
Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing

Abstract
:1. Introduction
2. Botulinum Neurotoxin Structure and Function
3. Peripheral Delivery of Botulinum Toxins and Nociception in Preclinical and Human Models

{kind=link}
{kind=link}
{kind=link}
| No | Type of Pain Condition/Model | BoNT Serotype | Outcome | Interpretations | References |
|---|---|---|---|---|---|
| 1) | Migraine | ||||
| Acute Migraine (<15 attacks/month) | BoNT/A1 | Positive | Reductions in migraine severity and headache frequency | [15,16,17] | |
| Negative | No significant differences observed between the placebo and BoNT treatment group. Few studies observed a trend however not significant | [23,24] | |||
| Chronic Migraine (>15 attacks/month) | BoNT/A1 | Positive | Pooled result of PREEMPT trials favored both primary and secondary endpoints. Reduction in cephalic allodynia associated with chronic migraine | [13,14,18,21,22] | |
| Negative | Mild or no effect was observed in this randomized controlled study | [25] | |||
| BoNT/B1 | Positive | Chronic migraineurs significantly responded to Rimabotulinumtoxin (BoNT/B1) | [19,20] | ||
| 2) | Chronic joint pain | BoNT/A1 | Positive | BoNT/A1 showed significant effect in treating refractive shoulder joint pain | [37,38] |
| Positive | Efficacy in painful knee and joint arthritis | [39,40] | |||
| 3) | Neuropathic pain | ||||
| Mononeuropathy | BoNT/A1 | Positive | BoNT treatment was effective in treating trigeminal neuralgia and peripheral nerve injury | [41,42,43,44,45,46,47,48] | |
| Polyneuropathy | BoNT/A1 | Positive | Effective in cases of post-herpetic neuralgia Efficacy reported for diabetic neuropathy | [49,50,51,52] | |
| 4) | Chronic low back pain | BoNT/A1 | Positive | Treatment in paraspinal muscles reduced pain in refractory low back pain patients | [53,54] |
| 5) | Myofascial pain | BoNT/A1 | Positive | BoNT reduced focal myofascial pain syndrome | [55,56] |
| 6) | Evoked nociception model | ||||
| Capsaicin | BoNT/A1 | Positive | BoNT inhibited capsaicin induced flare alone or both flare and evoked nociception | [57,58,59,60] | |
| Negative | Showed no effect on any end points | [61,62,63] | |||
| Glutamate | BoNT/A1 | Positive | Reduced glutamate evoked pain and local increase in skin blood flow | [64] | |
| Thermal Injury | BoNT/A1 | Negative | Evoked primary or secondary hyperalgesia was not altered by BoNT | [65] | |
| 7) | Acute thresholds | BoNT/A1 | Negative | No effect upon normal thermal and mechanical pain thresholds in quantitative testing paradigm | [57,61,62,63] |
| No | Type of Pain Condition/Model | BoNT Serotype | Species | Outcome | Interpretations | References |
|---|---|---|---|---|---|---|
| 1) | Inflammatory pain models | |||||
| Formalin | BoNT/A1/B1 | Rat/mouse | Positive | Peripheral BoNT shows little or no effect on phase I with long term inhibition in phase II formalin flinching and neuronal c-fos activation. Inhibition of neurotransmitter release. Cleavage of SNARES in DRG/TG and spinal cord | [66,67,68,69,70,71,72,73] | |
| Capsaicin | BoNT/A1/B1 | Rat/mouse | Positive | Peripheral BoNT reduces capsaicin evoked flare, nociceptive behavior and inhibition of neurotransmitter release. Cleavage of SNARES in TG and nucleus caudalis | [67,74,75] | |
| Carrageenan | BoNT/A1 | Rat | Negative | Lack of effect on peripheral inflammation and pain | [76,77] | |
| BoNT/A1/A2 | Rat | Negative | No effect of BoNT was observed in carrageenan evoked flare and plasma extravasation | [77,78,79] | ||
| Arthritis | BoNT/A1 | Rat | Positive | BoNT reduced allodynia in CFA induced knee arthritic animals | [80] | |
| BoNT/A1 | Dogs | Positive | Intraarticular BoNT reduced indices of pain | [81,82,83] | ||
| 2) | Neuropathic pain | |||||
| Mononeuropathy (peripheral and infraorbital nerve ligation, ventral root transection) | BoNT/A1/B1 | Rat/mouse | Positive | BoNT reduced allodynia following peripheral treatment. Suggests central action of BoNT | [74,84,85,86,87] | |
| Polyneuropathy (Chemotherapeutics, diabetes) | BoNT/A1 | Rat/mouse | Positive | Reduced thermal and mechanical hyperalgesia with bilateral effects | [78,84,88,89] | |
| 3) | Trigeminal pain models | BoNT/A1/B1 | Rat/mouse | Positive | Orofacial BoNT reduced capsaicin evoked nocifensive behavior, inhibited trafficking of TRPV1 to plasma membrane, cleavage of SNARES | [75,90] |
| Decreased mechanical sensitivity of temporal muscle nociceptors. Inhibited responses to mechanical stimulation of dura to supra threshold forces | [91] | |||||
| In vitro (cell/organ culture studies) | ||||||
| 1) | Dorsal root ganglion sensory neuron cell culture | BoNT/A1 | Rat | Positive | BoNT cleaves neuronal SNARES and prevents release of neurotransmitters | [92,93] |
| 2) | Trigeminal sensory neuron organ/cell culture | BoNT/A1 | Rat | Positive | BoNT inhibits CGRP secretion from TG neuronal culture | [94] |
| Modify expression of inflammatory markers both in neurons and glial cells | [95] | |||||
| 3) | Trigeminal satellite glial cell culture | BoNT/A1 | Rat | Positive | BoNT cleaves glial SNARES and inhibited glutamate release | [96] |
3.1. Acute Nociception
3.1.1. Mechanisms
3.1.2. Botulinum Toxins
3.2. Tissue Injury/Inflammation
3.2.1. Mechanisms
3.2.2. Botulinum Toxin
3.3. Peripheral Nerve Injury
3.3.1. Mechanisms
3.3.2. Botulinum Toxin
4. Effects of Peripherally Delivered Botulinum Toxins on Nociceptive Linkages
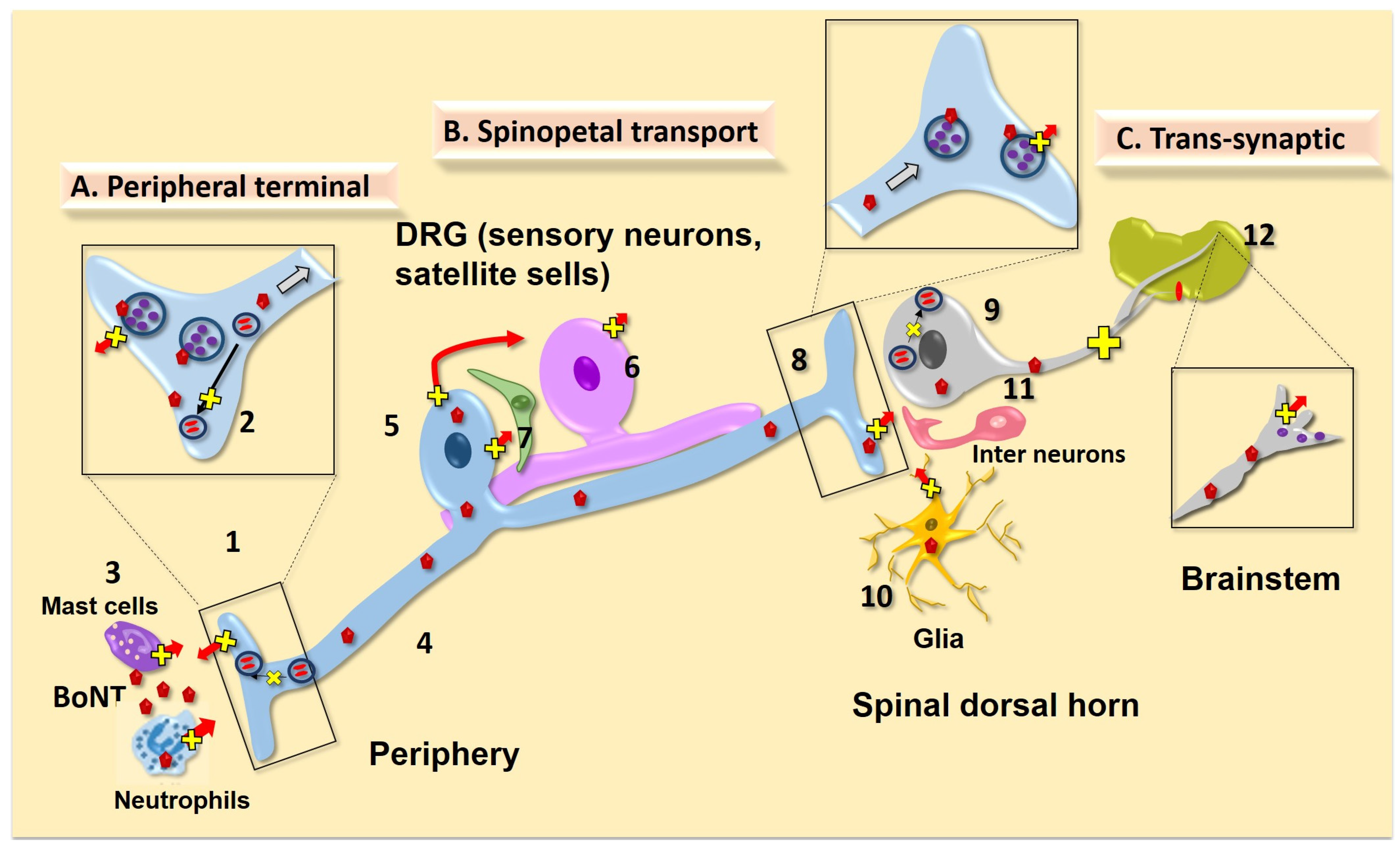
4.1. Local Action on Peripheral Afferent Terminal
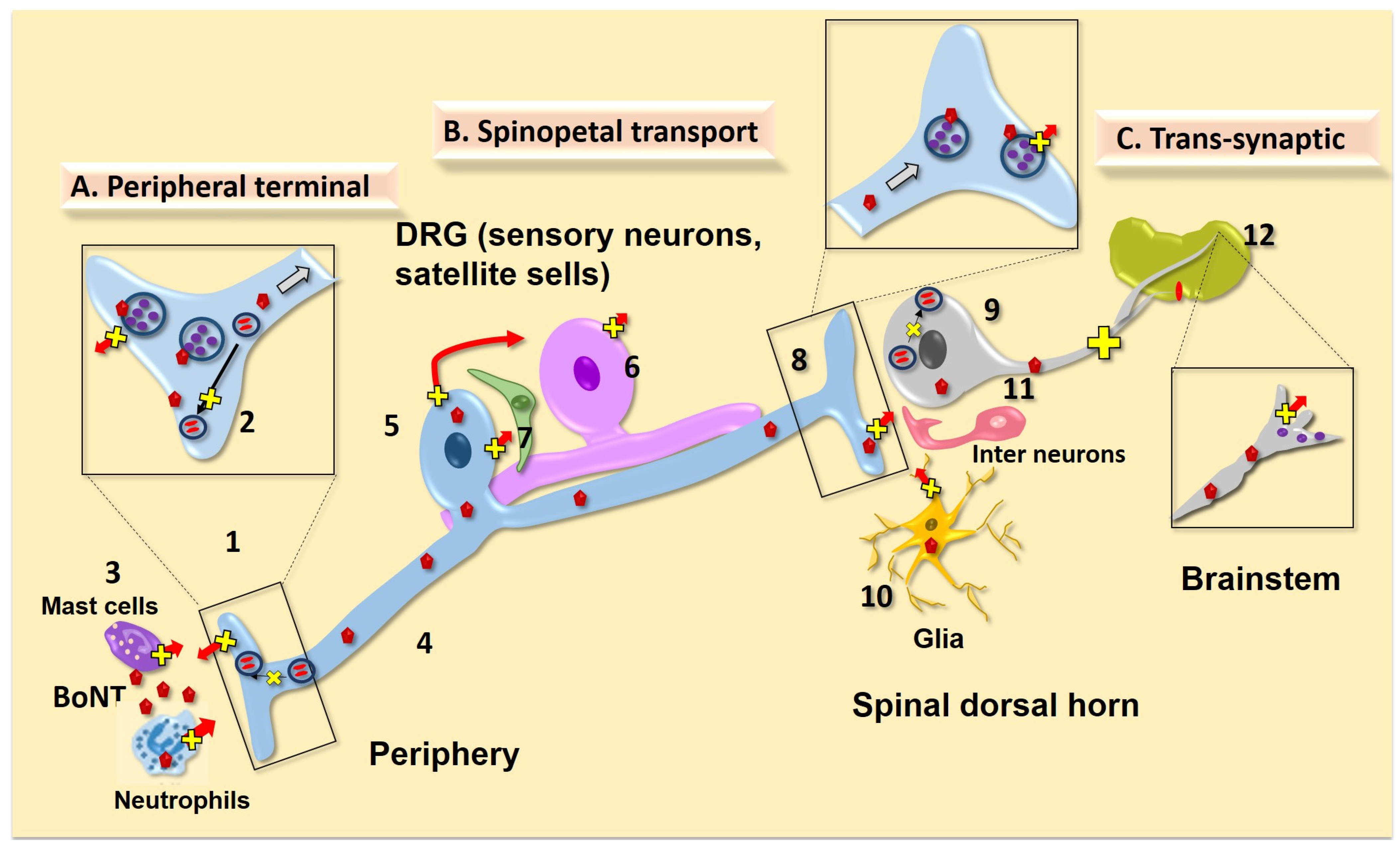
4.1.1. Afferent Terminal Uptake/Transport
4.1.2. Effects of Local BoNTs on Peripheral Non Afferent Cell Systems
4.1.3. BoNT Effects Upon Sensitized Afferent Terminals
4.1.4. Trafficking of Receptor Subunits in the Primary Afferent and DRG/TG
4.1.5. Role of Local (peripheral) BoNT Action in Spinal Nociceptive Transmission
4.2. Spinopetal Afferent Transport
4.2.1. Evidence of Transport of Active BoNT in the Primary Afferent
4.2.2. Role of Cell Body (DRG) Release and BoNT Action
4.2.3. Effect of BoNT on Central Trafficking
4.2.4. Effect of Intrathecally-Delivered BoNTs
4.3. Trans-Synaptic Effects
4.4. BoNT Actions on Glial Cells
5. Diversity of Botulinum Neurotoxins
| No | Mechanism of Action | BoNT Serotype | Species/Models | Interpretations | References |
|---|---|---|---|---|---|
| 1) | Local Peripheral effect | BoNT/A1/B1 | Human/rat/mouse | Blocks local flare, vasodilation and plasma extravasation evoked by local irritants in human and animal studies | [57,58,59,60,66,67,74,91,132] |
| Human/rat/mouse | Lack of effect on normal sensory thresholds. For e.g. acute thermal and mechanical pain thresholds | [57,61,62,63] | |||
| Rat/mouse | Effective only in facilitated pain states such as activation of c-fibers. Does not affect phase I of formalin flinching with significant effects on facilitated phase II | [67,68,69,70,71,72,73,117,181] | |||
| 2) | Axonal transport | BoNT/A1/B1 | Rat/Cell culture | Fast and slow long distance axonal transport in neurons | [190,191,192,193] |
| Cat/rat | Movement of radiolabeled toxin observed in motor and sensory pathways. However, transport of radiolabeled isotope per se were not evaluated | [194,195,196] | |||
| Rat/mouse | Cleavage of SNARES in cell body/soma (DRG/TG) i.e., away from the site of BoNT injection | [67,75,197,198] | |||
| Rat/mouse | Cleavage of SNARES in the central terminals in spinal cord following peripheral BoNT | [67,197] | |||
| 3) | Central actions | BoNT/A1/B1 | Rat/mouse | Inhibits substance P release evoked by intrathecal capsaicin in spinal cord following peripheral BoNT. Blocking neurotransmitter release after retrograde transport. Inhibition of nociceptive behavior and neuronal activation of c-fos | [67,192] |
| BoNT/A1 | Rat | Blocking the axonal transport of peripheral BoNT in trigeminal and sciatic nerve using colchicine inhibits hyperalgesia | [71,74,89,197] | ||
| Rat | Bilateral effect of unilateral BoNT in diabetic neuropathy and trigeminal neuropathy | [74,78,88,89] | |||
| 4) | Trans-synaptic actions | BoNT/A1 | Rat | BoNT is transported from retina to colliculus and transcytosed to tectal synapses as observed by SNARES cleavage | [191] |
| BoNT/A1/B1 | Rat/mouse | Peripheral BoNT inhibited intrathecal substance P induced and intracisternal NMDA induced neuronal activation. | [67,243] | ||
| Rat/mouse | Supraorbital BoNT reduced meningeally evoked activation of second order neurons and substance P release. BoNT in TMJ inhibits dural plasma extravasation | [74,75] | |||
| BoNT/A2 | Rat | Peripheral BoNT cleaved SNARES in spinal glial cells | [232] | ||
| BoNT/A1 | Rat | Unilateral BoNT produce contralateral muscle weakness and bilateral SNARE cleavage | [125,190] |
5.1. SNARE Target and Cleavage Sites
5.2. Membrane Sites Mediating Internalization
5.3. Species Specificity
5.4. Duration of Action
6. Future Directions in Botulinum Neurotoxins as Pain Pharmaceuticals
6.1. Recombinant Technology
6.2. Mining through the Diverse Family of BoNTs
6.3. BoNT/C1 and D1
6.4. Alternate Targeting Strategies
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Johnson, E.A.; Montecucco, C. BOTULISM. In Handbook of Clinical Neurology; Andrew, G.E., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 91, pp. 333–368. [Google Scholar]
- Drachman, D.B. Botulinum toxin as a tool for research on the nervous system. In Neuropoisons. Their Physiological Actions; Simpson, L.L., Ed.; Plenum: New York, NY, USA, 1971; Volume 1, pp. 325–347. [Google Scholar]
- Scott, A.B.; Rosenbaum, A.; Collins, C.C. Pharmacologic weakening of extraocular muscles. Investig. Ophthalmol. 1973, 12, 924–927. [Google Scholar]
