Breakdown of Phosphatidylserine Asymmetry Following Treatment of Erythrocytes with Lumefantrine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
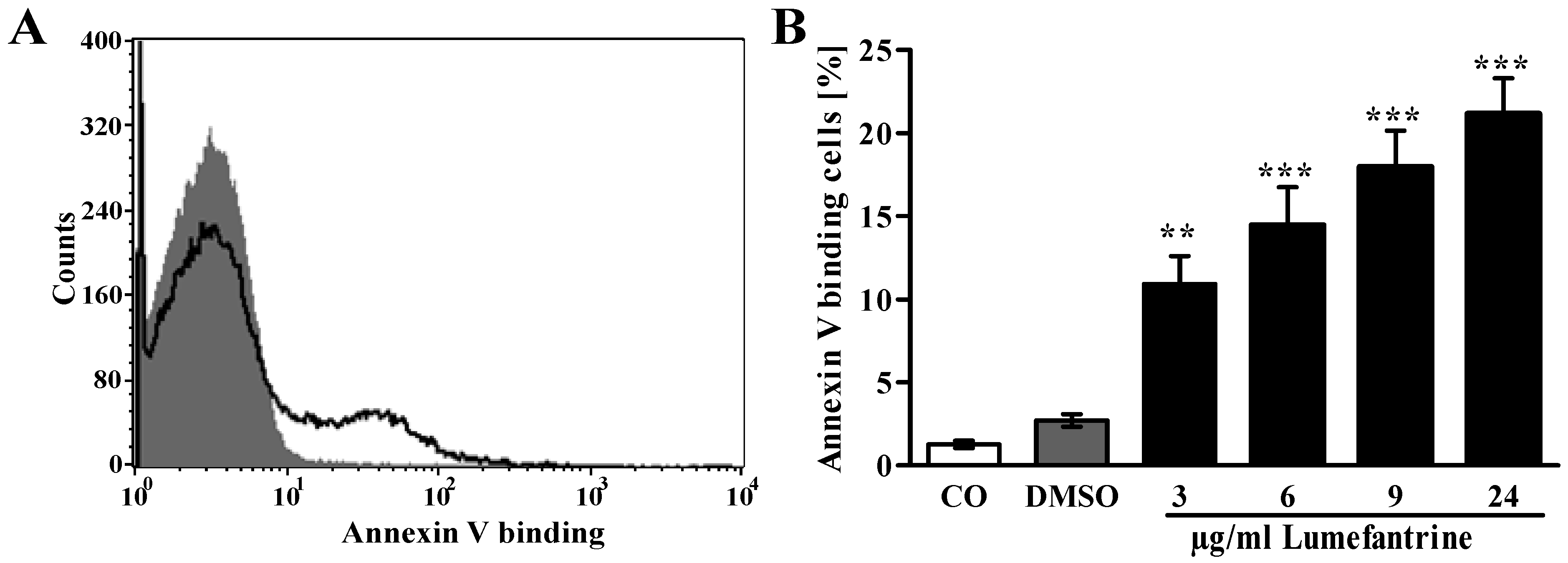
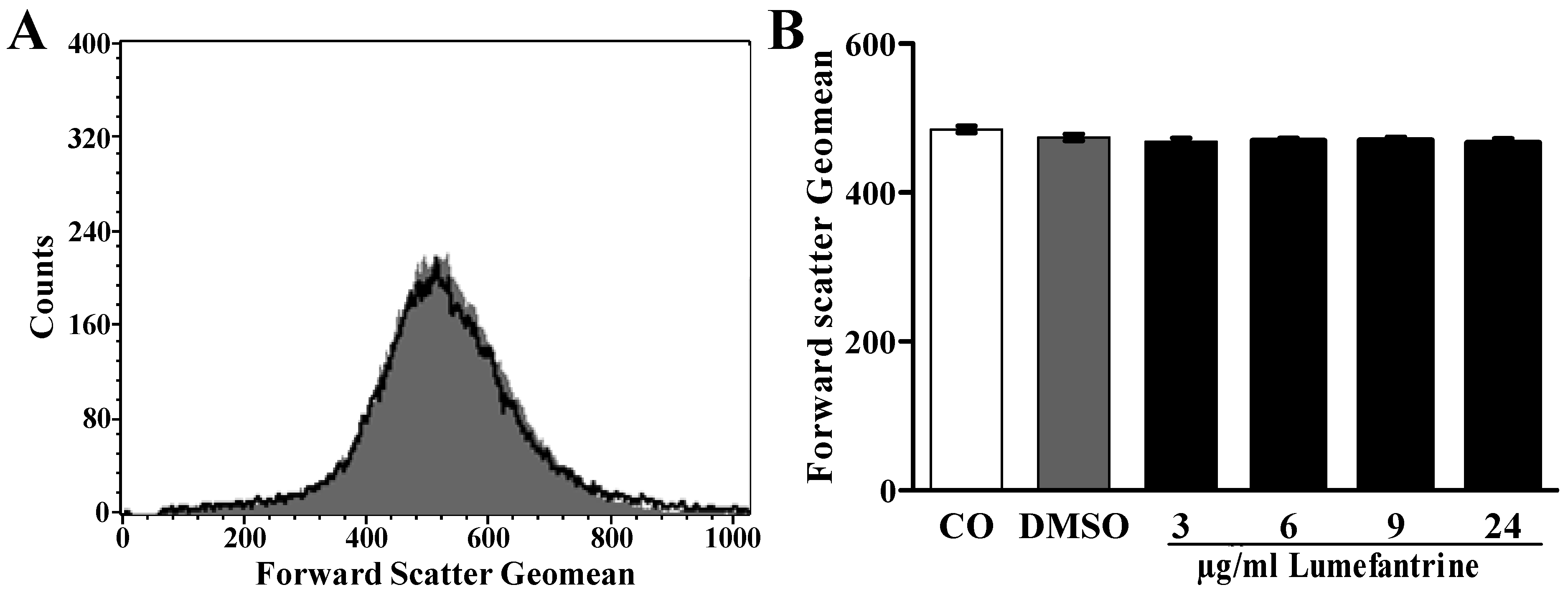
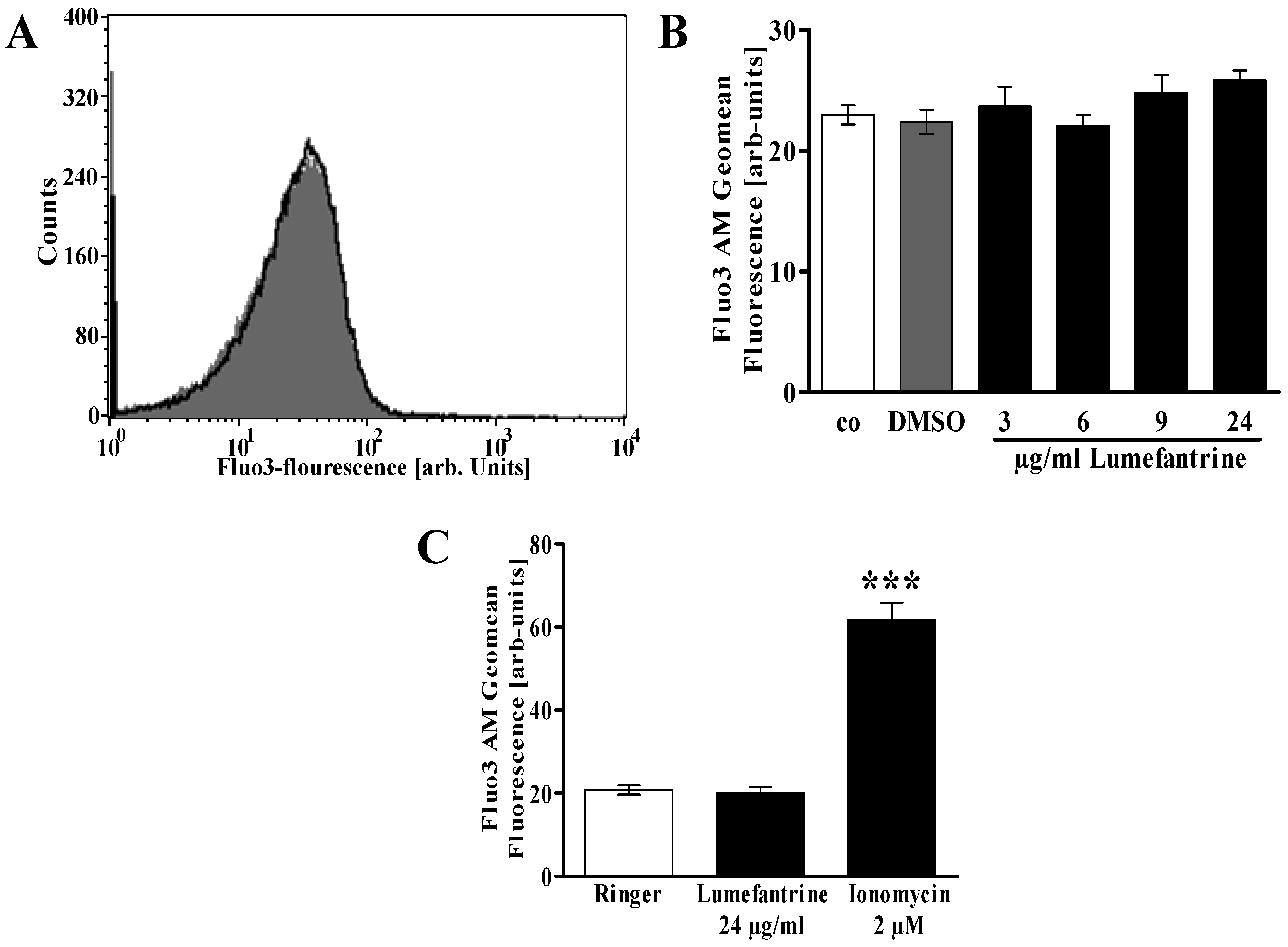
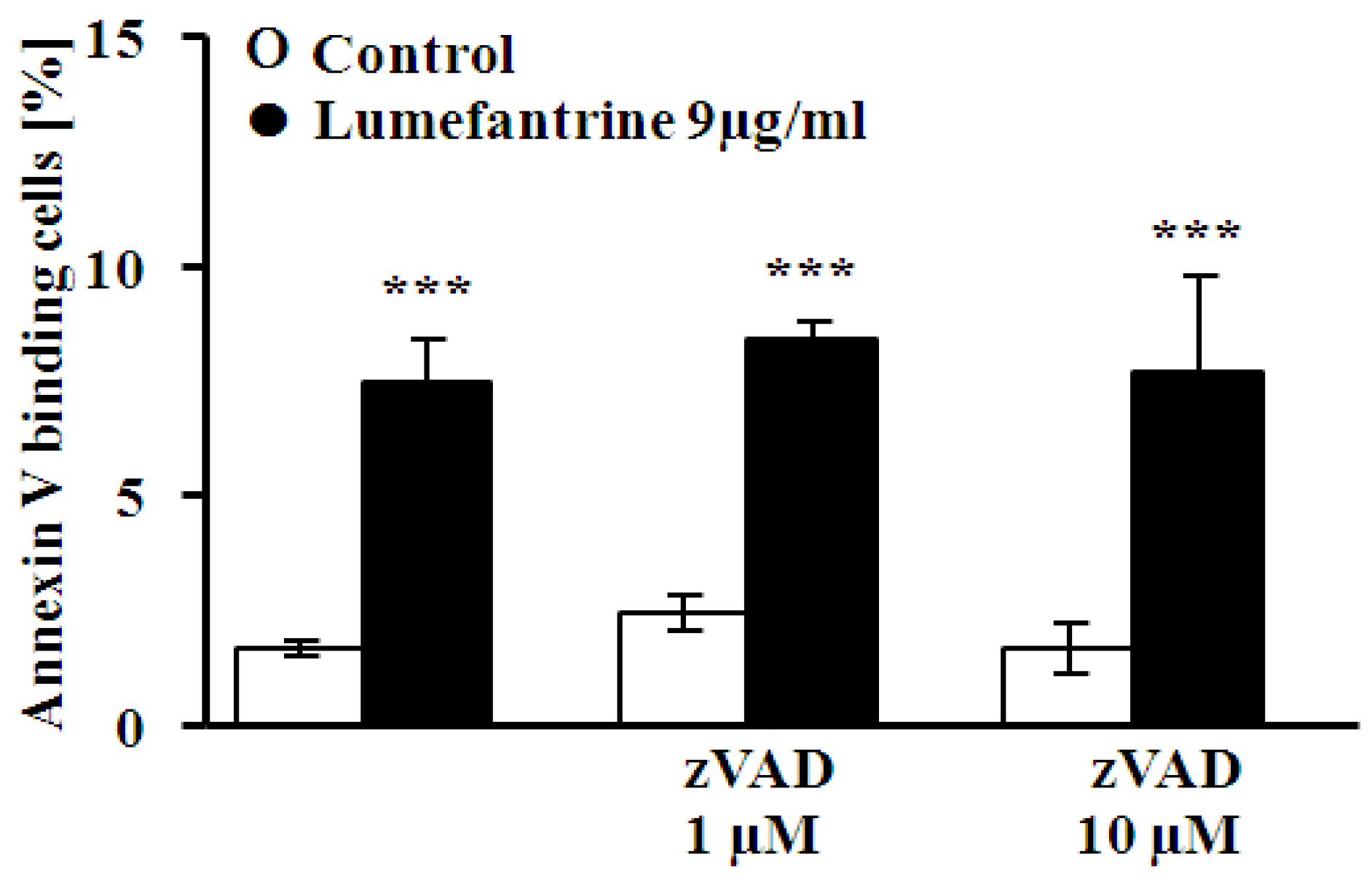
2. Results and Discussion
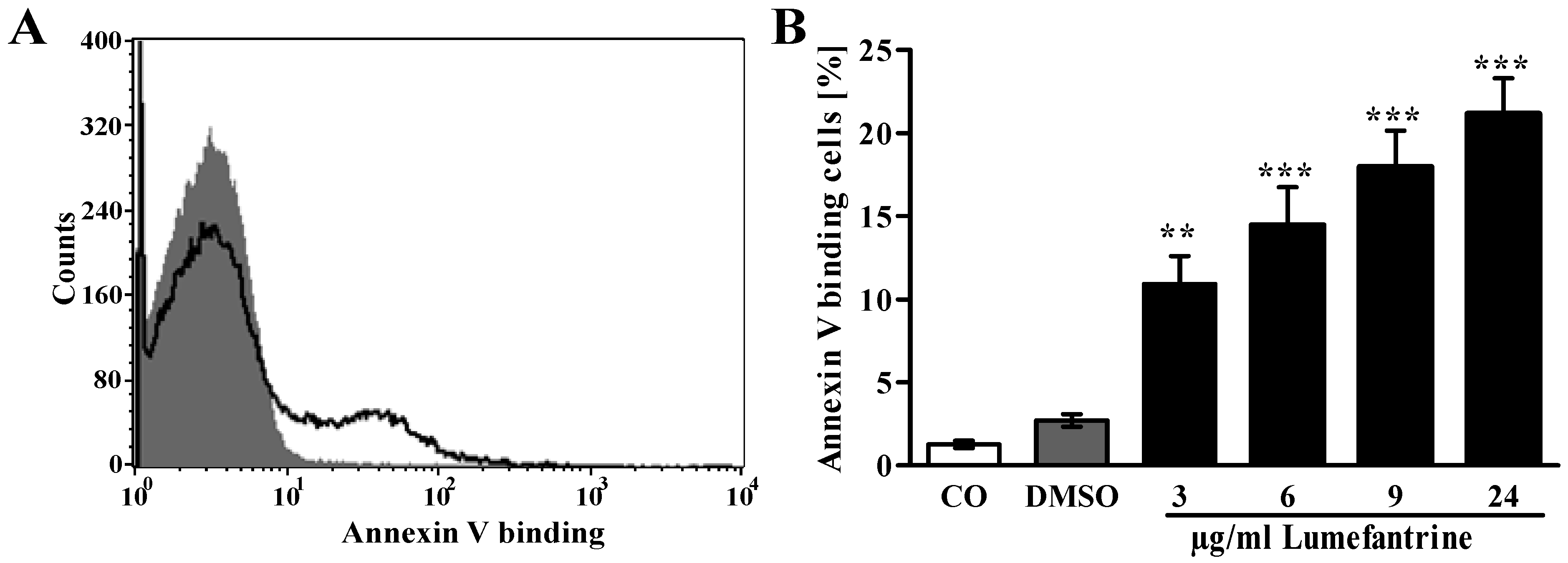
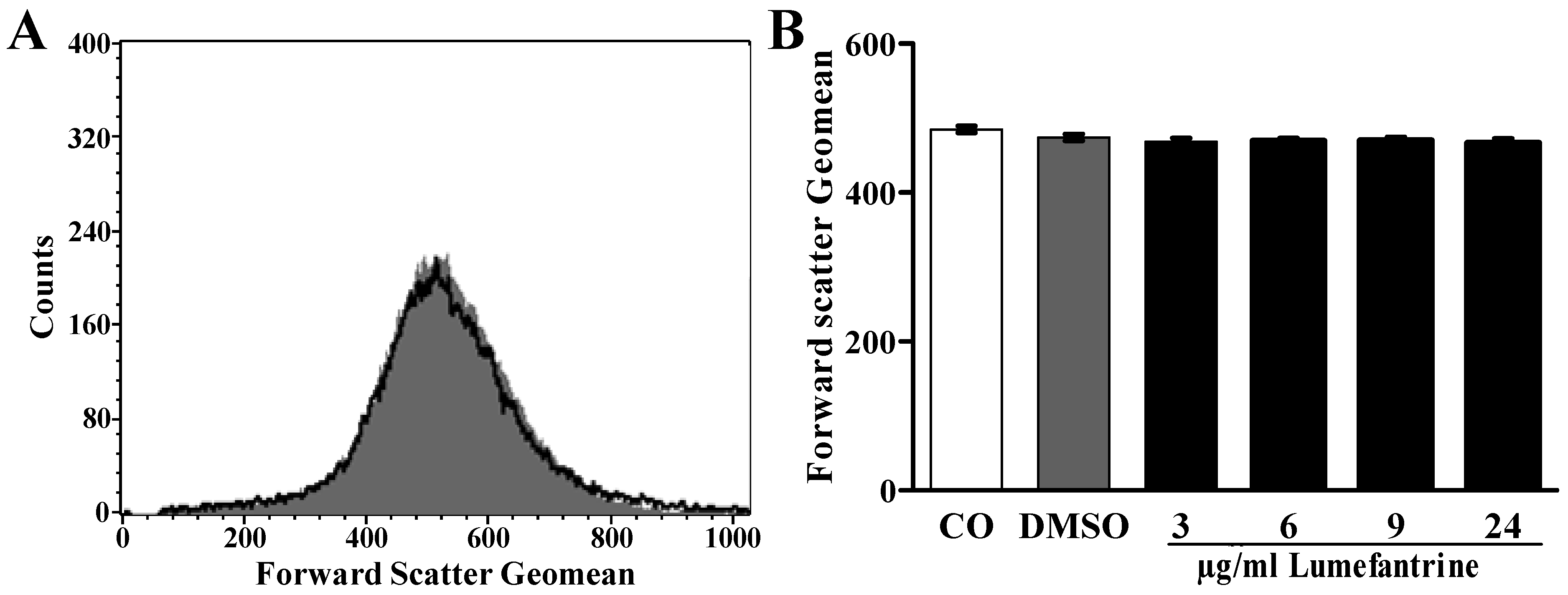
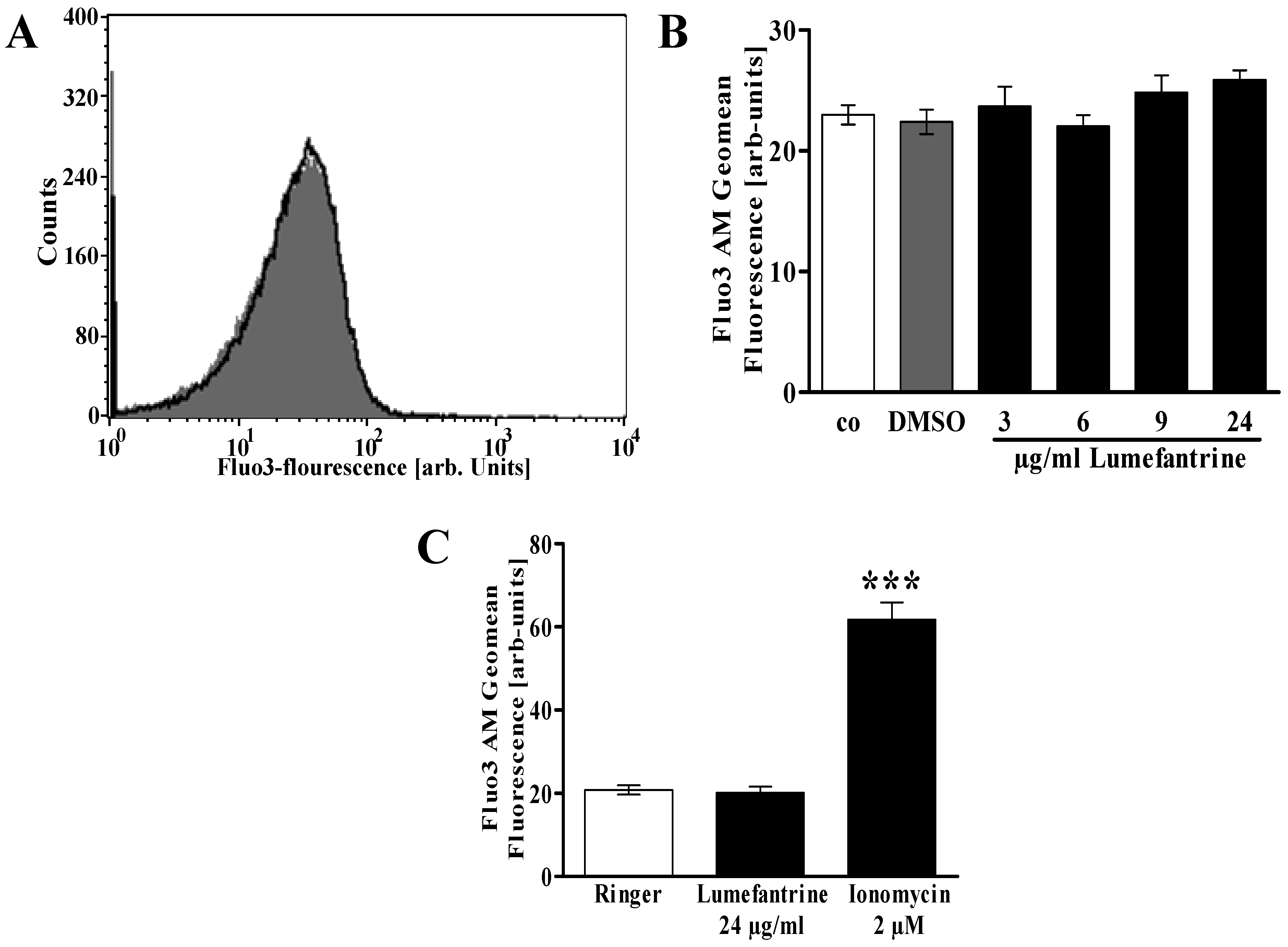
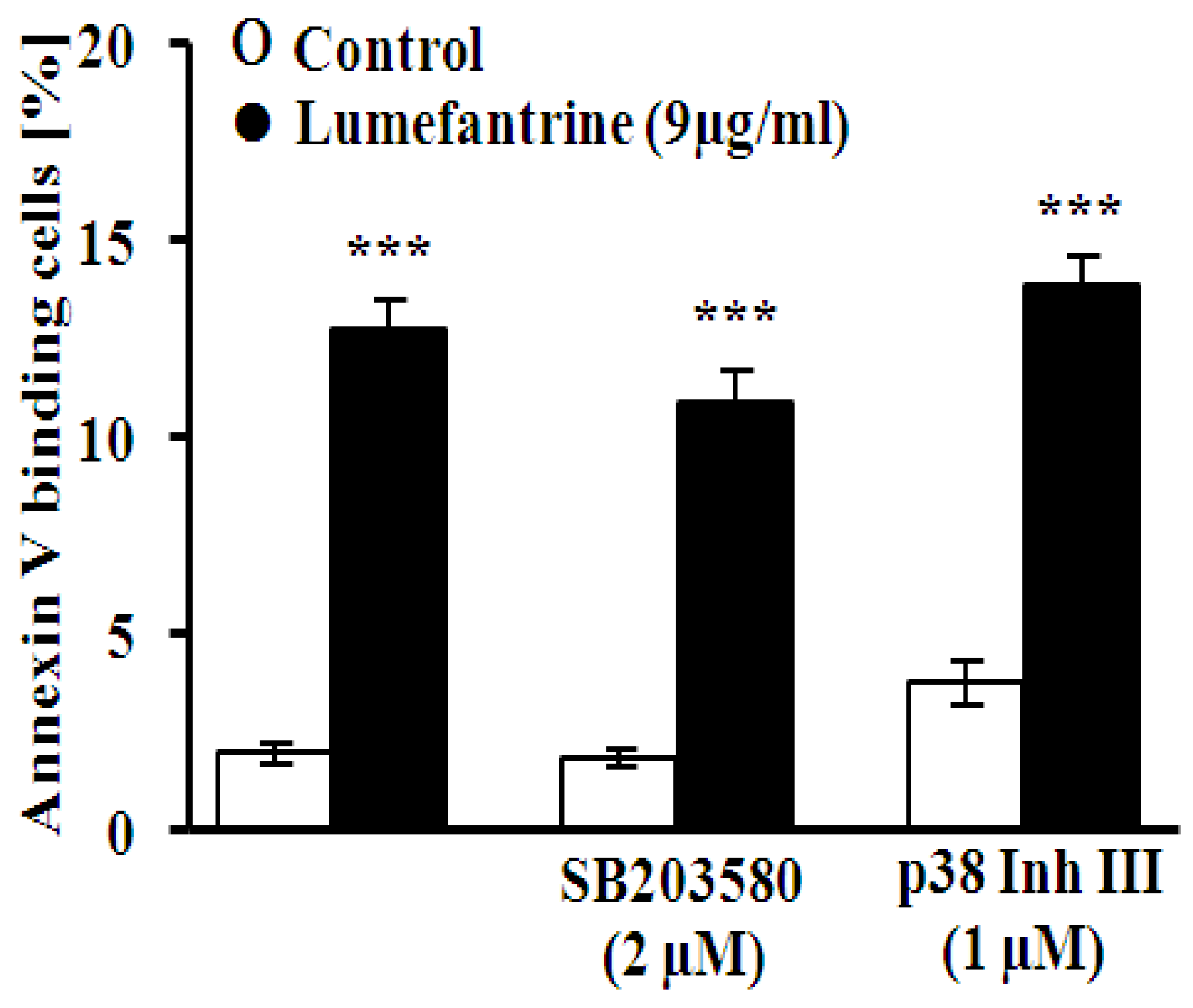
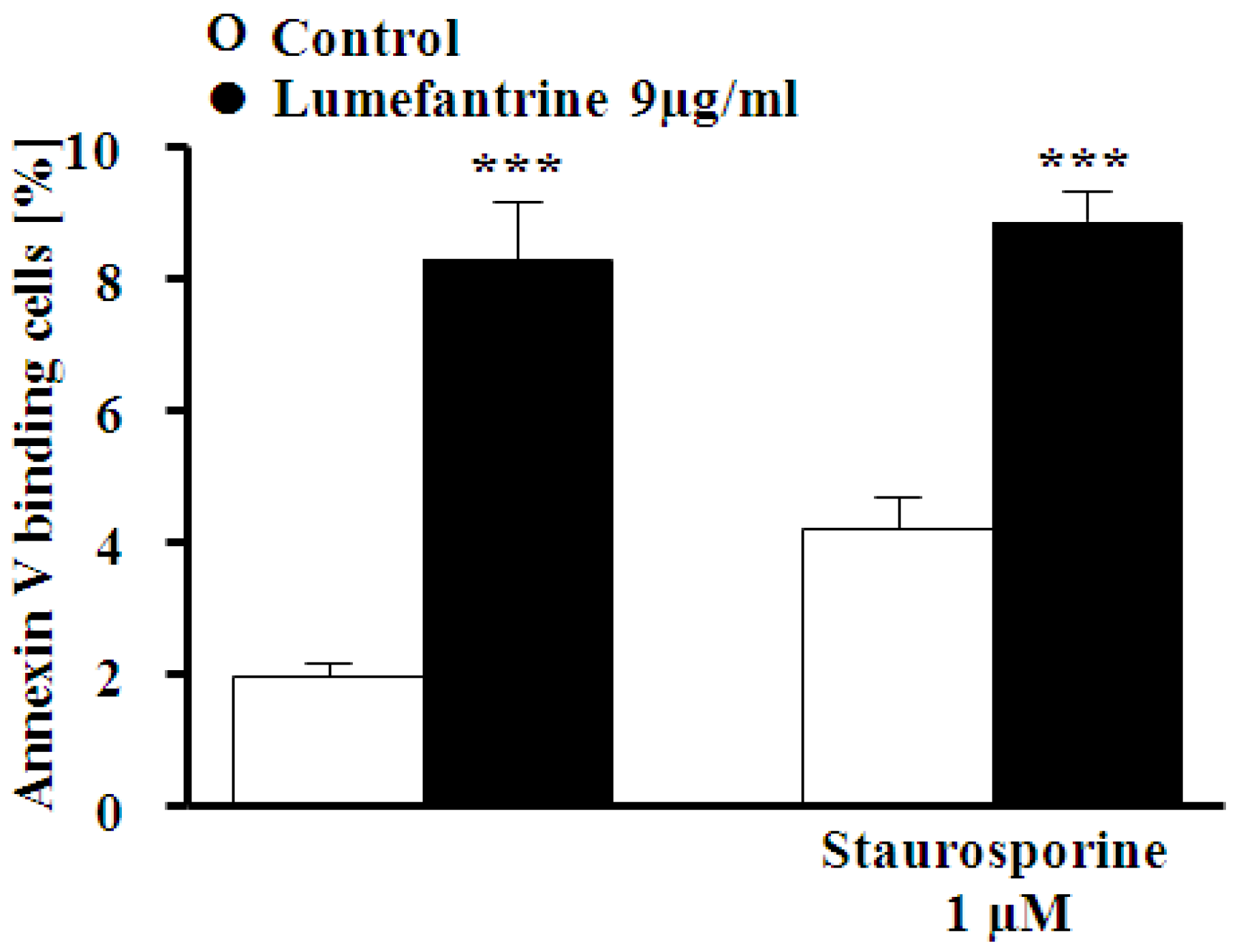
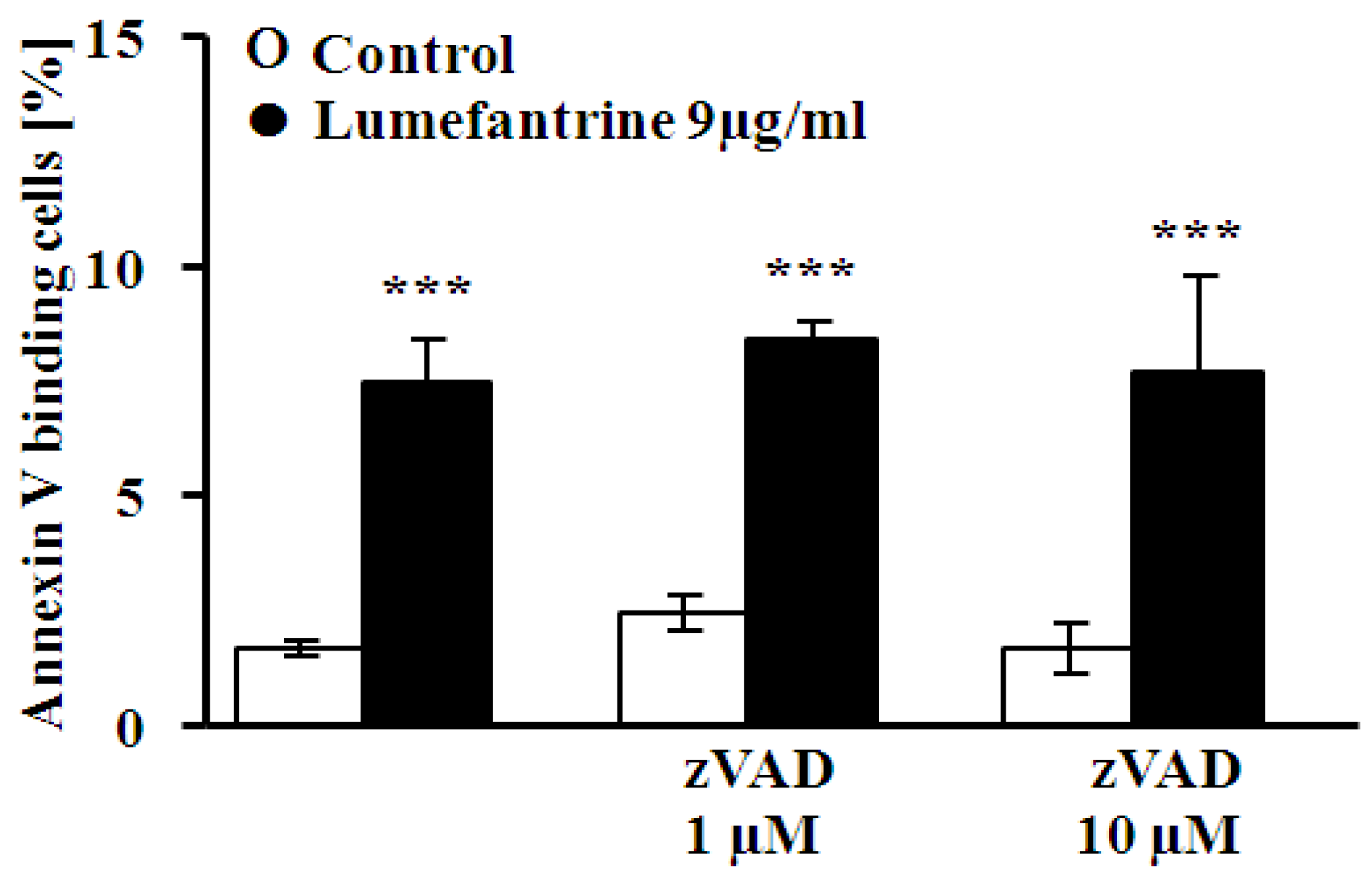
3. Experimental Section
3.1. Erythrocytes, Solutions, and Chemicals
3.2. FACS Analysis of Annexin-V-Binding and Forward Scatter
3.3. Measurement of Intracellular Ca2+
