Extracellular Hb Enhances Cardiac Toxicity in Endotoxemic Guinea Pigs: Protective Role of Haptoglobin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
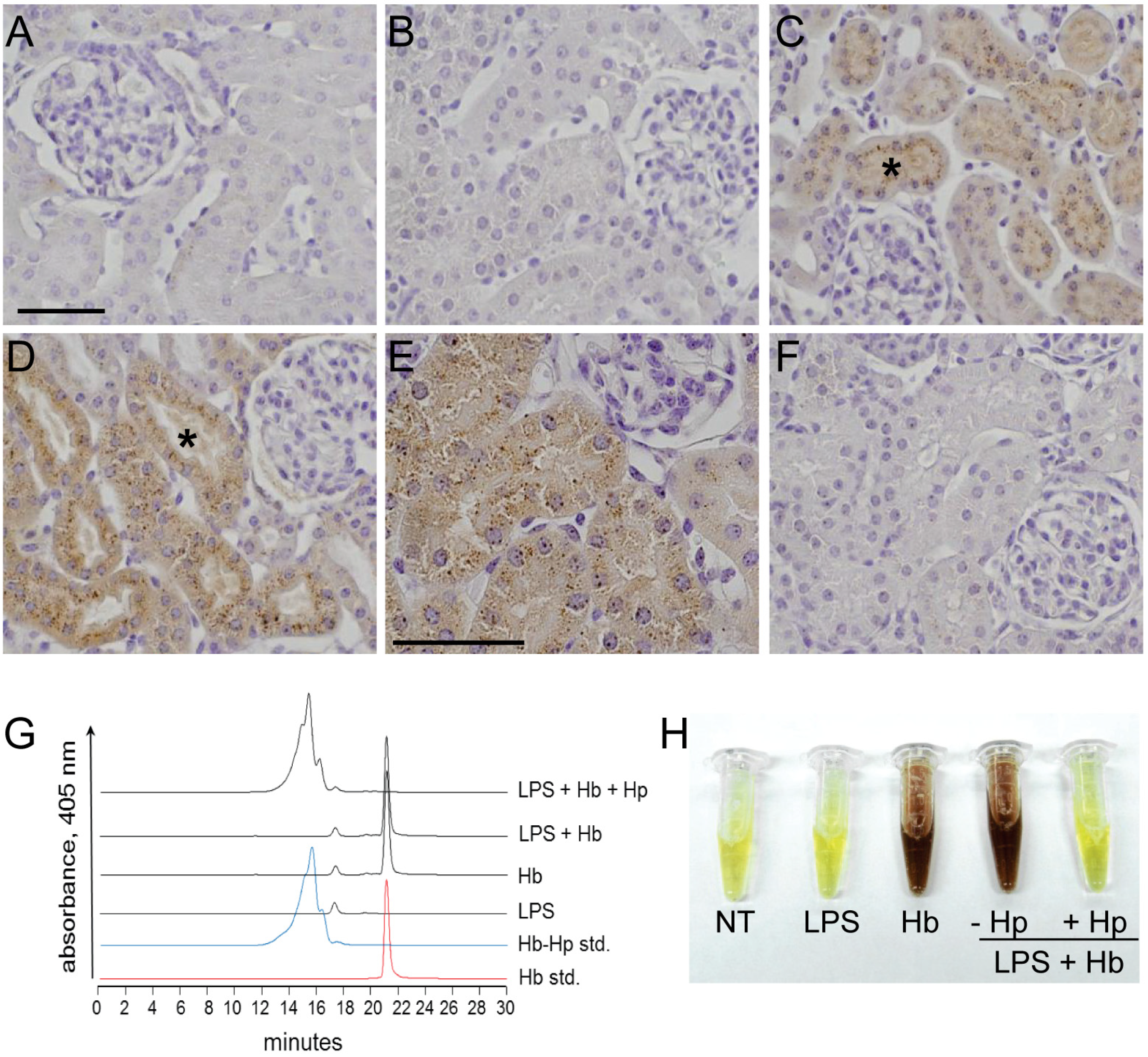
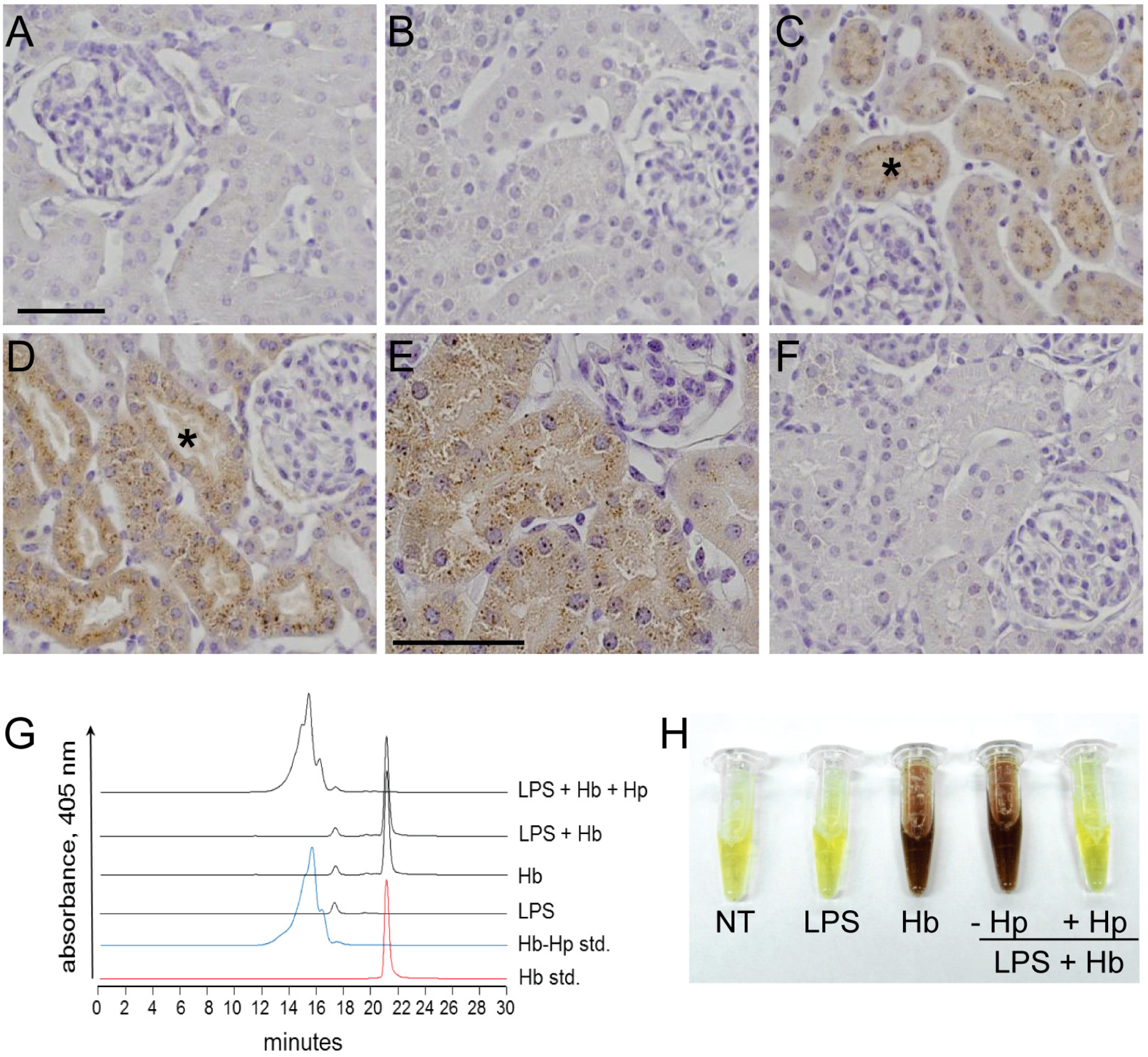
2.1. Sequestration of Hb by Hp Prevents Renal Filtration
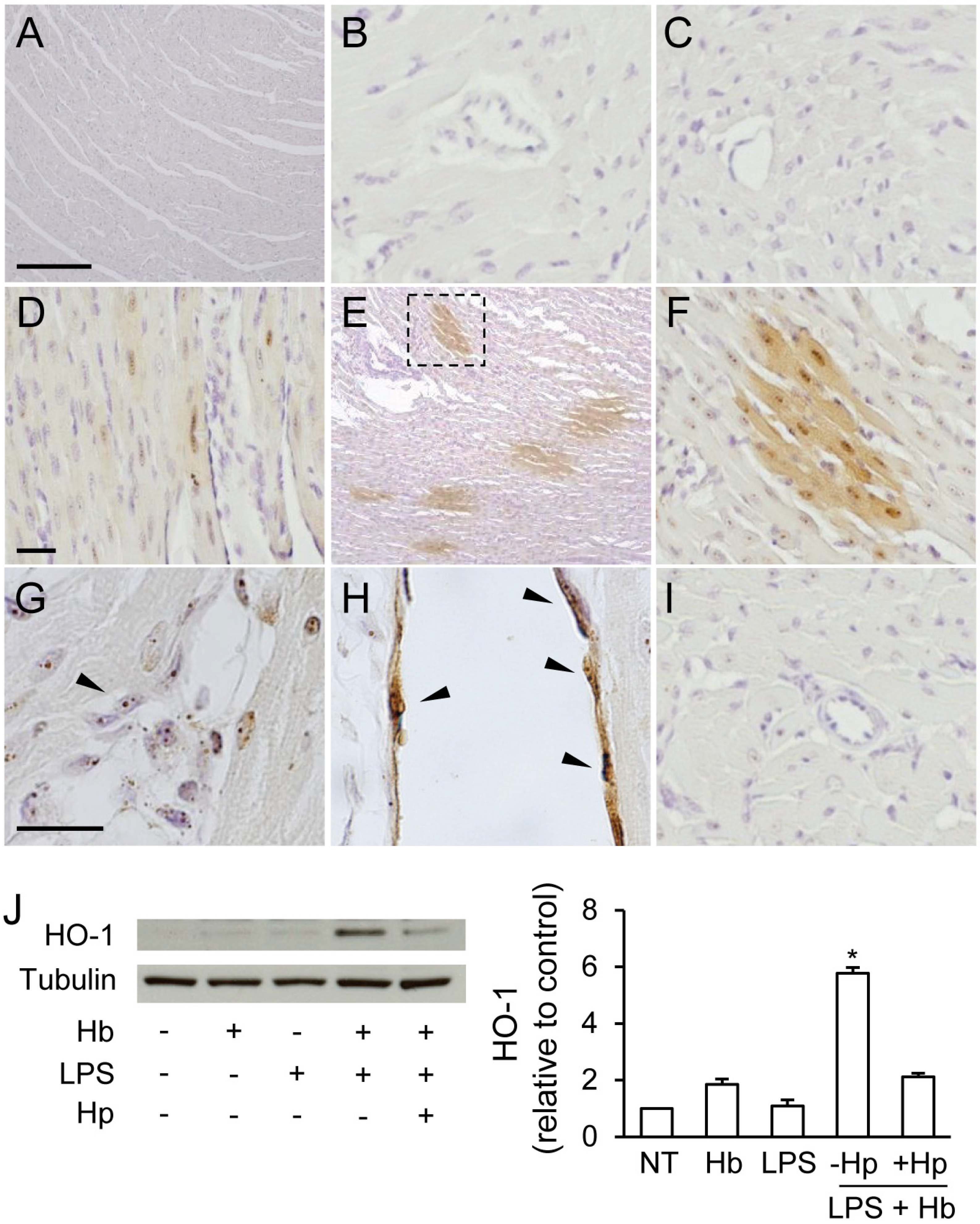
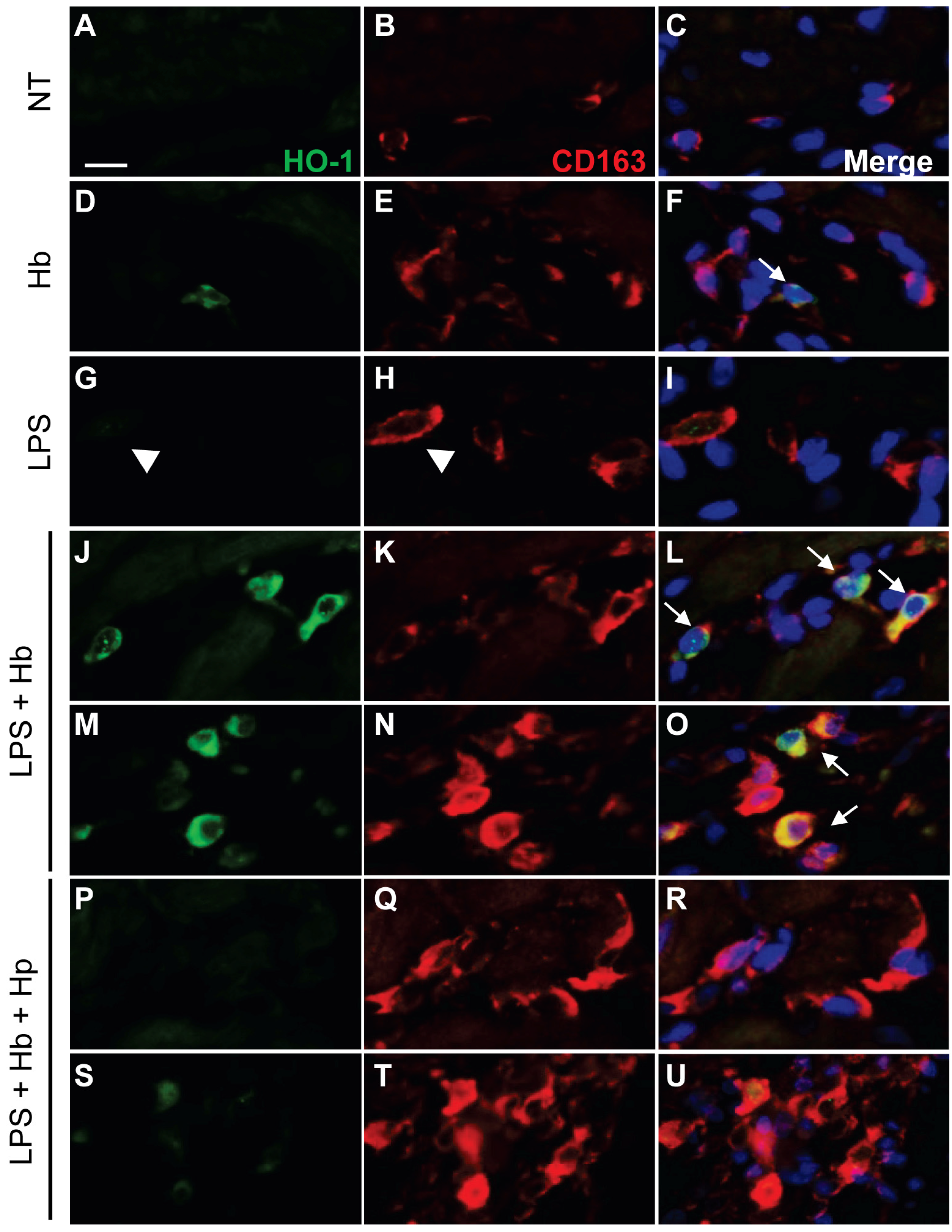
2.2. Hp Prevents Myocardial Iron Deposition and HO-1 Induction by LPS plus Hb
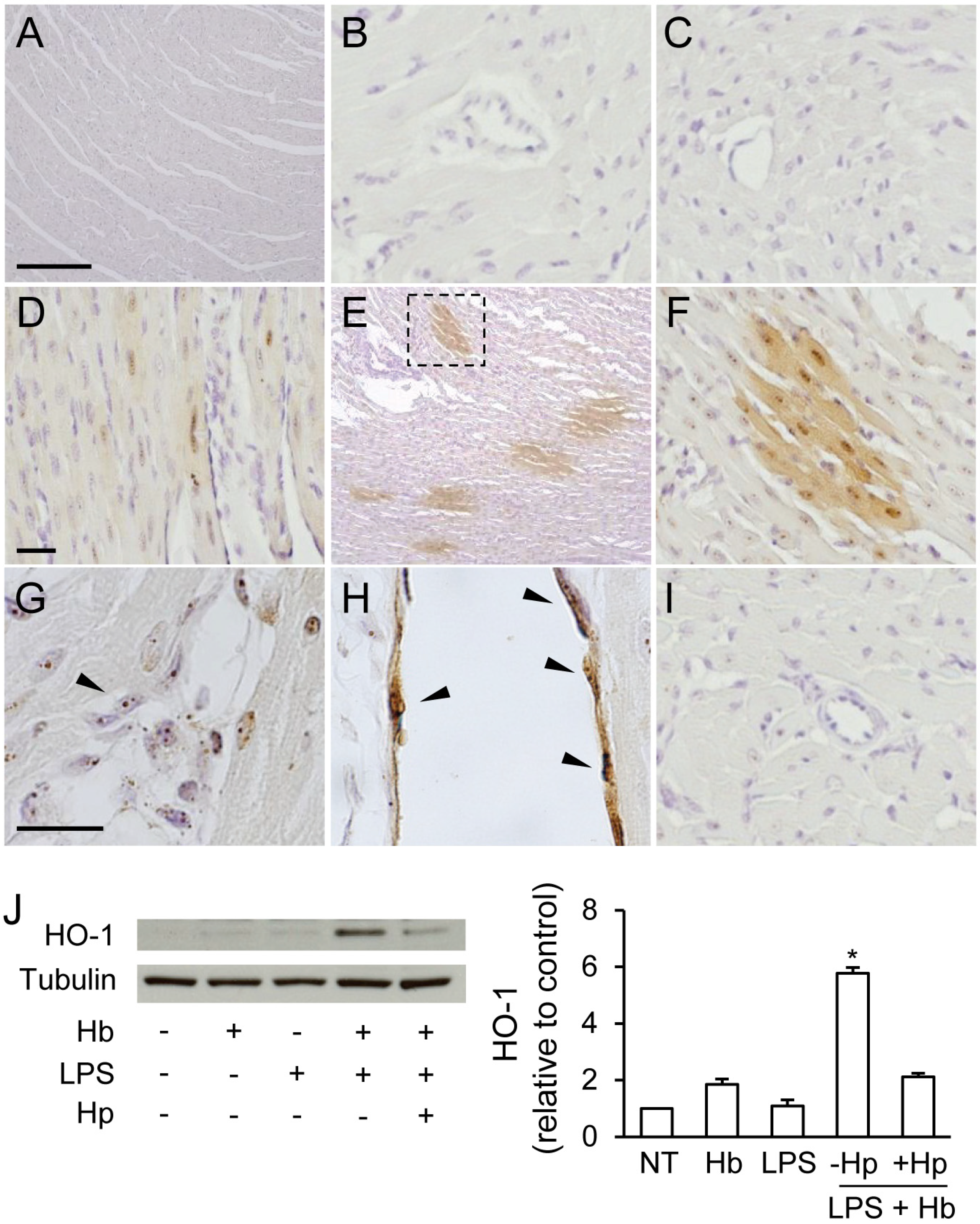
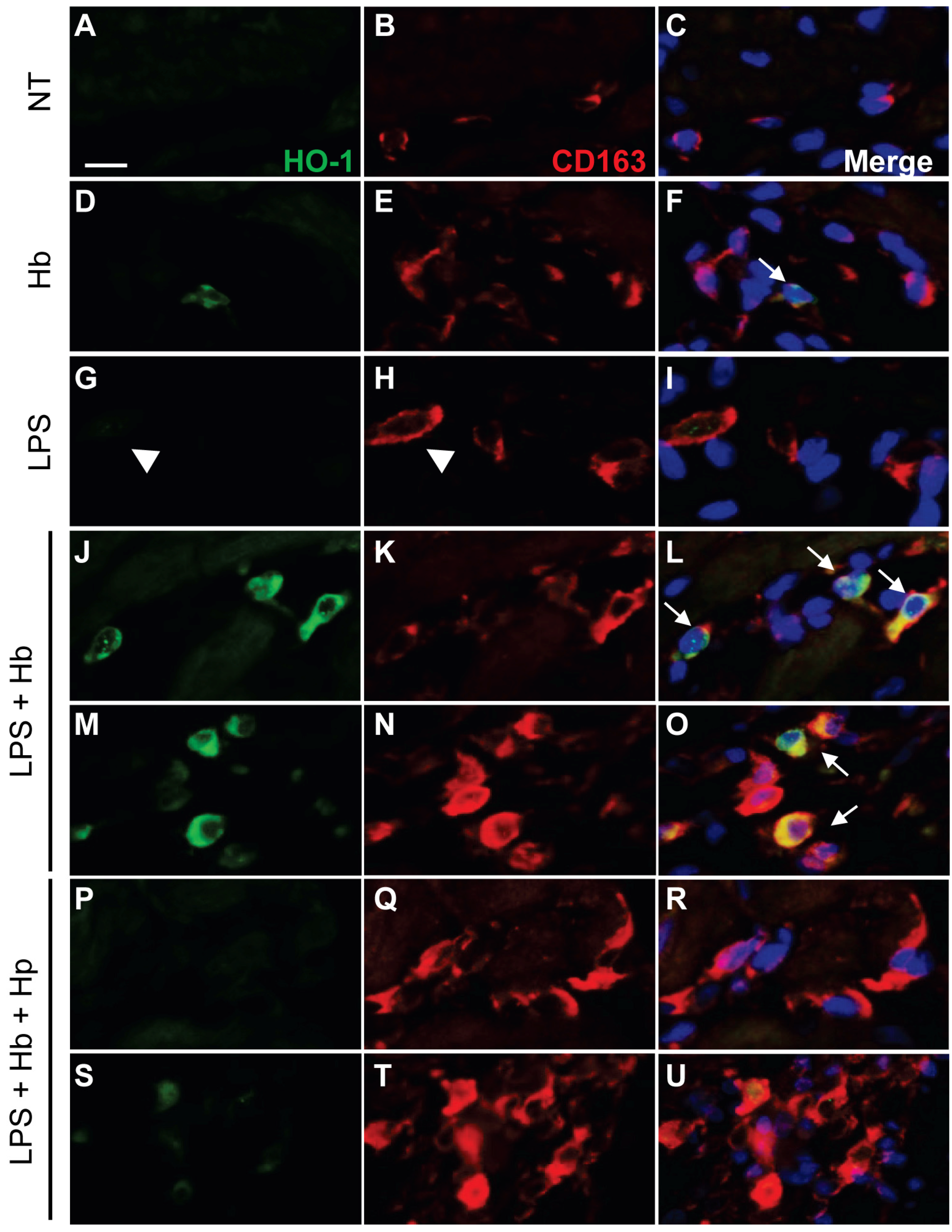
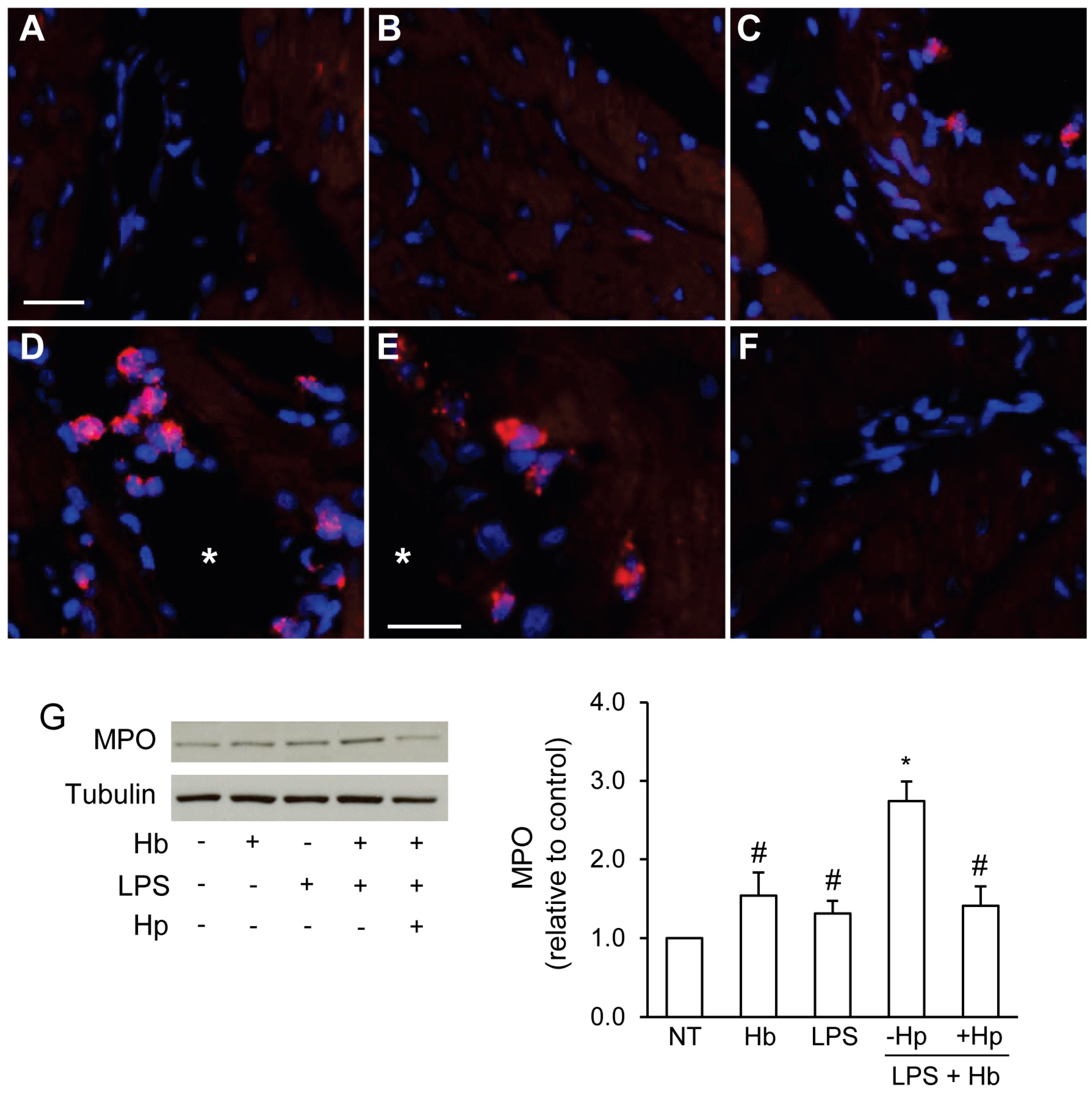
2.3. Hp Reduces Myocardial Myeloperoxidase Expression Induced by LPS plus Hb
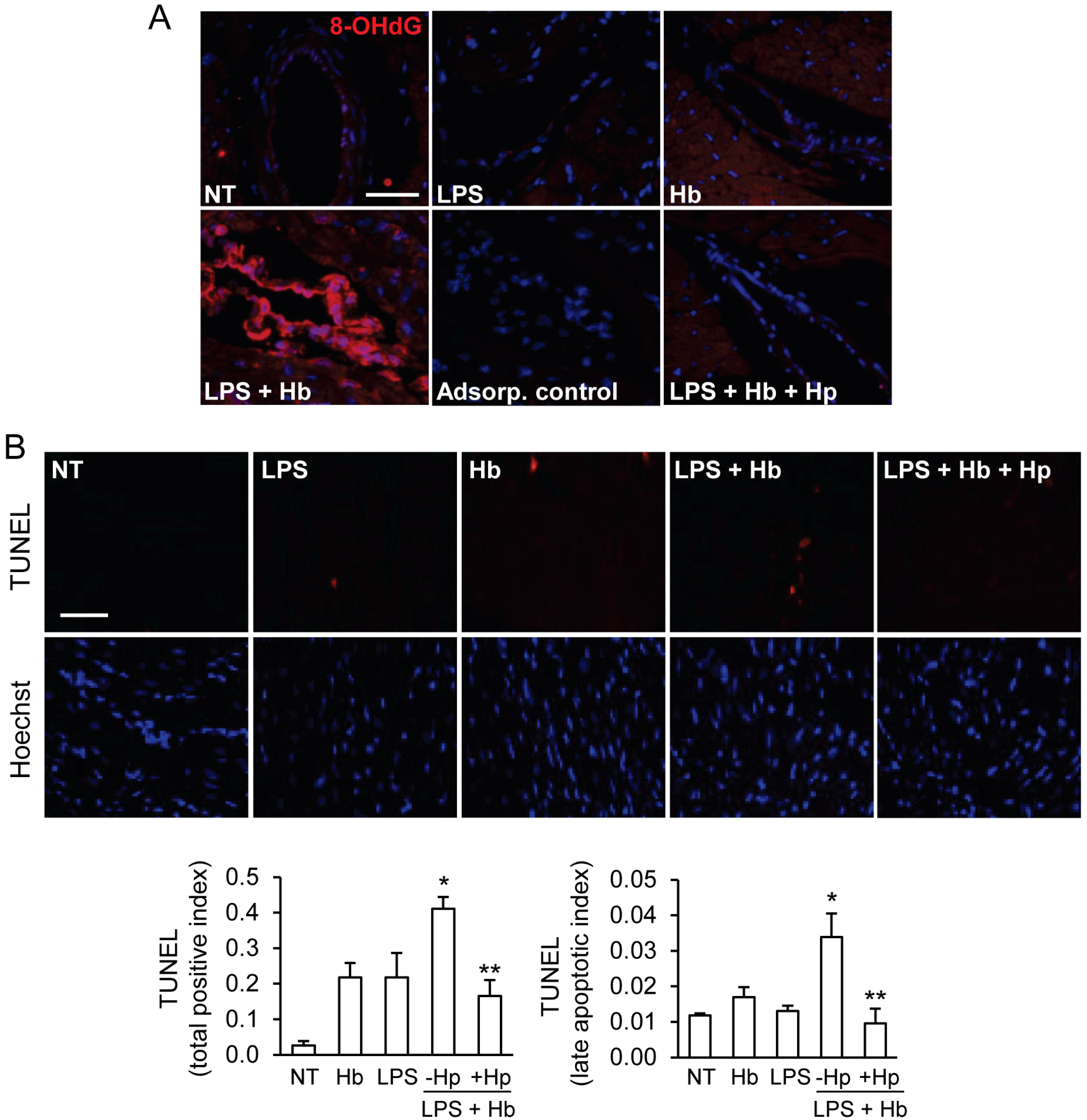
2.4. Hp Protects against Myocardial Oxidative DNA Damage and Apoptosis Induced by LPS plus Hb
3. Discussion
4. Experimental Section
4.1. Reagents and Antibodies
4.2. Animal Experimental Protocol
4.3. Plasma Hemoglobin Distribution
4.4. Non-heme Iron Immunohistochemistry
4.5. Preparation of Heart Tissue Lysates
4.6. Western Blot Analyses
4.7. Immunofluorescence
4.8. TUNEL Assay
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zanotti-Cavazzoni, S.L.; Hollenberg, S.M. Cardiac dysfunction in severe sepsis and septic shock. Curr. Opin. Crit Care 2009, 15, 392–397. [Google Scholar] [CrossRef]
