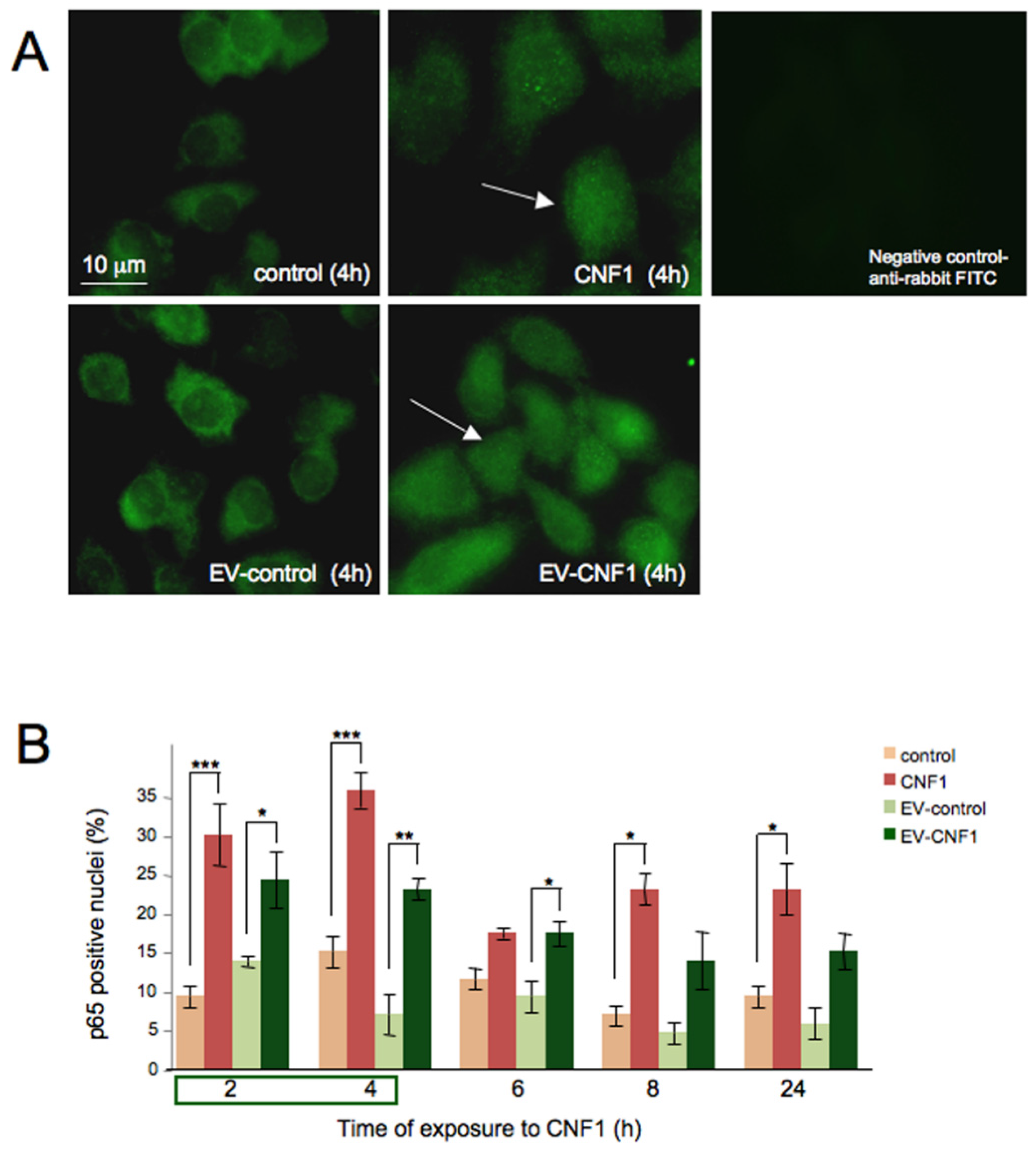
2.2. EV-CNF1 Stimulates NF-κB Nuclear Translocation
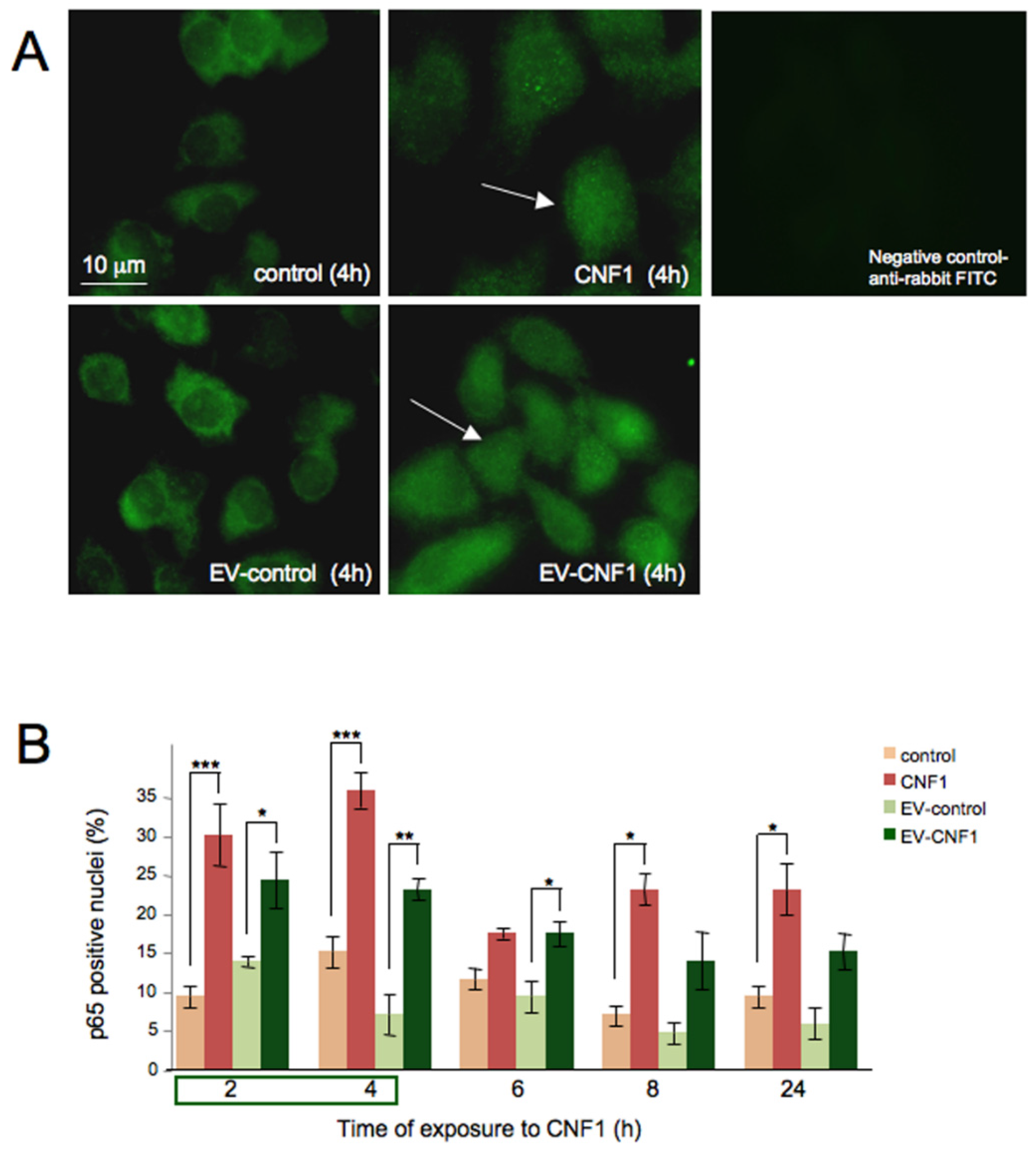
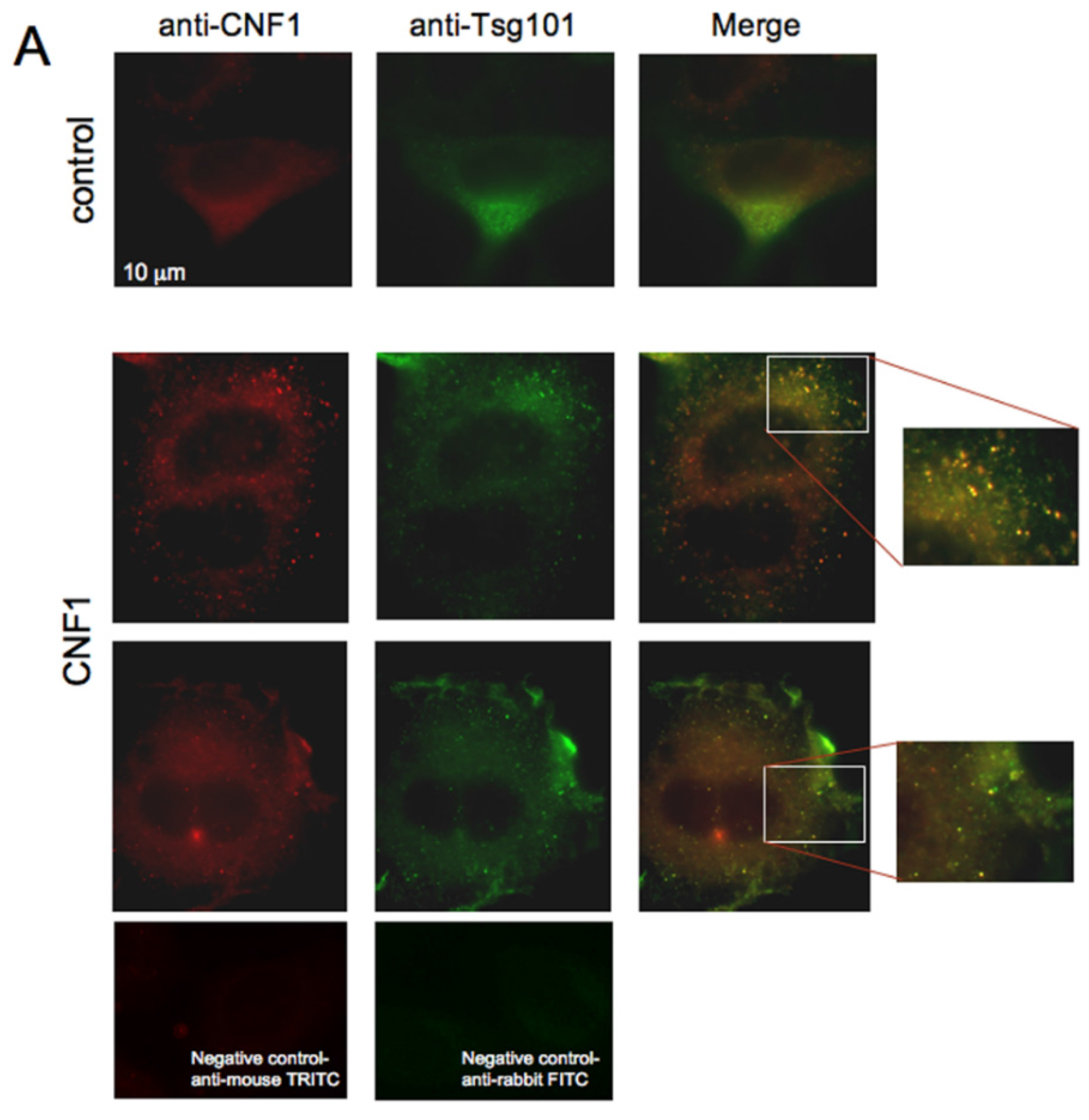
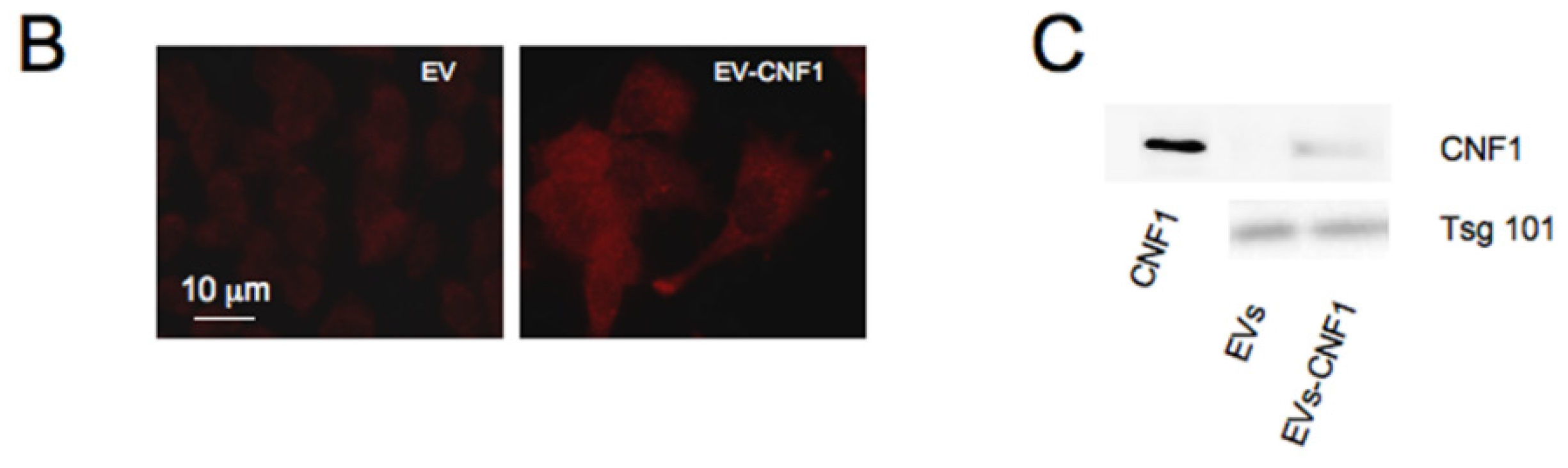
2.3. CNF1 Is Detectable in EVs Derived from CNF1-Intoxicated Cells
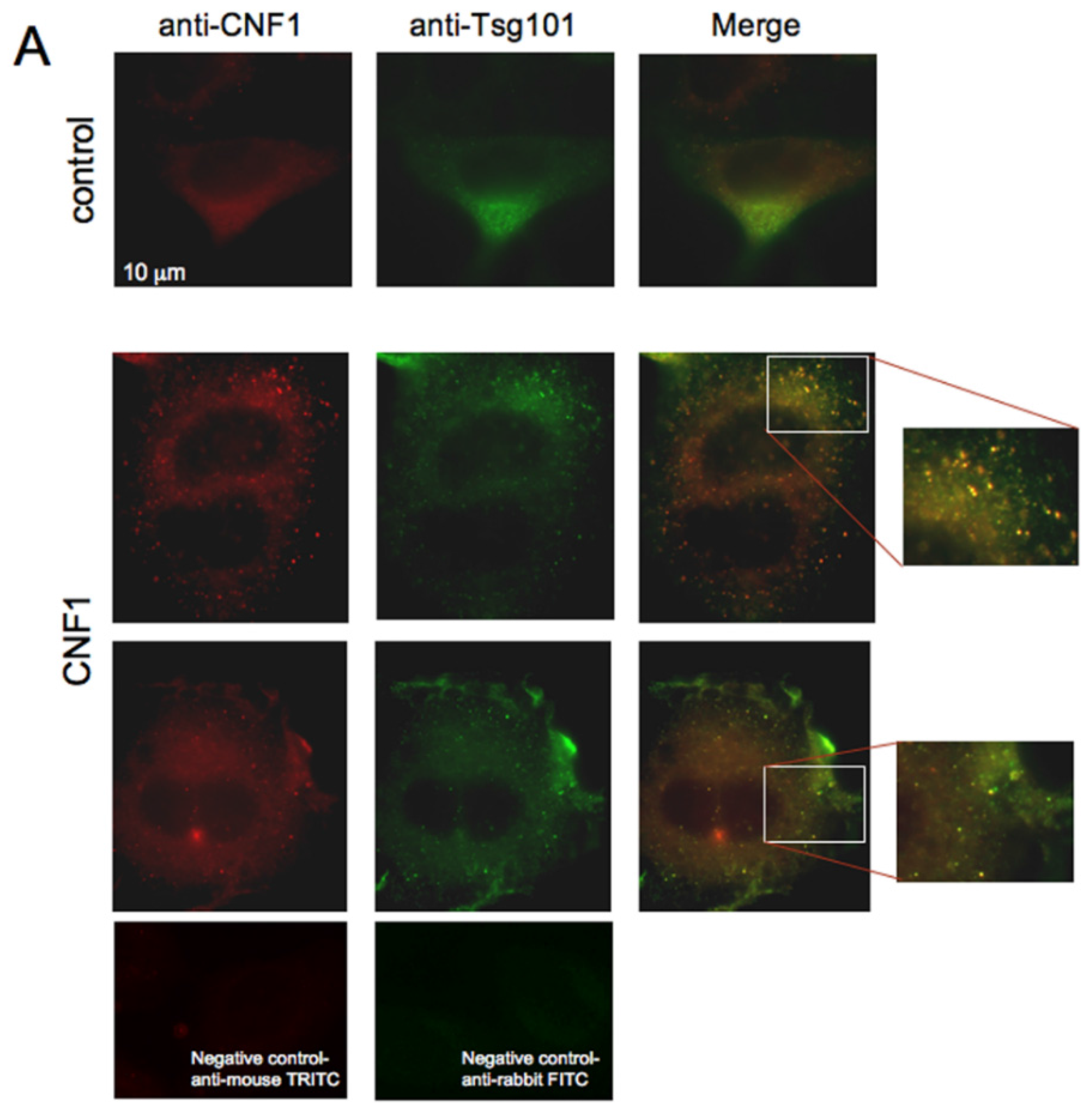
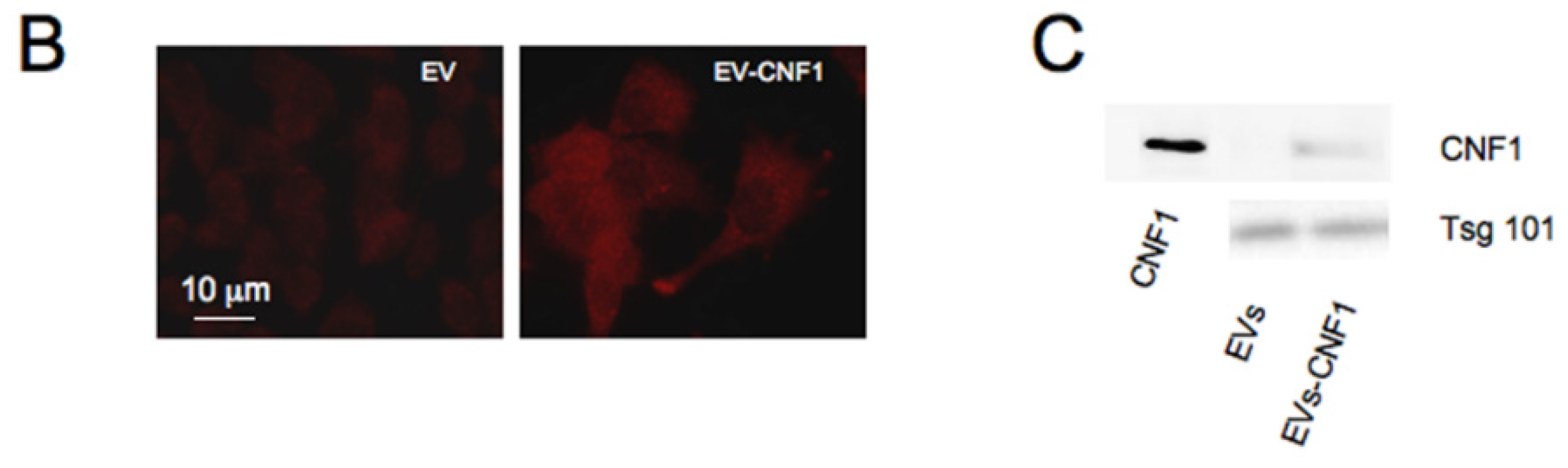
3. Discussion
4. Materials and Methods
4.1. CNF1 Purification
4.2. Cell Cultures and Treatments
4.3. Collection of Extracellular Vesicles
4.4. Amount Determination of the Collected EVs
4.5. Western Blot Analyses and Pull-Down Assay