3.4. Determination of Oxidative Status
3.5. Determination of Ceramide Formation
3.6. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Egunsola, O.; Oshikoya, K.A. Comparative safety of artemether-lumefantrine and other artemisinin-based combinations in children: A systematic review. Malar. J. 2013, 12, 385. [Google Scholar] [CrossRef]
- Ogutu, B. Artemether and lumefantrine for the treatment of uncomplicated plasmodium falciparum malaria in sub-saharan africa. Expert Opin. Pharmacother. 2013, 14, 643–654. [Google Scholar] [CrossRef]
- Kredo, T.; Mauff, K.; van der Walt, J.S.; Wiesner, L.; Maartens, G.; Cohen, K.; Smith, P.; Barnes, K.I. Interaction between artemether-lumefantrine and nevirapine-based antiretroviral therapy in hiv-1-infected patients. Antimicrob Agents Chemother. 2011, 55, 5616–5623. [Google Scholar] [CrossRef]
- Combrinck, J.M.; Mabotha, T.E.; Ncokazi, K.K.; Ambele, M.A.; Taylor, D.; Smith, P.J.; Hoppe, H.C.; Egan, T.J. Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol. 2013, 8, 133–137. [Google Scholar] [CrossRef]
- Foller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival—Death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef]
- Duranton, C.; Huber, S.; Tanneur, V.; Lang, K.; Brand, V.; Sandu, C.; Lang, F. Electrophysiological properties of the plasmodium falciparum-induced cation conductance of human erythrocytes. Cell Physiol. Biochem. 2003, 13, 189–198. [Google Scholar] [CrossRef]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar]