- Flynn, A.; Chokkalingam Mani, B.; Mather, P.J. Sepsis-induced cardiomyopathy: A review of pathophysiologic mechanisms. Heart Fail Rev. 2010, 15, 605–611. [Google Scholar] [CrossRef]
- Balija, T.M.; Lowry, S.F. Lipopolysaccharide and sepsis-associated myocardial dysfunction. Curr. Opin. Infect. Dis. 2011, 24, 248–253. [Google Scholar] [CrossRef]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288. [Google Scholar] [CrossRef]
- Su, D.; Roth, R.I.; Yoshida, M.; Levin, J. Hemoglobin increases mortality from bacterial endotoxin. Infect. Immun. 1997, 65, 1258–1266. [Google Scholar]
- Krishnamurti, C.; Carter, A.J.; Maglasang, P.; Hess, J.R.; Cutting, M.A.; Alving, B.M. Cardiovascular toxicity of human cross-linked hemoglobin in a rabbit endotoxemia model. Crit. Care Med. 1997, 25, 1874–1880. [Google Scholar] [CrossRef]
- McGahan, M.C.; Grimes, A.M.; Fleisher, L.N. Hemoglobin exacerbates the ocular inflammatory response to endotoxin. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 643–647. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassu, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Adamzik, M.; Hamburger, T.; Petrat, F.; Peters, J.; de Groot, H.; Hartmann, M. Free hemoglobin concentration in severe sepsis: Methods of measurement and prediction of outcome. Crit. Care 2012, 16, R125. [Google Scholar] [CrossRef]
- Janz, D.R.; Bastarache, J.A.; Peterson, J.F.; Sills, G.; Wickersham, N.; May, A.K.; Roberts, L.J., 2nd; Ware, L.B. Association between cell-free hemoglobin, acetaminophen, and mortality in patients with sepsis: An observational study. Crit. Care Med. 2013, 41, 784–790. [Google Scholar] [CrossRef]
- Janz, D.R.; Bastarache, J.A.; Sills, G.; Wickersham, N.; May, A.K.; Bernard, G.R.; Ware, L.B. Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit. Care 2013, 17, R272. [Google Scholar] [CrossRef]
- Smithies, O.; Connell, G.E.; Dixon, G.H. Inheritance of haptoglobin subtypes. Am. J. Hum. Genet. 1962, 14, 14–21. [Google Scholar]
- Nosslin, B.F.; Nyman, M. Haptoglobin determination in diagnosis of haemolytic diseases. Lancet 1958, 1, 1000–1001. [Google Scholar] [CrossRef]
- Andersen, C.B.; Torvund-Jensen, M.; Nielsen, M.J.; de Oliveira, C.L.; Hersleth, H.P.; Andersen, N.H.; Pedersen, J.S.; Andersen, G.R.; Moestrup, S.K. Structure of the haptoglobin-haemoglobin complex. Nature 2012, 489, 456–459. [Google Scholar] [CrossRef]