- Scott, A.B. Botulinum toxin injection into extraocular muscles as an alternative to strabismus surgery. J. Pediatr. Ophthalmol. Strabismus 1980, 17, 21–25. [Google Scholar] [CrossRef]
- Schantz, E.J.; Kautter, D.A. Standardized assay for clostridium botulinum toxins. J. AOAC. 1978, 61, 96–99. [Google Scholar]
- Dressler, D. Clinical applications of botulinum toxin. Curr. Opin. Microbiol. 2012, 15, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.D.; Stenner, A.; Reichel, G. Current clinical applications of botulinum toxin. Curr. Pharm. Des. 2009, 15, 3671–3680. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell Mol. Life Sci. 2014, 71, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Brin, M.F.; Fahn, S.; Moskowitz, C.; Friedman, A.; Shale, H.M.; Greene, P.E.; Blitzer, A.; List, T.; Lange, D.; Lovelace, R.E.; et al. Localized injections of botulinum toxin for the treatment of focal dystonia and hemifacial spasm. Mov. Disord. 1987, 2, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Whitcup, S.M.; Turkel, C.C.; DeGryse, R.E.; Brin, M.F. Development of onabotulinumtoxina for chronic migraine. Ann. N. Y. Acad. Sci. 2014, 1329, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.E.; Garza, I. Critical analysis of the use of onabotulinumtoxina (botulinum toxin type A) in migraine. Neuropsychiatr. Dis. Treat. 2012, 8, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. OnabotulinumtoxinA (BOTOX®): A review of its use in the prophylaxis of headaches in adults with chronic migraine. Drugs 2012, 72, 825–845. [Google Scholar] [CrossRef] [PubMed]
- Dodick, D.W.; Turkel, C.C.; DeGryse, R.E.; Aurora, S.K.; Silberstein, S.D.; Lipton, R.B.; Diener, H.C.; Brin, M.F. Onabotulinumtoxina for treatment of chronic migraine: Pooled results from the double-blind, randomized, placebo-controlled phases of the preempt clinical program. Headache 2010, 50, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Dodick, D.W.; Aurora, S.K.; Turkel, C.C.; DeGryse, R.E.; Lipton, R.B.; Silberstein, S.D.; Brin, M.F. Onabotulinumtoxina for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the preempt 2 trial. Cephalalgia Int. J. Headache 2010, 30, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, N.; Chana, P. Botulinum toxin type A in prophylactic treatment of migraine headaches: A preliminary study. J. Headache Pain 2003, 4, 146–151. [Google Scholar] [CrossRef]
- Silberstein, S.; Mathew, N.; Saper, J.; Jenkins, S. Botulinum toxin type A as a migraine preventive treatment. For the BoTOX migraine clinical research group. Headache 2000, 40, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.S.; Prasad, A.; Singh, M.M.; Sharma, S.; Bala, K. Botulinum toxin type A in prophylactic treatment of migraine. Am. J. Therapeutics 2006, 13, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Aurora, S.K.; Winner, P.; Freeman, M.C.; Spierings, E.L.; Heiring, J.O.; DeGryse, R.E.; VanDenburgh, A.M.; Nolan, M.E.; Turkel, C.C. Onabotulinumtoxina for treatment of chronic migraine: Pooled analyses of the 56-week preempt clinical program. Headache 2011, 51, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Fadeyi, M.O.; Adams, Q.M. Use of botulinum toxin type B for migraine and tension headaches. Am. J. Health-Syst. Pharm. 2002, 59, 1860–1862. [Google Scholar] [PubMed]
- Grogan, P.M.; Alvarez, M.V.; Jones, L. Headache direction and aura predict migraine responsiveness to rimabotulinumtoxin B. Headache 2013, 53, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lipton, R.B.; Varon, S.F.; Grosberg, B.; McAllister, P.J.; Freitag, F.; Aurora, S.K.; Dodick, D.W.; Silberstein, S.D.; Diener, H.C.; DeGryse, R.E.; et al. Onabotulinumtoxina improves quality of life and reduces impact of chronic migraine. Neurology 2011, 77, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Hollanda, L.; Monteiro, L.; Melo, A. Botulinum toxin type A for cephalic cutaneous allodynia in chronic migraine: A randomized, double-blinded, placebo-controlled trial. Neurol. Int. 2014, 6, 5133. [Google Scholar] [CrossRef] [PubMed]
- Elkind, A.H.; O’Carroll, P.; Blumenfeld, A.; DeGryse, R.; Dimitrova, R. A series of three sequential, randomized, controlled studies of repeated treatments with botulinum toxin type A for migraine prophylaxis. J. Pain 2006, 7, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Relja, M.; Poole, A.C.; Schoenen, J.; Pascual, J.; Lei, X.; Thompson, C. A multicentre, double-blind, randomized, placebo-controlled, parallel group study of multiple treatments of botulinum toxin type A (BoNTA) for the prophylaxis of episodic migraine headaches. Cephalalgia Int. J. Headache 2007, 27, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Gady, J.; Ferneini, E.M. Botulinum toxin A and headache treatment. Conn. Med. 2013, 77, 165–166. [Google Scholar] [PubMed]
- Peck, M.W. Biology and genomic analysis of clostridium botulinum. Adv. Microb. Physiol. 2009, 55, 183–265. [Google Scholar] [PubMed]
- Stringer, S.C.; Carter, A.T.; Webb, M.D.; Wachnicka, E.; Crossman, L.C.; Sebaihia, M.; Peck, M.W. Genomic and physiological variability within group II (non-proteolytic) clostridium botulinum. BMC genomics 2013, 14, 333. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.K.; Smith, T.J. Genetic diversity within clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Curr. Top. Microbiol. Immunol. 2013, 364, 1–20. [Google Scholar] [PubMed]
- Gimenez, D.F.; Gimenez, J.A. The typing of botulinal neurotoxins. Int. J. Food Microbiol. 1995, 27, 1–9. [Google Scholar] [CrossRef]
- Montal, M. Botulinum neurotoxin: A marvel of protein design. Ann. Rev. Biochem. 2010, 79, 591–617. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Montal, M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 10447–10452. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Nakai, Y.; Eubanks, L.M.; Clancy, C.M.; Tepp, W.H.; Pellett, S.; Dickerson, T.J.; Johnson, E.A.; Janda, K.D.; Montal, M. Bimodal modulation of the botulinum neurotoxin protein-conducting channel. Proc. Natl. Acad. Sci. USA 2009, 106, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Megighian, A.; Scorzeto, M.; Fillo, S.; Shone, C.C.; Binz, T.; Rossetto, O.; Lista, F.; et al. Thioredoxin and its reductase are present on synaptic vesicles, and their inhibition prevents the paralysis induced by botulinum neurotoxins. Cell Rep. 2014, 8, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Schiavo, G. Mechanism of action of tetanus and botulinum neurotoxins. Mol. Microbiol. 1994, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Whitemarsh, R.C.; Tepp, W.H.; Johnson, E.A.; Pellett, S. Persistence of botulinum neurotoxin a subtypes 1–5 in primary rat spinal cord cells. PLoS ONE 2014, 9, e90252. [Google Scholar] [CrossRef] [PubMed]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Mahowald, M.L.; Singh, J.A.; Dykstra, D. Long term effects of intra-articular botulinum toxin A for refractory joint pain. Neurotox. Res. 2006, 9, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, A.; Bagnato, S.; Boccagni, C.; Romano, M.C.; Galardi, G. Efficacy of intra-articular injection of botulinum toxin type A in refractory hemiplegic shoulder pain. Arch. Phys. Med. Rehabil. 2011, 92, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Fitzgerald, P.M. Botulinum toxin for shoulder pain: A cochrane systematic review. J. Rheumatol. 2011, 38, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.J.; Smith, J.; Dahm, D.L.; Sorenson, E.J.; Larson, D.R.; Fitz-Gibbon, P.D.; Dykstra, D.D.; Singh, J.A. Efficacy of intra-articular botulinum toxin type A in painful knee osteoarthritis: A pilot study. PM & R J. Injury Funct. Rehabil. 2010, 2, 268–276. [Google Scholar] [CrossRef]
- Ranoux, D.; Attal, N.; Morain, F.; Bouhassira, D. Botulinum toxin type a induces direct analgesic effects in chronic neuropathic pain. Ann. Neurol. 2008, 64, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, G.; Asensio-Samper, J.M.; Palmisani, S.; Villanueva-Perez, V.L.; de Andres, J. Subcutaneous botulinum toxin for chronic post-thoracotomy pain. Pain Pract. 2013, 13, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Matsuka, Y.; Spigelman, I.; Ishihara, Y.; Yamamoto, Y.; Sonoyama, W.; Kamioka, H.; Yamashiro, T.; Kuboki, T.; Oguma, K. Botulinum toxin type A (150 kda) decreases exaggerated neurotransmitter release from trigeminal ganglion neurons and relieves neuropathy behaviors induced by infraorbital nerve constriction. Neuroscience 2009, 159, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, E.J.; Teive, H.G.; Kowacs, P.A.; Della Coletta, M.V.; Werneck, L.C.; Silberstein, S.D. An open study of botulinum-A toxin treatment of trigeminal neuralgia. Neurology 2005, 65, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, C.; Diaz, S.; Piedimonte, F.; Micheli, F. Beneficial effects of botulinum toxin type A in trigeminal neuralgia. Arquivos Neuro-Psiquiatria 2008, 66, 500–503. [Google Scholar] [CrossRef]
- Turk, U.; Ilhan, S.; Alp, R.; Sur, H. Botulinum toxin and intractable trigeminal neuralgia. Clin. Neuropharmacol. 2005, 28, 161–162. [Google Scholar] [PubMed]
- Wu, C.J.; Lian, Y.J.; Zheng, Y.K.; Zhang, H.F.; Chen, Y.; Xie, N.C.; Wang, L.J. Botulinum toxin type A for the treatment of trigeminal neuralgia: Results from a randomized, double-blind, placebo-controlled trial. Cephalalgia Int. J. Headache 2012, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Guardiani, E.; Sadoughi, B.; Blitzer, A.; Sirois, D. A new treatment paradigm for trigeminal neuralgia using botulinum toxin type A. Laryngoscope 2014, 124, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T.; Tsai, S.K.; Kao, M.C.; Hu, J.S. Botulinum toxin A relieved neuropathic pain in a case of post-herpetic neuralgia. Pain Med. 2006, 7, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Huete, C.; Bermejo, P. Botulinum toxin type A in the treatment of neuropathic pain in a case of postherpetic neuralgia. Neurologia 2008, 23, 259–262. [Google Scholar] [PubMed]