- Lang, F.; Lang, P.A.; Lang, K.S.; Brand, V.; Tanneur, V.; Duranton, C.; Wieder, T.; Huber, S.M. Channel-induced apoptosis of infected host cells-the case of malaria. Pflugers Arch. 2004, 448, 319–324. [Google Scholar] [CrossRef]
- Arruda, M.M.; Mecabo, G.; Rodrigues, C.A.; Matsuda, S.S.; Rabelo, I.B.; Figueiredo, M.S. Antioxidant vitamins C and E supplementation increases markers of haemolysis in sickle cell anaemia patients: A randomized, double-blind, placebo-controlled trial. Br. J. Haematol. 2013, 160, 688–700. [Google Scholar] [CrossRef]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar] [CrossRef]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (g6pd)-deficient erythrocytes parasitized by plasmodium falciparum may explain malaria protection in g6pd deficiency. Blood 1998, 92, 2527–2534. [Google Scholar]
- De Franceschi, L.; Turrini, F.; Honczarenko, M.; Ayi, K.; Rivera, A.; Fleming, M.D.; Law, T.; Mannu, F.; Kuypers, F.A.; Bast, A.; et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica 2004, 89, 1287–1298. [Google Scholar]
- De Jong, K.; Emerson, R.K.; Butler, J.; Bastacky, J.; Mohandas, N.; Kuypers, F.A. Short survival of phosphatidylserine-exposing red blood cells in murine sickle cell anemia. Blood 2001, 98, 1577–1584. [Google Scholar] [CrossRef]
- Kean, L.S.; Brown, L.E.; Nichols, J.W.; Mohandas, N.; Archer, D.R.; Hsu, L.L. Comparison of mechanisms of anemia in mice with sickle cell disease and beta-thalassemia: Peripheral destruction, ineffective erythropoiesis, and phospholipid scramblase-mediated phosphatidylserine exposure. Exp. Hematol. 2002, 30, 394–402. [Google Scholar] [CrossRef]