- Connell, G.E.; Dixon, G.H.; Smithies, O. Subdivision of the three common haptoglobin types based on “hidden” differences. Nature 1962, 193, 505–506. [Google Scholar] [CrossRef]
- Boretti, F.S.; Buehler, P.W.; D’Agnillo, F.; Kluge, K.; Glaus, T.; Butt, O.I.; Jia, Y.; Goede, J.; Pereira, C.P.; Maggiorini, M.; et al. Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J. Clin. Invest 2009, 119, 2271–2280. [Google Scholar]
- Lim, S.K.; Kim, H.; Lim, S.K.; bin Ali, A.; Lim, Y.K.; Wang, Y.; Chong, S.M.; Costantini, F.; Baumman, H. Increased susceptibility in Hp knockout mice during acute hemolysis. Blood 1998, 92, 1870–1877. [Google Scholar]
- Hashimoto, K.; Nomura, K.; Nakano, M.; Sasaki, T.; Kurosawa, H. Pharmacological intervention for renal protection during cardiopulmonary bypass. Heart Vessel. 1993, 8, 203–210. [Google Scholar] [CrossRef]
- Bunn, H.F.; Jandl, J.H. The renal handling of hemoglobin. II. Catabolism. J. Exp. Med. 1969, 129, 925–934. [Google Scholar] [CrossRef]
- Schindhelm, R.K.; van der Zwan, L.P.; Teerlink, T.; Scheffer, P.G. Myeloperoxidase: A useful biomarker for cardiovascular disease risk stratification? Clin. Chem. 2009, 55, 1462–1470. [Google Scholar]