- Xiao, L.; Mackey, S.; Hui, H.; Xong, D.; Zhang, Q.; Zhang, D. Subcutaneous injection of botulinum toxin A is beneficial in postherpetic neuralgia. Pain Med. 2010, 11, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.Y.; Sheu, J.J.; Yu, J.M.; Chen, W.T.; Tseng, I.J.; Chang, H.H.; Hu, C.J. Botulinum toxin for diabetic neuropathic pain: A randomized double-blind crossover trial. Neurology 2009, 72, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, B. Evidence based medicine in the use of botulinum toxin for back pain. J. Neural Transm. 2008, 115, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, B. Treatment of chronic low back pain with botulinum neurotoxins. Curr. Pain Headache Rep. 2007, 11, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Cheshire, W.P.; Abashian, S.W.; Mann, J.D. Botulinum toxin in the treatment of myofascial pain syndrome. Pain 1994, 59, 65–69. [Google Scholar] [CrossRef]
- Wheeler, A.H.; Goolkasian, P.; Gretz, S.S. A randomized, double-blind, prospective pilot study of botulinum toxin injection for refractory, unilateral, cervicothoracic, paraspinal, myofascial pain syndrome. Spine 1998, 23, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Pedersen, N.S.; Staahl, C.; Drewes, A.M.; Arendt-Nielsen, L. Subcutaneous botulinum toxin type A reduces capsaicin-induced trigeminal pain and vasomotor reactions in human skin. Pain 2009, 141, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Staahl, C.; Drewes, A.M.; Arendt-Nielsen, L. The effects of botulinum toxin type A on capsaicin-evoked pain, flare, and secondary hyperalgesia in an experimental human model of trigeminal sensitization. Pain 2006, 122, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, N.M.; Dostrovsky, J.O.; Charlton, M.P. Peptide-mediated transdermal delivery of botulinum neurotoxin type A reduces neurogenic inflammation in the skin. Pain 2010, 149, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Tugnoli, V.; Capone, J.G.; Eleopra, R.; Quatrale, R.; Sensi, M.; Gastaldo, E.; Tola, M.R.; Geppetti, P. Botulinum toxin type A reduces capsaicin-evoked pain and neurogenic vasodilatation in human skin. Pain 2007, 130, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Mattler, W.J.; Opatz, O.; Blersch, W.; May, A.; Bigalke, H.; Wohlfahrt, K. Botulinum toxin A does not alter capsaicin-induced pain perception in human skin. J. Neurol. Sci. 2007, 260, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Blersch, W.; Schulte-Mattler, W.J.; Przywara, S.; May, A.; Bigalke, H.; Wohlfarth, K. Botulinum toxin A and the cutaneous nociception in humans: A prospective, double-blind, placebo-controlled, randomized study. J. Neurol. Sci. 2002, 205, 59–63. [Google Scholar] [CrossRef]
- Voller, B.; Sycha, T.; Gustorff, B.; Schmetterer, L.; Lehr, S.; Eichler, H.G.; Auff, E.; Schnider, P. A randomized, double-blind, placebo controlled study on analgesic effects of botulinum toxin A. Neurology 2003, 61, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.B.; Kulas, D.; Karshenas, A.; Cairns, B.E.; Bach, F.W.; Arendt-Nielsen, L.; Gazerani, P. Time course analysis of the effects of botulinum neurotoxin type A on pain and vasomotor responses evoked by glutamate injection into human temporalis muscles. Toxins 2014, 6, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Sycha, T.; Samal, D.; Chizh, B.; Lehr, S.; Gustorff, B.; Schnider, P.; Auff, E. A lack of antinociceptive or antiinflammatory effect of botulinum toxin A in an inflammatory human pain model. Anesth. Analg. 2006, 102, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.J.; Terashima, T.; Steinauer, J.J.; Eddinger, K.A.; Yaksh, T.L.; Xu, Q. Botulinum toxin B in the sensory afferent: Transmitter release, spinal activation, and pain behavior. Pain 2014, 155, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Nagai, J.; Sekino, Y.; Goto, Y.; Nakahira, S.; Ueda, H. Single application of A2 NTX, a botulinum toxin A2 subunit, prevents chronic pain over long periods in both diabetic and spinal cord injury-induced neuropathic pain models. J. Pharmacol. Sci. 2012, 119, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Lackovic, Z. Botulinum toxin A, brain and pain. Prog. Neurobiol. 2014, 119–120, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Bach-Rojecky, L.; Filipovic, B.; Lackovic, Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience 2011, 186, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Vacca, V.; Marinelli, S.; Eleuteri, C.; Luvisetto, S.; Pavone, F. Botulinum neurotoxin A enhances the analgesic effects on inflammatory pain and antagonizes tolerance induced by morphine in mice. Brain Behav. Immun. 2012, 26, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S.; Marinelli, S.; Lucchetti, F.; Marchi, F.; Cobianchi, S.; Rossetto, O.; Montecucco, C.; Pavone, F. Botulinum neurotoxins and formalin-induced pain: Central vs. Peripheral effects in mice. Brain Res. 2006, 1082, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, B.; Matak, I.; Bach-Rojecky, L.; Lackovic, Z. Central action of peripherally applied botulinum toxin type A on pain and dural protein extravasation in rat model of trigeminal neuropathy. PLoS ONE 2012, 7, e29803. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Lam, C.; Yaksh, T.L. Botulinum toxin in migraine: Role of transport in trigemino-somatic and trigemino-vascular afferents. Neurobiol. Dis. 2015, 79, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lackovic, Z. Antinociceptive effect of botulinum toxin type A in rat model of carrageenan and capsaicin induced pain. Croat. Med. J. 2005, 46, 201–208. [Google Scholar] [PubMed]
- Bach-Rojecky, L.; Dominis, M.; Lackovic, Z. Lack of anti-inflammatory effect of botulinum toxin type A in experimental models of inflammation. Fundam. Clin. Pharmacol. 2008, 22, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Favre-Guilmard, C.; Auguet, M.; Chabrier, P.E. Different antinociceptive effects of botulinum toxin type A in inflammatory and peripheral polyneuropathic rat models. Eur. J. Pharmacol. 2009, 617, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Yukihira, T.; Ito, Y.; Akaike, N. Antinociceptive effects of A1 and A2 type botulinum toxins on carrageenan-induced hyperalgesia in rat. Toxicon 2013, 64, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Krug, H.E.; Frizelle, S.; McGarraugh, P.; Mahowald, M.L. Pain behavior measures to quantitate joint pain and response to neurotoxin treatment in murine models of arthritis. Pain Med. 2009, 10, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, H.M.; Hielm-Bjorkman, A.K.; Morelius, M.; Larsen, S.; Honkavaara, J.; Innes, J.F.; Laitinen-Vapaavuori, O.M. Intra-articular botulinum toxin A for the treatment of osteoarthritic joint pain in dogs: A randomized, double-blinded, placebo-controlled clinical trial. Vet. J. 2014, 200, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Hadley, H.S.; Wheeler, J.L.; Petersen, S.W. Effects of intra-articular botulinum toxin type A (Botox®) in dogs with chronic osteoarthritis. Vet. Comp. Orthop. Traumatol. 2010, 23, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Rialland, P.; Authier, S.; Guillot, M.; del Castillo, J.R.; Veilleux-Lemieux, D.; Frank, D.; Gauvin, D.; Troncy, E. Validation of orthopedic postoperative pain assessment methods for dogs: A prospective, blinded, randomized, placebo-controlled study. PLoS ONE 2012, 7, e49480. [Google Scholar] [CrossRef]
- Park, H.J.; Marino, M.J.; Rondon, E.S.; Xu, Q.; Yaksh, T.L. The effects of intraplantar and intrathecal botulinum toxin type B on tactile allodynia in mono and polyneuropathy in the mouse. Anesth. Analg. 2015, 121, 229–238. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, Y.; Lee, J.; Park, C.; Moon, D.E. The effects of botulinum toxin A on mechanical and cold allodynia in a rat model of neuropathic pain. Can. J. Anaesth. 2006, 53, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Dai, J.; Zhang, D. Botulinum toxin decreases hyperalgesia and inhibits P2X3 receptor over-expression in sensory neurons induced by ventral root transection in rats. Pain Med. 2011, 12, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Zhuang, Y.; Qu, W.; Muir, J.; Liang, H.; Zhang, D. Botulinum toxin type A reduces hyperalgesia and trpv1 expression in rats with neuropathic pain. Pain Med. 2013, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Salkovic-Petrisic, M.; Lackovic, Z. Botulinum toxin type A reduces pain supersensitivity in experimental diabetic neuropathy: Bilateral effect after unilateral injection. Eur. J. Pharmacol. 2010, 633, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lackovic, Z. Central origin of the antinociceptive action of botulinum toxin type A. Pharmacol. Biochem. Behav. 2009, 94, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Shibata, M.; Toriumi, H.; Iwashita, T.; Funakubo, M.; Sato, H.; Kuroi, T.; Ebine, T.; Koizumi, K.; Suzuki, N. Reduction of trpv1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol. Dis. 2012, 48, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Au, S.; Dong, X.; Kumar, U.; Arendt-Nielsen, L.; Cairns, B.E. Botulinum neurotoxin type A (BoNTA) decreases the mechanical sensitivity of nociceptors and inhibits neurogenic vasodilation in a craniofacial muscle targeted for migraine prophylaxis. Pain 2010, 151, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, J.; Lawrence, G.; Dolly, J.O. Synaptobrevin i mediates exocytosis of cgrp from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J. Cell Sci. 2007, 120, 2864–2874. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; O’Connell, M.A. Neurotherapeutics to inhibit exocytosis from sensory neurons for the control of chronic pain. Curr. Opin. Pharmacol. 2012, 12, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.L.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache 2004, 44, 35–42; discussion 42–43. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, J.; Warfvinge, K.; Edvinsson, L. Modulation of inflammatory mediators in the trigeminal ganglion by botulinum neurotoxin type A: An organ culture study. J. Headache Pain 2015, 16, 555. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.B.; Poulsen, J.N.; Arendt-Nielsen, L.; Gazerani, P. Botulinum neurotoxin type a modulates vesicular release of glutamate from satellite glial cells. J. Cell Mol. Med. 2015, 19, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.D., Jr. The somatosensory system, with emphasis on structures important for pain. Brain Res. Rev. 2007, 55, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Relja, M.; Lackovic, Z. Botulinum toxin type A in experimental neuropathic pain. J. Neural Transm. 2005, 112, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.O.; Helveston, E.M. Botulinum in the treatment of adult motility disorders. Int. Ophthalmol. Clin. 1986, 26, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.; Woller, S.; Ramachandran, R.; Sorkin, L. The search for novel analgesics: Targets and mechanisms. F1000 Biol. Rep. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Davis, C.L.; Burgess, G.M. Prostaglandin E2-induced sensitization of bradykinin-evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur. J. Neurosci. 2000, 12, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Quirion, R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: Therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin. Ther. Targets 2007, 11, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Chahine, M.; O’Leary, M.E. Regulation/modulation of sensory neuron sodium channels. Handb. Exp. Pharmacol. 2014, 221, 111–135. [Google Scholar] [PubMed]
- Gold, M.S.; Levine, J.D.; Correa, A.M. Modulation of TTX-R ina by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 10345–10355. [Google Scholar]
- Aley, K.O.; Levine, J.D. Role of protein kinase A in the maintenance of inflammatory pain. J. Neurosci. 1999, 19, 2181–2186. [Google Scholar] [PubMed]
- Waxman, S.G.; Cummins, T.R.; Dib-Hajj, S.D.; Black, J.A. Voltage-gated sodium channels and the molecular pathogenesis of pain: A review. J. Rehabil. Res. Dev. 2000, 37, 517–528. [Google Scholar] [PubMed]
- Suzuki, R.; Dickenson, A. Spinal and supraspinal contributions to central sensitization in peripheral neuropathy. Neuro-Signals 2005, 14, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Kawasaki, Y.; Zhuang, Z.Y.; Wen, Y.R.; Zhang, Y.Q. Protein kinases as potential targets for the treatment of pathological pain. Handb. Exp. Pharmacol. 2007, 177, 359–389. [Google Scholar] [PubMed]
- Crown, E.D. The role of mitogen activated protein kinase signaling in microglia and neurons in the initiation and maintenance of chronic pain. Exp. Neurol. 2012, 234, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Edelmayer, R.M.; Brederson, J.D.; Jarvis, M.F.; Bitner, R.S. Biochemical and pharmacological assessment of map-kinase signaling along pain pathways in experimental rodent models: A potential tool for the discovery of novel antinociceptive therapeutics. Biochem. Pharmacol. 2014, 87, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Svensson, C.I.; Koehrn, F.J.; Bhuskute, A.; Sorkin, L.S. Peripheral inflammation induces tumor necrosis factor dependent ampa receptor trafficking and akt phosphorylation in spinal cord in addition to pain behavior. Pain 2010, 149, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Quirion, R. Targeting cell surface trafficking of pain-facilitating receptors to treat chronic pain conditions. Expert Opin. Ther. Targets 2014, 18, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Krotov, V.; Belan, P.; Voitenko, N. Inflammatory-induced changes in synaptic drive and postsynaptic ampars in lamina ii dorsal horn neurons are cell-type specific. Pain 2015, 156, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Taves, S.; Berta, T.; Chen, G.; Ji, R.R. Microglia and spinal cord synaptic plasticity in persistent pain. Neural Plast. 2013, 2013, 753656. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2014, 20, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Yaksh, T.L. Therapeutic use of botulinum toxin in migraine: Mechanisms of action. Br. J. Pharmacol. 2014, 171, 4177–4192. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.A.; Corr, M.; Firestein, G.S.; Mobargha, A.; Yaksh, T.L.; Svensson, C.I. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain 2010, 151, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.B.; Su, J.; Sandor, K.; Agalave, N.M.; Lundberg, J.; Codeluppi, S.; Baharpoor, A.; Nandakumar, K.S.; Holmdahl, R.; Svensson, C.I. Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum. 2012, 64, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Andrade, J.M.; Mantyh, P.W. Sensory and sympathetic nerve fibers undergo sprouting and neuroma formation in the painful arthritic joint of geriatric mice. Arthritis Res. Therapy 2012, 14, R101. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yaksh, T.L. A brief comparison of the pathophysiology of inflammatory versus neuropathic pain. Curr. Opin. Anaesthesiol. 2011, 24, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Manger, B.; Alvaro-Gracia, J.; Johnstone, R.; Gomez-Reino, J.; Eberhardt, E.; Wolfe, F.; Schwartzman, S.; Furfaro, N.; Kavanaugh, A. Patient perceptions concerning pain management in the treatment of rheumatoid arthritis. J. Int. Med. Res. 2010, 38, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Michaud, K. Assessment of pain in rheumatoid arthritis: Minimal clinically significant difference, predictors, and the effect of anti-tumor necrosis factor therapy. J. Rheumatol. 2007, 34, 1674–1683. [Google Scholar] [PubMed]
- DePuy, T.; Howard, R.; Keegan, K.; Wilson, D.; Kramer, J.; Cook, J.L.; Childers, M.K. Effects of intra-articular botulinum toxin type A in an equine model of acute synovitis: A pilot study. Am. J. Phys. Med. Rehabil./Assoc. Acad. Physiatr. 2007, 86, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Akaike, N.; Shin, M.C.; Wakita, M.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kato, K.; Kaji, R.; Kozaki, S. Transsynaptic inhibition of spinal transmission by A2 botulinum toxin. J. Physiol. 2013, 591, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchio, R.; Caleo, M. More than at the neuromuscular synapse: Actions of botulinum neurotoxin A in the central nervous system. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2015, 21, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Gerwin, R. Botulinum toxin treatment of myofascial pain: A critical review of the literature. Curr. Pain Headache Rep. 2012, 16, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Avendano-Coy, J.; Gomez-Soriano, J.; Valencia, M.; Estrada, J.; Leal, F.; Ruiz-Campa, R. Botulinum toxin type A and myofascial pain syndrome: A retrospective study of 301 patients. J. Back Musculoskelet. Rehabil. 2014, 27, 485–492. [Google Scholar] [PubMed]
- Zhou, J.Y.; Wang, D. An update on botulinum toxin A injections of trigger points for myofascial pain. Curr. Pain Headache Rep. 2014, 18, 386. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.J.; Shkolnikova, T.; Nava, A.; Inwald, D. A critical appraisal of the evidence for botulinum toxin type A in the treatment for cervico-thoracic myofascial pain syndrome. Pain Pract. Off. J. World Inst. Pain 2014, 14, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Mahowald, M.L.; Kushnaryov, A.; Goelz, E.; Dykstra, D. Repeat injections of intra-articular botulinum toxin A for the treatment of chronic arthritis joint pain. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2009, 15, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.H.; Angerer, C.; Erbguth, F.; Schmelz, M.; Birklein, F. Botulinum toxin A reduces neurogenic flare but has almost no effect on pain and hyperalgesia in human skin. J. Neurol. 2003, 250, 188–193. [Google Scholar] [PubMed]
- Sorkin, L.S.; Yaksh, T.L. Behavioral models of pain states evoked by physical injury to the peripheral nerve. NeuroTherapeutics 2009, 6, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Challa, S.R. Surgical animal models of neuropathic pain: Pros and Cons. Int. J. Neurosci. 2015, 125, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Siau, C.; Xiao, W.; Bennett, G.J. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: Loss of epidermal innervation and activation of langerhans cells. Exp. Neurol. 2006, 201, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Allen, T.J. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology 2007, 12, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Devor, M. Sodium channels and mechanisms of neuropathic pain. J. Pain. 2006, 7, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.; Devor, M. Extra spike formation in sensory neurons and the disruption of afferent spike patterning. Biophys. J. 2003, 84, 2700–2708. [Google Scholar] [CrossRef]