4.6. Immunofluorescence Microscopy
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [PubMed]
- Simpson, R.J.; Mathivanan, S. Extracellular microvesicles: The need for internationally recognised nomenclature and stringent purification criteria. J. Proteomics Bioinform. 2012. [Google Scholar] [CrossRef]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Choi, K.H.; Kim, Y.S.; Hong, B.S.; Kim, O.Y.; Kim, J.H.; Yoon, C.M.; Koh, G.Y.; Kim, Y.K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE 2010, 5, e11334. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Kim, M.R.; Lee, E.Y.; Kim, J.H.; Kim, Y.S.; Jeon, S.G.; Yang, J.M.; Lee, B.J.; Pyun, B.Y.; Gho, Y.S.; et al. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 2011, 66, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.R.; Hong, S.W.; Choi, E.B.; Lee, W.H.; Kim, Y.S.; Jeon, S.G.; Jang, M.H.; Gho, Y.S.; Kim, Y.K. Staphylococcus aureus-derived extracellular vesicles induce neutrophilic pulmonary inflammation via both Th1 and Th17 cell responses. Allergy 2012, 67, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Perelle, S.; Gibert, M.; Boquet, P.; Popoff, M.R. Characterization of Clostridium perfringens iota-toxin genes and expression in Escherichia coli. Infect. Immun. 1993, 61, 5147–5156. [Google Scholar] [PubMed]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Remaley, A.T. Lipid-based carriers of microRNAs and intercellular communication. Curr. Opin. Lipidol. 2012, 23, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [PubMed]
- Travaglione, S.; Loizzo, S.; Ballan, G.; Fiorentini, C.; Fabbri, A. The E. coli CNF1 as a pioneering therapy for the central nervous system diseases. Toxins 2014, 6, 270–682. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 increases brain energy level, counteracts neuroinflammatory markers and rescues cognitive deficits in a murine model of Alzheimer’s disease. PLoS ONE 2013, 8, e65898. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, C.; Gallina, A.; Fabbri, A.; Parolini, I.; Boussadia, Z.; Coscia, C.; Biffoni, M.; Palermo, A.; Fiorentini, C.; Sargiacomo, M. Exosomes are effectual vehicles for cell-to-cell bacterial protein toxin propagation. In Proceedings of the Second International Meeting of ISEV, Boston, MA, USA, 17–20 April 2013.
- Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 (CNF1): Toxin biology, in vivo applications and therapeutic potential. Toxins 2010, 2, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Pavone, F.; Luvisetto, S.; Marinelli, S.; Straface, E.; Fabbri, A.; Falzano, L.; Fiorentini, C.; Malorni, W. The Rac GTPase-activating bacterial protein toxin CNF1 induces analgesia up-regulating mu-opioid receptors. Pain 2009, 145, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Cerri, C.; Fabbri, A.; Vannini, E.; Spolidoro, M.; Costa, M.; Maffei, L.; Fiorentini, C.; Caleo, M. Activation of Rho GTPases triggers structural remodeling and functional plasticity in the adult rat visual cortex. J. Neurosci. 2011, 31, 15163–15172. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase instructs nuclear factor-kappaB activation by conveying the SCF complex and IkBalpha to the ruffling membranes. Mol. Biol. Cell 2004, 15, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Giamboi-Miraglia, A.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: Role of nuclear factor-κB and Bcl-2. Mol. Biol. Cell 2007, 18, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Torrecilhas, A.C.; Schumacher, R.I.; Alves, M.J.; Colli, W. Vesicles as carriers of virulence factors in parasitic protozoan diseases. Microbes Infect. 2012, 14, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G. Exosomal Communication Goes Viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, F.; Errico, M.C.; Bottero, L.; Segnalino, P.; Stoppacciaro, A.; Biffoni, M.; Felli, N.; Mattia, G.; Petrini, M.; Colombo, M.P.; et al. The promyelocytic leucemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008, 68, 2745–2754. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Travaglione, S.; Messina, G.; Fabbri, A.; Falzano, L.; Giammarioli, A.M.; Grossi, M.; Rufini, S.; Fiorentini, C. Cytotoxic necrotizing factor 1 hinders skeletal muscle differentiation in vitro by perturbing the activation/deactivation balance of Rho GTPases. Cell Death Differ. 2005, 12, 78–86. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabbri, A.; Cori, S.; Zanetti, C.; Guidotti, M.; Sargiacomo, M.; Loizzo, S.; Fiorentini, C. Cell-to-Cell Propagation of the Bacterial Toxin CNF1 via Extracellular Vesicles: Potential Impact on the Therapeutic Use of the Toxin. Toxins 2015, 7, 4610-4621. https://doi.org/10.3390/toxins7114610
Fabbri A, Cori S, Zanetti C, Guidotti M, Sargiacomo M, Loizzo S, Fiorentini C. Cell-to-Cell Propagation of the Bacterial Toxin CNF1 via Extracellular Vesicles: Potential Impact on the Therapeutic Use of the Toxin. Toxins. 2015; 7(11):4610-4621. https://doi.org/10.3390/toxins7114610
Chicago/Turabian StyleFabbri, Alessia, Sara Cori, Cristiana Zanetti, Marco Guidotti, Massimo Sargiacomo, Stefano Loizzo, and Carla Fiorentini. 2015. "Cell-to-Cell Propagation of the Bacterial Toxin CNF1 via Extracellular Vesicles: Potential Impact on the Therapeutic Use of the Toxin" Toxins 7, no. 11: 4610-4621. https://doi.org/10.3390/toxins7114610
APA StyleFabbri, A., Cori, S., Zanetti, C., Guidotti, M., Sargiacomo, M., Loizzo, S., & Fiorentini, C. (2015). Cell-to-Cell Propagation of the Bacterial Toxin CNF1 via Extracellular Vesicles: Potential Impact on the Therapeutic Use of the Toxin. Toxins, 7(11), 4610-4621. https://doi.org/10.3390/toxins7114610