- Kaca, W.; Roth, R.I.; Levin, J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J. Biol. Chem. 1994, 269, 25078–25084. [Google Scholar]
- Bahl, N.; Du, R.; Winarsih, I.; Ho, B.; Tucker-Kellogg, L.; Tidor, B.; Ding, D.L. Delineation of lipopolysaccharide (LPS)-binding sites on hemoglobin: From in silico predictions to biophysical characterization. J. Biol. Chem. 2011, 286, 37793–37803. [Google Scholar]
- Bodet, C.; Chandad, F.; Grenier, D. Hemoglobin and LPS act in synergy to amplify the inflammatory response. J. Dent. Res. 2007, 86, 878–882. [Google Scholar] [CrossRef]
- Gorczynski, R.M.; Alexander, C.; Bessler, W.; Fournier, K.; Hoffmann, P.; Mach, J.P.; Manuel, J.; Ramakrishna, V.; Rietschel, E.T.; Song, L.; et al. Characterization of an interaction between fetal hemoglobin and lipid A of LPS resulting in augmented induction of cytokine production in vivo and in vitro. Int. Immunopharmacol. 2004, 4, 1859–1872. [Google Scholar] [CrossRef]
- Luchtemberg, M.N.; Petronilho, F.; Constantino, L.; Gelain, D.P.; Andrades, M.; Ritter, C.; Moreira, J.C.; Streck, E.L.; Dal-Pizzol, F. Xanthine oxidase activity in patients with sepsis. Clin. Biochem. 2008, 41, 1186–1190. [Google Scholar] [CrossRef]