- Tsujino, H.; Kondo, E.; Fukuoka, T.; Dai, Y.; Tokunaga, A.; Miki, K.; Yonenobu, K.; Ochi, T.; Noguchi, K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 2000, 15, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 2008, 12, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.Y.; Cheng, J.K.; Chu, D.; Cheng, C.F.; Tsaur, M.L. Reduced expression of a-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J. Neurosci. 2007, 27, 9855–9865. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Krugel, U.; Abbracchio, M.P.; Illes, P. Purinergic signalling: From normal behaviour to pathological brain function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Winkelstein, B.A. Schwann cell proliferation and macrophage infiltration are evident at day 14 after painful cervical nerve root compression in the rat. J. Neurotrauma 2011, 28, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Lindia, J.A.; Cumiskey, A.M.; Peterson, L.B.; Mudgett, J.S.; Bayne, E.K.; DeMartino, J.A.; MacIntyre, D.E.; Forrest, M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad Sci. USA 2003, 100, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Patterson, P.H.; Jessen, K.R.; Mirsky, R. Denervated schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J. Neurosci. 2002, 22, 6696–6703. [Google Scholar] [PubMed]
- McLachlan, E.M.; Hu, P. Inflammation in dorsal root ganglia after peripheral nerve injury: Effects of the sympathetic innervation. Auton. Neurosci. Basic Clin. 2014, 182, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Blum, E.; Procacci, P.; Conte, V.; Hanani, M. Systemic inflammation alters satellite glial cell function and structure. A possible contribution to pain. Neuroscience 2014, 274, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Shu, Y.; Zheng, Z.; Chen, Y.; Yao, H.; Greenquist, K.W.; White, F.A.; LaMotte, R.H. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J. Neurophysiol. 2003, 89, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Sukhotinsky, I.; Ben-Dor, E.; Raber, P.; Devor, M. Key role of the dorsal root ganglion in neuropathic tactile hypersensibility. Eur. J. Pain 2004, 8, 135–143. [Google Scholar] [CrossRef]
- Ellis, A.; Bennett, D.L. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 2013, 111, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. 1), S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Gobel, H.; Heinze, A.; Heinze-Kuhn, K.; Jost, W.H. Evidence-based medicine: Botulinum toxin A in migraine and tension-type headache. J. Neurol. 2001, 248 (Suppl. 1), 34–38. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.; Smith, H.S. Botulinum toxins: Mechanisms of action, antinociception and clinical applications. Toxicology 2013, 306, 124–146. [Google Scholar] [CrossRef]
- Quick, M.W. The role of snare proteins in trafficking and function of neurotransmitter transporters. Handb. Exp. Pharmacol. 2006, 181–196. [Google Scholar]
- Schrattenholz, A.; Soskic, V. Nmda receptors are not alone: Dynamic regulation of nmda receptor structure and function by neuregulins and transient cholesterol-rich membrane domains leads to disease-specific nuances of glutamate-signalling. Curr. Top. Med. Chem. 2006, 6, 663–686. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Huang, Y.; Amato, S.; Snyder, S.H.; Huganir, R.L.; Man, H.Y. Regulation of ampa receptor localization in lipid rafts. Mol. Cell. Neurosci. 2008, 38, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Murray, F.; Insel, P.A. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharmacol. 2008, 167–184. [Google Scholar]
- Delint-Ramirez, I.; Fernandez, E.; Bayes, A.; Kicsi, E.; Komiyama, N.H.; Grant, S.G. In vivo composition of nmda receptor signaling complexes differs between membrane subdomains and is modulated by PSD-95 and PSD-93. J. Neurosci. 2010, 30, 8162–8170. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B.; Parks, J.S. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J. Immunol. 2011, 187, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Kakegawa, W.; Yuzaki, M. A mechanism underlying ampa receptor trafficking during cerebellar long-term potentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 17846–17851. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Pozzi, D.; Pravettoni, E.; Inverardi, F.; Schenk, U.; Coco, S.; Proux-Gillardeaux, V.; Galli, T.; Rossetto, O.; Frassoni, C.; et al. Snap-25 modulation of calcium dynamics underlies differences in gabaergic and glutamatergic responsiveness to depolarization. Neuron 2004, 41, 599–610. [Google Scholar] [CrossRef]
- Pozzi, D.; Condliffe, S.; Bozzi, Y.; Chikhladze, M.; Grumelli, C.; Proux-Gillardeaux, V.; Takahashi, M.; Franceschetti, S.; Verderio, C.; Matteoli, M. Activity-dependent phosphorylation of SER187 is required for snap-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Gerachshenko, T.; Blackmer, T.; Yoon, E.J.; Bartleson, C.; Hamm, H.E.; Alford, S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat. Neurosci. 2005, 8, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Morenilla-Palao, C.; Planells-Cases, R.; García-Sanz, N.; Ferrer-Montiel, A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J. Biol. Chem. 2004, 279, 25665–25672. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S. Progress in cell based assays for botulinum neurotoxin detection. Curr. Top. Microbiol. Immunol. 2013, 364, 257–285. [Google Scholar] [PubMed]
- Richardson, J.D.; Vasko, M.R. Cellular mechanisms of neurogenic inflammation. J. Pharmacol. Exp. Ther. 2002, 302, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Ma, Q. Nociceptors-noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Hucho, T.; Levine, J.D. Signaling pathways in sensitization: Toward a nociceptor cell biology. Neuron 2007, 55, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. Bacterial inhibition of phagocytosis. Cell. Microbiol. 2000, 2, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Calafat, J.; Janssen, H.; Martin-Martin, B.; Canchado, J.; Nabokina, S.M.; Gajate, C. Combinatorial snare complexes modulate the secretion of cytoplasmic granules in human neutrophils. J. Immunol. 2006, 177, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.Z.; Stow, J.L. Cytokine secretion in macrophages: Snares, rabs, and membrane trafficking. Front. Immunol. 2014, 5, 538. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [PubMed]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed]
- Logan, M.R.; Odemuyiwa, S.O.; Moqbel, R. Understanding exocytosis in immune and inflammatory cells: The molecular basis of mediator secretion. J. Allergyd Clin. Immunol. 2003, 111, 923–932; quiz 933. [Google Scholar] [CrossRef]
- Lorentz, A.; Baumann, A.; Vitte, J.; Blank, U. The snare machinery in mast cell secretion. Front. Immunol. 2012, 3, 143. [Google Scholar] [CrossRef] [PubMed]
- Park, T.H. The effects of botulinum toxin A on mast cell activity: Preliminary results. Burns J. Int. Soc. Burn Inj. 2013, 39, 816–817. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Brooks, P. The use of botulinum toxin in the management of burns itching: Preliminary results. Burns 2012, 38, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Yamamura, H.; Malick, A.; Strassman, A.M. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J. Neurophysiol. 1998, 79, 964–982. [Google Scholar] [PubMed]
- Burstein, R.; Zhang, X.; Levy, D.; Aoki, K.R.; Brin, M.F. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia Int. J. Headache 2014, 34, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Salaun, C.; Gould, G.W.; Chamberlain, L.H. Lipid raft association of snare proteins regulates exocytosis in PC12 cells. J. Biol. Chem. 2005, 280, 19449–19453. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Magenau, A.; Williamson, D.; Gaus, K. The lipid raft hypothesis revisited—New insights on raft composition and function from super-resolution fluorescence microscopy. BioEssays 2012, 34, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Ovsepian, S.V.; Wang, J.; Pickering, M.; Sasse, A.; Aoki, K.R.; Lawrence, G.W.; Dolly, J.O. Activation of trpv1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J. Neurosci. 2009, 29, 4981–4992. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. Exciting trips for TRPS. Nat. Cell Biol. 2004, 6, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.Y.; Skeberdis, V.A.; Jover, T.; Grooms, S.Y.; Lin, Y.; Araneda, R.C.; Zheng, X.; Bennett, M.V.; Zukin, R.S. Protein kinase C modulates nmda receptor trafficking and gating. Nat. Neurosci. 2001, 4, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Planells-Cases, R.; Ferrer-Montiel, A. TRP Channel Trafficking. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007; Chapter 23. [Google Scholar]
- Ferrandiz-Huertas, C.; Mathivanan, S.; Wolf, C.J.; Devesa, I.; Ferrer-Montiel, A. Trafficking of thermotrp channels. Membranes 2014, 4, 525–564. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L.; Ozaki, G.; McCumber, D.; Rathbun, M.; Svensson, C.; Malkmus, S.; Yaksh, M.C. An automated flinch detecting system for use in the formalin nociceptive bioassay. J. Appl. Physiol. 2001, 90, 2386–2402. [Google Scholar] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Antonucci, F.; Gianfranceschi, L.; Rossi, C.; Rossetto, O.; Caleo, M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J. Neurosci. 2011, 31, 15650–15659. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Giribaldi, F.; Manich, M.; Bercsenyi, K.; Menendez, G.; Rossetto, O.; Caleo, M.; Schiavo, G. Botulinum neurotoxins A and e undergo retrograde axonal transport in primary motor neurons. PLoS Pathog. 2012, 8, e1003087. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, G.W.; Ovsepian, S.V.; Wang, J.; Aoki, K.R.; Dolly, J.O. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochem. J. 2012, 441, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. 125I-labeled neurotoxin from clostridium botulinum a: Preparation, binding to synaptosomes and ascent to the spinal cord. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1974, 281, 47–56. [Google Scholar] [CrossRef]
- Wiegand, H.; Erdmann, G.; Wellhoner, H.H. 125I-labelled botulinum A neurotoxin: Pharmacokinetics in cats after intramuscular injection. Naunyn-Schmiedeberg's Arch. Pharmacol. 1976, 292, 161–165. [Google Scholar] [CrossRef]