- Wu, J.; Xu, H.; Yang, M.; Martin, C.M.; Kvietys, P.R.; Rui, T. NADPH oxidase contributes to conversion of cardiac myocytes to a proinflammatory phenotype in sepsis. Free Radic. Biol. Med. 2009, 46, 1338–1345. [Google Scholar] [CrossRef]
- Kothari, N.; Keshari, R.S.; Bogra, J.; Kohli, M.; Abbas, H.; Malik, A.; Dikshit, M.; Barthwal, M.K. Increased myeloperoxidase enzyme activity in plasma is an indicator of inflammation and onset of sepsis. J. Crit. Care 2011, 26, 435.e1–435.e7. [Google Scholar]
- Gao, M.; Ha, T.; Zhang, X.; Liu, L.; Wang, X.; Kelley, J.; Singh, K.; Kao, R.; Gao, X.; Williams, D.; et al. Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit. Care Med. 2012, 40, 2390–2399. [Google Scholar] [CrossRef]
- Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative stress and endothelial dysfunction during sepsis. Front. Biosci. 2011, 16, 1986–1995. [Google Scholar] [CrossRef]
- Cheng, W.E.; Shih, C.M.; Hang, L.W.; Wu, K.Y.; Yang, H.L.; Hsu, W.H.; Hsia, T.C. Urinary biomarker of oxidative stress correlating with outcome in critically septic patients. Intensive Care Med. 2007, 33, 1187–1190. [Google Scholar] [CrossRef]
- Buehler, P.W.; D’Agnillo, F. Toxicological consequences of extracellular hemoglobin: biochemical and physiological perspectives. Antioxid. Redox. Signal. 2010, 12, 275–291. [Google Scholar] [CrossRef]