- Black, J.D.; Dolly, J.O. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. I. Ultrastructural autoradiographic localization and quantitation of distinct membrane acceptors for types A and B on motor nerves. J. Cell Biol. 1986, 103, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Riederer, P.; Lackovic, Z. Botulinum toxin’s axonal transport from periphery to the spinal cord. Neurochem. Int. 2012, 61, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.R. Evidence for antinociceptive activity of botulinum toxin type A in pain management. Headache 2003, 43 (Suppl. 1), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Goto, S.; Okita, S.; Morigaki, R.; Akaike, N.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kaji, R. Spinal central effects of peripherally applied botulinum neurotoxin A in comparison between its subtypes A1 and A2. Front. Neurol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Novelli, E.; Bottari, D.; Leone, P.; Barone, I.; Galli-Resta, L.; Strettoi, E.; Caleo, M. Botulinum neurotoxin A impairs neurotransmission following retrograde transynaptic transport. Traffic 2012, 13, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Alexiades-Armenakas, M. Retrograde transport and transcytosis of botulinum toxin serotypes to the brain: Analysis of potential neurotoxicity. J. Drugs Dermatol. JDD 2008, 7, 1006–1007. [Google Scholar] [PubMed]
- Marchand-Pauvert, V.; Aymard, C.; Giboin, L.S.; Dominici, F.; Rossi, A.; Mazzocchio, R. Beyond muscular effects: Depression of spinal recurrent inhibition after botulinum neurotoxin A. J. Physiol. 2013, 591, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Takasusuki, T.; Yaksh, T.L. The effects of intrathecal and systemic gabapentin on spinal substance p release. Anesth. Analg. 2011, 112, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L. Calcium channels as therapeutic targets in neuropathic pain. J. Pain 2006, 7, S13–30. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, A.B.; Yaksh, T.L. Effect of continuous intrathecal infusion of omega-conopeptides, n-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995, 60, 83–90. [Google Scholar] [CrossRef]
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic conotoxins: Block and G protein-coupled receptor modulation of N-type (CaV 2.2) calcium channels. Br. J. Pharmacol. 2012, 166, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Xu, Q.; Yamaguchi, S.; Yaksh, T.L. Intrathecal P/Q- and R-type calcium channel blockade of spinal substance P release and c-Fos expression. Neuropharmacology 2013, 75, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Pogrel, J.W.; Yaksh, T.L. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J. Pharmacol. Exp. Ther. 1994, 269, 1117–1123. [Google Scholar] [PubMed]
- Xiao, Y.; Richter, J.A.; Hurley, J.H. Release of glutamate and cgrp from trigeminal ganglion neurons: Role of calcium channels and 5-ht1 receptor signaling. Mol. Pain 2008, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.G.; MacDermott, A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 1997, 389, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Deng, P.; Matthews, E.A.; Kim, D.S.; Feng, G.; Dickenson, A.H.; Xu, Z.C.; Luo, Z.D. Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel alpha-2-delta-1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol. Pain 2009, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Shinder, V.; Amir, R.; Devor, M. Cross-excitation in dorsal root ganglia does not depend on close cell-to-cell apposition. Neuroreport 1998, 9, 3997–4000. [Google Scholar] [CrossRef] [PubMed]
- Devor, M.; Wall, P.D. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J. Neurophysiol. 1990, 64, 1733–1746. [Google Scholar] [PubMed]
- Amir, R.; Devor, M. Functional cross-excitation between afferent a- and c-neurons in dorsal root ganglia. Neuroscience 2000, 95, 189–195. [Google Scholar] [CrossRef]
- Kung, L.H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for glutamate as a neuroglial transmitter within sensory ganglia. PLoS ONE 2013, 8, e68312. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, L.F.; Lotufo, C.M.; Araldi, D.; Rodrigues, M.A.; Macedo, L.P.; Ferreira, S.H.; Parada, C.A. Inflammatory sensitization of nociceptors depends on activation of nmda receptors in drg satellite cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18363–18368. [Google Scholar] [CrossRef] [PubMed]
- Omoto, K.; Maruhama, K.; Terayama, R.; Yamamoto, Y.; Matsushita, O.; Sugimoto, T.; Oguma, K.; Matsuka, Y. Cross-excitation in peripheral sensory ganglia associated with pain transmission. Toxins 2015, 7, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.P.; Huganir, R.L.; Linden, D.J. N-ethylmaleimide-sensitive factor is required for the synaptic incorporation and removal of ampa receptors during cerebellar long-term depression. Proc. Natl. Acad. Sci. USA 2004, 101, 18212–18216. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.G.; Takayasu, Y.; Rodenas-Ruano, A.; Paternain, A.V.; Lerma, J.; Bennett, M.V.; Zukin, R.S. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 2010, 30, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Broman, J. Synaptic plasticity and pain: Role of ionotropic glutamate receptors. Neuroscientist 2011, 17, 256–273. [Google Scholar] [CrossRef] [PubMed]
- Bardoni, R. Role of presynaptic glutamate receptors in pain transmission at the spinal cord level. Curr. Neuropharmacol. 2013, 11, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.M.; Beggs, S.; Baccei, M.L. Persistent changes in peripheral and spinal nociceptive processing after early tissue injury. Exp. Neurol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.P.; Khan, I.; Suhail, M.S.; Malkmus, S.; Yaksh, T.L. Spinal botulinum neurotoxin B: Effects on afferent transmitter release and nociceptive processing. PLoS ONE 2011, 6, e19126. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Shin, T.J.; Kim, H.J.; Lee, J.K.; Suh, H.W.; Lee, S.C.; Seo, K. Intrathecal administration of botulinum neurotoxin type A attenuates formalin-induced nociceptive responses in mice. Anesth. Analg. 2011, 112, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.; Oliveira, R.; Rossetto, O.; Cruz, C.D.; Cruz, F.; Avelino, A. Intrathecal administration of botulinum toxin type A improves urinary bladder function and reduces pain in rats with cystitis. Eur. J. Pain 2014, 18, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino de Laureto, P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Grumelli, C.; Raiteri, L.; Coco, S.; Paluzzi, S.; Caccin, P.; Rossetto, O.; Bonanno, G.; Montecucco, C.; Matteoli, M. Traffic of botulinum toxins A and e in excitatory and inhibitory neurons. Traffic 2007, 8, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; Lawrence, G.W.; Meng, J.; Wang, J.; Ovsepian, S.V. Neuro-exocytosis: Botulinum toxins as inhibitory probes and versatile therapeutics. Curr. Opin. Pharmacol. 2009, 9, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E. Inhibition by tetanus and botulinum A toxin of the release of [3H]noradrenaline and [3H]gaba from rat brain homogenate. Experientia 1988, 44, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Carroll, I.; Fischbein, N.; Barad, M.; Mackey, S. Human response to unintended intrathecal injection of botulinum toxin. Pain Med. 2011, 12, 1094–1097. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marinelli, S.; Vacca, V.; Ricordy, R.; Uggenti, C.; Tata, A.M.; Luvisetto, S.; Pavone, F. The analgesic effect on neuropathic pain of retrogradely transported botulinum neurotoxin a involves schwann cells and astrocytes. PLoS ONE 2012, 7, e47977. [Google Scholar] [CrossRef] [PubMed]
- Littlewood, N.K.; Todd, A.J.; Spike, R.C.; Watt, C.; Shehab, S.A. The types of neuron in spinal dorsal horn which possess neurokinin-1 receptors. Neuroscience 1995, 66, 597–608. [Google Scholar] [CrossRef]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Kang, N.; Xu, J.; Stanton, P.K.; Kang, J. Two distinct modes of exocytotic fusion pore expansion in large astrocytic vesicles. J. Biol. Chem. 2013, 288, 16872–16881. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Cagnoli, C.; Bergami, M.; Francolini, M.; Schenk, U.; Colombo, A.; Riganti, L.; Frassoni, C.; Zuccaro, E.; Danglot, L.; et al. Ti-VAMP/VAMP7 is the snare of secretory lysosomes contributing to ATP secretion from astrocytes. Biol. Cell 2012, 104, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Prada, I.; Marchaland, J.; Podini, P.; Magrassi, L.; D'Alessandro, R.; Bezzi, P.; Meldolesi, J. REST/NRSF governs the expression of dense-core vesicle gliosecretion in astrocytes. J. Cell Biol. 2011, 193, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Hepp, R.; Perraut, M.; Chasserot-Golaz, S.; Galli, T.; Aunis, D.; Langley, K.; Grant, N.J. Cultured glial cells express the SNAP-25 analogue SNAP-23. Glia 1999, 27, 181–187. [Google Scholar] [CrossRef]
- Parpura, V.; Fang, Y.; Basarsky, T.; Jahn, R.; Haydon, P.G. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 1995, 377, 489–492. [Google Scholar] [CrossRef]
- Schubert, V.; Bouvier, D.; Volterra, A. Snare protein expression in synaptic terminals and astrocytes in the adult hippocampus: A comparative analysis. Glia 2011, 59, 1472–1488. [Google Scholar] [CrossRef] [PubMed]
- Jeftinija, S.D.; Jeftinija, K.V.; Stefanovic, G. Cultured astrocytes express proteins involved in vesicular glutamate release. Brain Res. 1997, 750, 41–47. [Google Scholar] [CrossRef]
- Maienschein, V.; Marxen, M.; Volknandt, W.; Zimmermann, H. A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia 1999, 26, 233–244. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, G.W.; Kim, M.J.; Yang, K.Y.; Kim, S.T.; Bae, Y.C.; Ahn, D.K. Antinociceptive effects of transcytosed botulinum neurotoxin type A on trigeminal nociception in rats. Korean J. Physiol. Pharmacol. 2015, 19, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Dover, N.; Barash, J.R.; Hill, K.K.; Xie, G.; Arnon, S.S. Molecular characterization of a novel botulinum neurotoxin type H gene. J. Infect. Dis. 2014, 209, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Barash, J.R.; Arnon, S.S. A novel strain of clostridium botulinum that produces type B and type H botulinum toxins. J. Infect. Dis. 2014, 209, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Baudys, J.; Raphael, B.H.; Dykes, J.K.; Luquez, C.; Maslanka, S.E.; Barr, J.R. Functional characterization of botulinum neurotoxin serotype H as a hybrid of known serotypes F and A (BoNT F/A). Anal. Chem. 2015, 87, 3911–3917. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Baudys, J.; Webb, R.P.; Wright, P.; Smith, T.J.; Smith, L.A.; Fernandez, R.; Raphael, B.H.; Maslanka, S.E.; Pirkle, J.L.; et al. Discovery of a novel enzymatic cleavage site for botulinum neurotoxin F5. FEBS Lett. 2012, 586, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A. Double receptor anchorage of botulinum neurotoxins accounts for their exquisite neurospecificity. Curr. Top. Microbiol. Immunol. 2013, 364, 61–90. [Google Scholar] [PubMed]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. Sv2 is the protein receptor for botulinum neurotoxin a. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Jacky, B.P.; Garay, P.E.; Dupuy, J.; Nelson, J.B.; Cai, B.; Molina, Y.; Wang, J.; Steward, L.E.; Broide, R.S.; Francis, J.; et al. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A). PLoS Pathog. 2013, 9, e1003369. [Google Scholar] [CrossRef] [PubMed]
- Grothe, C.; Meisinger, C.; Claus, P. In vivo expression and localization of the fibroblast growth factor system in the intact and lesioned rat peripheral nerve and spinal ganglia. J. Comp. Neurol. 2001, 434, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Jungnickel, J.; Gransalke, K.; Timmer, M.; Grothe, C. Fibroblast growth factor receptor 3 signaling regulates injury-related effects in the peripheral nervous system. Mol. Cell. Neurosci. 2004, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pringle, N.P.; Yu, W.P.; Howell, M.; Colvin, J.S.; Ornitz, D.M.; Richardson, W.D. FGFR3 expression by astrocytes and their precursors: Evidence that astrocytes and oligodendrocytes originate in distinct neuroepithelial domains. Development 2003, 130, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Nonaka, H.; Akaike, A.; Kihara, T.; Sugimoto, H. Basic fibroblast growth factor promotes the generation of microtubule-associated protein 2-positive cells from microglia. Biochem. Biophys. Res. Commun. 2009, 390, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Adler, M.; Demogines, A.; Borrell, A.; Liu, H.; Tao, L.; Tepp, W.H.; Zhang, S.C.; Johnson, E.A.; Sawyer, S.L.; et al. Widespread sequence variations in vamp1 across vertebrates suggest a potential selective pressure from botulinum neurotoxins. PLoS Pathog. 2014, 10, e1004177. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Baumeister, A.; Binz, T.; Blasi, J.; Link, E.; Cornille, F.; Roques, B.; Fykse, E.M.; Sudhof, T.C.; Jahn, R. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J. Biol. Chem. 1994, 269, 12764–12772. [Google Scholar] [PubMed]
- Yamamoto, H.; Ida, T.; Tsutsuki, H.; Mori, M.; Matsumoto, T.; Kohda, T.; Mukamoto, M.; Goshima, N.; Kozaki, S.; Ihara, H. Specificity of botulinum protease for human vamp family proteins. Microbiol. Immunol. 2012, 56, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Strotmeier, J.; Willjes, G.; Binz, T.; Rummel, A. Human synaptotagmin-ii is not a high affinity receptor for botulinum neurotoxin B and G: Increased therapeutic dosage and immunogenicity. FEBS Lett. 2012, 586, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Berntsson, R.P.; Tepp, W.H.; Pitkin, R.M.; Johnson, E.A.; Stenmark, P.; Dong, M. Botulinum neurotoxin D-C uses synaptotagmin I/II as receptors and human synaptotagmin II is not an effective receptor for type B, D-C, and G toxins. J. Cell Sci. 2012, 125, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.A. Clostridium botulinum and clostridium tetani. In Topley and Wilson’s Microbiology and Microbial Infections; Borriello, S.P., Murray, P.R., Funke, G., Eds.; Hodder Arnold: London, UK, 2005; Volume 8, pp. 1035–1088. [Google Scholar]
- Eleopra, R.; Tugnoli, V.; Quatrale, R.; Rossetto, O.; Montecucco, C. Different types of botulinum toxin in humans. Mov. Disord. 2004, 19 (Suppl. 8), S53–S59. [Google Scholar] [CrossRef] [PubMed]
- Sloop, R.R.; Cole, B.A.; Escutin, R.O. Human response to botulinum toxin injection: Type B compared with type A. Neurology 1997, 49, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Foran, P.G.; Mohammed, N.; Lisk, G.O.; Nagwaney, S.; Lawrence, G.W.; Johnson, E.; Smith, L.; Aoki, K.R.; Dolly, J.O. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 2003, 278, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.E. Recovery from botulinum neurotoxin poisoning in vivo. Neuroscience 2006, 139, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Eleopra, R.; Tugnoli, V.; Quatrale, R.; Rossetto, O.; Montecucco, C.; Dressler, D. Clinical use of non-A botulinum toxins: Botulinum toxin type c and botulinum toxin type F. Neurotox. Res. 2006, 9, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Eleopra, R.; Tugnoli, V.; Rossetto, O.; Montecucco, C.; de Grandis, D. Botulinum neurotoxin serotype C: A novel effective botulinum toxin therapy in human. Neurosci. Lett. 1997, 224, 91–94. [Google Scholar] [CrossRef]
- Houser, M.K.; Sheean, G.L.; Lees, A.J. Further studies using higher doses of botulinum toxin type F for torticollis resistant to botulinum toxin type A. J. Neurol. Neurosurg. Psychiatry 1998, 64, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Sheean, G.L.; Lees, A.J. Botulinum toxin F in the treatment of torticollis clinically resistant to botulinum toxin A. J. Neurol. Neurosurg. Psychiatry 1995, 59, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Greene, P.E.; Fahn, S. Use of botulinum toxin type F injections to treat torticollis in patients with immunity to botulinum toxin type A. Mov. Disord. 1993, 8, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Eleopra, R.; Montecucco, C.; Devigili, G.; Lettieri, C.; Rinaldo, S.; Verriello, L.; Pirazzini, M.; Caccin, P.; Rossetto, O. Botulinum neurotoxin serotype D is poorly effective in humans: An in vivo electrophysiological study. Clin. Neurophysiol. 2013, 124, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Scherf, J.M.; Pier, C.L.; Johnson, E.A. Activity of botulinum neurotoxin type d (strain 1873) in human neurons. Toxicon 2015, 101, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Whitemarsh, R.C.; Tepp, W.H.; Bradshaw, M.; Lin, G.; Pier, C.L.; Scherf, J.M.; Johnson, E.A.; Pellett, S. Characterization of botulinum neurotoxin a subtypes 1 through 5 by investigation of activities in mice, neuronal cell cultures, and in vitro. Infect. Immun. 2013, 81, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Krilich, J.; Pellett, S.; Baudys, J.; Tepp, W.H.; Barr, J.R.; Johnson, E.A.; Kalb, S.R. Comparison of the catalytic properties of the botulinum neurotoxin subtypes A1 and A5. Biochim. Biophys. Acta 2013, 1834, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Pier, C.L.; Chen, C.; Tepp, W.H.; Lin, G.; Janda, K.D.; Barbieri, J.T.; Pellett, S.; Johnson, E.A. Botulinum neurotoxin subtype A2 enters neuronal cells faster than subtype a1. FEBS Lett. 2011, 585, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tepp, W.H.; Lin, G.; Johnson, E.A. Purification and characterization of a novel subtype A3 botulinum neurotoxin. Appl. Environ. Microbiol. 2012, 78, 3108–3113. [Google Scholar] [CrossRef] [PubMed]
- Kull, S.; Schulz, K.M.; Weisemann, J.; Kirchner, S.; Schreiber, T.; Bollenbach, A.; Dabrowski, P.W.; Nitsche, A.; Kalb, S.R.; Dorner, M.B.; et al. Isolation and functional characterization of the novel clostridium botulinum neurotoxin A8 subtype. PLoS ONE 2015, 10, e0116381. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Whitemarsh, R.C.; Bradshaw, M.; Johnson, E.A. In vivo onset and duration of action varies for botulinum neurotoxin A subtypes 1–5. Toxicon 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Torii, Y.; Goto, Y.; Nakahira, S.; Kozaki, S.; Kaji, R.; Ginnaga, A. Comparison of systemic toxicity between botulinum toxin subtypes A1 and A2 in mice and rats. Basic Clin. Pharmacol. Toxicol. 2014, 116, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Torii, Y.; Akaike, N.; Harakawa, T.; Kato, K.; Sugimoto, N.; Goto, Y.; Nakahira, S.; Kohda, T.; Kozaki, S.; Kaji, R.; et al. Type A1 but not type A2 botulinum toxin decreases the grip strength of the contralateral foreleg through axonal transport from the toxin-treated foreleg of rats. J. Pharmacol. Sci. 2011, 117, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Torii, Y.; Kiyota, N.; Sugimoto, N.; Mori, Y.; Goto, Y.; Harakawa, T.; Nakahira, S.; Kaji, R.; Kozaki, S.; Ginnaga, A. Comparison of effects of botulinum toxin subtype A1 and A2 using twitch tension assay and rat grip strength test. Toxicon Off. J. Int. Soc. Toxinol. 2010, 57, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Shimatani, Y.; Sako, W.; Asanuma, K.; Nodera, H.; Sakamoto, T.; Izumi, Y.; Kohda, T.; Kozaki, S.; Kaji, R. Comparison between botulinum neurotoxin type A2 and type A1 by electrophysiological study in healthy individuals. Toxicon 2014, 81, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Tepp, W.H.; Pier, C.L.; Jacobson, M.J.; Johnson, E.A. Expression of the clostridium botulinum A2 neurotoxin gene cluster proteins and characterization of the A2 complex. Appl. Environ. Microbiol. 2010, 76, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Akaike, N.; Ito, Y.; Shin, M.C.; Nonaka, K.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kozaki, S.; Kaji, R. Effects of A2 type botulinum toxin on spontaneous miniature and evoked transmitter release from the rat spinal excitatory and inhibitory synapses. Toxicon 2010, 56, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Jacobson, M.; Tepp, W.; Pier, C.; Johnson, E.A.; Barbieri, J.T. Catalytic properties of botulinum neurotoxin subtypes A3 and A4 (dagger). Biochemistry 2009, 48, 2522. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Meng, J.; Lawrence, G.W.; Zurawski, T.H.; Sasse, A.; Bodeker, M.O.; Gilmore, M.A.; Fernandez-Salas, E.; Francis, J.; Steward, L.E.; et al. Novel chimeras of botulinum neurotoxins a and e unveil contributions from the binding, translocation, and protease domains to their functional characteristics. J. Biol. Chem. 2008, 283, 16993–17002. [Google Scholar] [CrossRef] [PubMed]