- D’Agnillo, F. Redox active hemoglobin enhances lipopolysaccharide-induced injury to cultured bovine endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1875–H1882. [Google Scholar] [CrossRef]
- Langheinrich, A.C.; Ritman, E.L. Quantitative imaging of microvascular permeability in a rat model of lipopolysaccharide-induced sepsis: Evaluation using cryostatic micro-computed tomography. Invest Radiol. 2006, 41, 645–650. [Google Scholar] [CrossRef]
- Darwish, I.; Liles, W.C. Emerging therapeutic strategies to prevent infection-related microvascular endothelial activation and dysfunction. Virulence 2013, 4, 572–582. [Google Scholar] [CrossRef]
- Sankar, V.; Webster, N.R. Clinical application of sepsis biomarkers. J. Anesth. 2013, 27, 269–283. [Google Scholar] [CrossRef]
- Banerjee, S.; Jia, Y.; Siburt, C.J.; Abraham, B.; Wood, F.; Bonaventura, C.; Henkens, R.; Crumbliss, A.L.; Alayash, A.I. Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic. Biol. Med. 2013, 53, 1317–1326. [Google Scholar]
- Buehler, P.W.; Abraham, B.; Vallelian, F.; Linnemayr, C.; Pereira, C.P.; Cipollo, J.F.; Jia, Y.; Mikolajczyk, M.; Boretti, F.S.; Schoedon, G.; et al. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 2009, 113, 2578–2586. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baek, J.H.; Zhang, X.; Williams, M.C.; Schaer, D.J.; Buehler, P.W.; D'Agnillo, F. Extracellular Hb Enhances Cardiac Toxicity in Endotoxemic Guinea Pigs: Protective Role of Haptoglobin. Toxins 2014, 6, 1244-1259. https://doi.org/10.3390/toxins6041244
Baek JH, Zhang X, Williams MC, Schaer DJ, Buehler PW, D'Agnillo F. Extracellular Hb Enhances Cardiac Toxicity in Endotoxemic Guinea Pigs: Protective Role of Haptoglobin. Toxins. 2014; 6(4):1244-1259. https://doi.org/10.3390/toxins6041244
Chicago/Turabian StyleBaek, Jin Hyen, Xiaoyuan Zhang, Matthew C. Williams, Dominik J. Schaer, Paul W. Buehler, and Felice D'Agnillo. 2014. "Extracellular Hb Enhances Cardiac Toxicity in Endotoxemic Guinea Pigs: Protective Role of Haptoglobin" Toxins 6, no. 4: 1244-1259. https://doi.org/10.3390/toxins6041244
APA StyleBaek, J. H., Zhang, X., Williams, M. C., Schaer, D. J., Buehler, P. W., & D'Agnillo, F. (2014). Extracellular Hb Enhances Cardiac Toxicity in Endotoxemic Guinea Pigs: Protective Role of Haptoglobin. Toxins, 6(4), 1244-1259. https://doi.org/10.3390/toxins6041244