- Strotmeier, J.; Mahrhold, S.; Krez, N.; Janzen, C.; Lou, J.; Marks, J.D.; Binz, T.; Rummel, A. Identification of the synaptic vesicle glycoprotein 2 receptor binding site in botulinum neurotoxin A. FEBS Lett. 2014, 588, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Mahrhold, S.; Bigalke, H.; Binz, T. Exchange of the HCC domain mediating double receptor recognition improves the pharmacodynamic properties of botulinum neurotoxin. FEBS J. 2011, 278, 4506–4515. [Google Scholar] [CrossRef] [PubMed]
- Band, P.A.; Blais, S.; Neubert, T.A.; Cardozo, T.J.; Ichtchenko, K. Recombinant derivatives of botulinum neurotoxin A engineered for trafficking studies and neuronal delivery. Protein Expr. Purif. 2010, 71, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Vessely, C.; Estey, T.; Randolph, T.W.; Henderson, I.; Cooper, J.; Nayar, R.; Braun, L.J.; Carpenter, J.F. Stability of a trivalent recombinant protein vaccine formulation against botulinum neurotoxin during storage in aqueous solution. J. Pharm. Sci. 2009, 98, 2970–2993. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Cintron, E.J.; Vakulenko, M.; Band, P.A.; Stanker, L.H.; Johnson, E.A.; Ichtchenko, K. Atoxic derivative of botulinum neurotoxin A as a prototype molecular vehicle for targeted delivery to the neuronal cytoplasm. PLoS ONE 2014, 9, e85517. [Google Scholar] [CrossRef] [PubMed]
- Pier, C.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Barbieri, J.T.; Baldwin, M.R. Recombinant holotoxoid vaccine against botulism. Infect. Immun. 2008, 76, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, M.; Tepp, W.H.; Whitemarsh, R.C.; Pellett, S.; Johnson, E.A. Holotoxin Activity of Botulinum Neurotoxin Subtype A4 Originating from a Nontoxigenic Clostridium botulinum Expression System. Appl. Environ. Microbiol. 2014, 80, 7415–7422. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Barbieri, J.T. Engineering botulinum neurotoxin to extend therapeutic intervention. Proc. Natl. Acad. Sci. USA 2009, 106, 9180–9184. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L. The life history of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Rasotto, M.B. On botulinum neurotoxin variability. mBio 2015, 6, e02131–14. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.; Myllykoski, J.; Sivela, S.; Korkeala, H. Clostridium botulinum in cattle and dairy products. Crit. Rev. Food Sci. Nutr. 2010, 50, 281–304. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.; Grosse-Herrenthey, A.; Schrodl, W.; Gerlach, A.; Rodloff, A. Visceral botulism at dairy farms in schleswig holstein, germany: Prevalence of clostridium botulinum in feces of cows, in animal feeds, in feces of the farmers, and in house dust. Anaerobe 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Galey, F.D. Botulism in the horse. Equine Pract. 2001, 17, 579–588. [Google Scholar]
- Elad, D.; Yas-Natan, E.; Aroch, I.; Shamir, M.H.; Kleinbart, S.; Hadash, D.; Chaffer, M.; Greenberg, K.; Shlosberg, A. Natural clostridium botulinum type C toxicosis in a group of cats. J. Clin. Microbiol. 2004, 42, 5406–5408. [Google Scholar] [CrossRef] [PubMed]
- Borst, G.H.; Lambers, G.M.; Haagsma, J. Type-C botulism in dogs. Tijdschr. Diergeneeskd. 1986, 111, 1104–1105. [Google Scholar] [PubMed]
- Whitemarsh, R.C.; Strathman, M.J.; Chase, L.G.; Stankewicz, C.; Tepp, W.H.; Johnson, E.A.; Pellett, S. Novel application of human neurons derived from induced pluripotent stem cells for highly sensitive botulinum neurotoxin detection. Toxicol. Sci. 2012, 126, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Raptis, A.; Torrejon-Escribano, B.; Gomez de Aranda, I.; Blasi, J. Distribution of synaptobrevin/VAMP 1 and 2 in rat brain. J. Chem. Neuroanat. 2005, 30, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Karalewitz, A.P.; Fu, Z.; Baldwin, M.R.; Kim, J.J.; Barbieri, J.T. Botulinum neurotoxin serotype C associates with dual ganglioside receptors to facilitate cell entry. J. Biol. Chem. 2012, 287, 40806–40816. [Google Scholar] [CrossRef] [PubMed]
- Blasi, J.; Chapman, E.R.; Yamasaki, S.; Binz, T.; Niemann, H.; Jahn, R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993, 12, 4821–4828. [Google Scholar] [PubMed]
- Blasi, J.; Chapman, E.R.; Link, E.; Binz, T.; Yamasaki, S.; de Camilli, P.; Sudhof, T.C.; Niemann, H.; Jahn, R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Liu, H.; Ruan, H.; Tepp, W.H.; Stoothoff, W.H.; Brown, R.H.; Johnson, E.A.; Yao, W.D.; Zhang, S.C.; Dong, M. Cytotoxicity of botulinum neurotoxins reveals a direct role of syntaxin 1 and SNAP-25 in neuron survival. Nat. Commun. 2013, 4, 1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Z.; Dong, M.; Sun, S.; Chapman, E.R.; Jackson, M.B. Syntaxin requirement for Ca2+-triggered exocytosis in neurons and endocrine cells demonstrated with an engineered neurotoxin. Biochemistry 2011, 50, 2711–2713. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.G.; Kline, R.H.t.; Vierck, C.J., Jr. Anti-nociceptive effects of selectively destroying substance P receptor-expressing dorsal horn neurons using [Sar9,Met(O2)11]-substance P-saporin: Behavioral and anatomical analyses. Neuroscience 2007, 146, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.G.; Lappi, D.A. Targeted toxins in pain. Adv. Drug Deliv. Rev. 2003, 55, 1043–1054. [Google Scholar] [CrossRef]
- Wiley, R.G.; Lappi, D.A. Destruction of neurokinin-1 receptor expressing cells in vitro and in vivo using substance p-saporin in rats. Neurosci. Lett. 1997, 230, 97–100. [Google Scholar] [CrossRef]
- Wiese, A.J.; Rathbun, M.; Butt, M.T.; Malkmus, S.A.; Richter, P.J.; Osborn, K.G.; Xu, Q.; Veesart, S.L.; Steinauer, J.J.; Higgins, D.; et al. Intrathecal substance p-saporin in the dog: Distribution, safety, and spinal neurokinin-1 receptor ablation. Anesthesiology 2013, 119, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Mantyh, P.W.; Rogers, S.D.; Honore, P.; Allen, B.J.; Ghilardi, J.R.; Li, J.; Daughters, R.S.; Lappi, D.A.; Wiley, R.G.; Simone, D.A. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance p receptor. Science 1997, 278, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Koehrn, F.J.; Sorkin, L.S. Carrageenan induced phosphorylation of akt is dependent on neurokinin-1 expressing neurons in the superficial dorsal horn. Mol. Pain 2012, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Anderson, E.M.; Bokrand-Donatelli, Y.; Neubert, J.K.; Caudle, R.M. Anti-nociceptive effect of a conjugate of substance p and light chain of botulinum neurotoxin type A. Pain 2013, 154, 2547–2553. [Google Scholar] [CrossRef] [PubMed]
- Keith, F.; John, C. Targeted secretion inhibitors-innovative protein therapeutics. Toxins 2010, 2, 2795–2815. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.G. TRPV1 in the central nervous system: Synaptic plasticity, function, and pharmacological implications. Prog. Drug Res. 2014, 68, 77–104. [Google Scholar] [PubMed]
- Duggan, M.J.; Quinn, C.P.; Chaddock, J.A.; Purkiss, J.R.; Alexander, F.C.; Doward, S.; Fooks, S.J.; Friis, L.M.; Hall, Y.H.; Kirby, E.R.; et al. Inhibition of release of neurotransmitters from rat dorsal root ganglia by a novel conjugate of a clostridium botulinum toxin a endopeptidase fragment and erythrina cristagalli lectin. J. Biol. Chem. 2002, 277, 34846–34852. [Google Scholar] [CrossRef] [PubMed]
- Apland, J.P.; Adler, M.; Oyler, G.A. Inhibition of neurotransmitter release by peptides that mimic the n-terminal domain of SNAP-25. J. Protein Chem. 2003, 22, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Park, T.Y.; Shin, M.J.; Park, S.D.; Lee, S.K. Alleviation of abnormal synaptic neurotransmitter release by cell-permeable form of the truncated SNAP-25 upon transcutaneous delivery. Neurosci. Lett. 2013, 543, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.S. The occurrence of bacillus botulinus in nature. J. Bacteriol. 1919, 4, 541–553. [Google Scholar] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellett, S.; Yaksh, T.L.; Ramachandran, R. Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing. Toxins 2015, 7, 4519-4563. https://doi.org/10.3390/toxins7114519
Pellett S, Yaksh TL, Ramachandran R. Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing. Toxins. 2015; 7(11):4519-4563. https://doi.org/10.3390/toxins7114519
Chicago/Turabian StylePellett, Sabine, Tony L. Yaksh, and Roshni Ramachandran. 2015. "Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing" Toxins 7, no. 11: 4519-4563. https://doi.org/10.3390/toxins7114519
APA StylePellett, S., Yaksh, T. L., & Ramachandran, R. (2015). Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing. Toxins, 7(11), 4519-4563. https://doi.org/10.3390/toxins7114519