- Kuypers, F.A.; Yuan, J.; Lewis, R.A.; Snyder, L.M.; Kiefer, C.R.; Bunyaratvej, A.; Fucharoen, S.; Ma, L.; Styles, L.; de Jong, K.; et al. Membrane phospholipid asymmetry in human thalassemia. Blood 1998, 91, 3044–3051. [Google Scholar]
- Lang, K.S.; Roll, B.; Myssina, S.; Schittenhelm, M.; Scheel-Walter, H.G.; Kanz, L.; Fritz, J.; Lang, F.; Huber, S.M.; Wieder, T. Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell. Physiol. Biochem. 2002, 12, 365–372. [Google Scholar]
- Ayi, K.; Giribaldi, G.; Skorokhod, A.; Schwarzer, E.; Prendergast, P.T.; Arese, P. 16alpha-bromoepiandrosterone, an antimalarial analogue of the hormone dehydroepiandrosterone, enhances phagocytosis of ring stage parasitized erythrocytes: A novel mechanism for antimalarial activity. Antimicrob Agents Chemother. 2002, 46, 3180–3184. [Google Scholar] [CrossRef]
- Koka, S.; Foller, M.; Lamprecht, G.; Boini, K.M.; Lang, C.; Huber, S.M.; Lang, F. Iron deficiency influences the course of malaria in plasmodium berghei infected mice. Biochem. Biophys. Res. Commun. 2007, 357, 608–614. [Google Scholar] [CrossRef]
- Koka, S.; Huber, S.M.; Boini, K.M.; Lang, C.; Foller, M.; Lang, F. Lead decreases parasitemia and enhances survival of plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 2007, 363, 484–489. [Google Scholar] [CrossRef]
- Koka, S.; Lang, C.; Boini, K.M.; Bobbala, D.; Huber, S.M.; Lang, F. Influence of chlorpromazine on eryptosis, parasitemia and survival of plasmodium berghe infected mice. Cell. Physiol. Biochem. 2008, 22, 261–268. [Google Scholar] [CrossRef]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Influence of no synthase inhibitor l-name on parasitemia and survival of plasmodium berghei infected mice. Cell Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell. Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef]
- Bhavsar, S.K.; Bobbala, D.; Xuan, N.T.; Foller, M.; Lang, F. Stimulation of suicidal erythrocyte death by alpha-lipoic acid. Cell. Physiol. Biochem. 2010, 26, 859–868. [Google Scholar] [CrossRef]
- Foller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Foller, M.; Mahmud, H.; Gu, S.; Wang, K.; Floride, E.; Kucherenko, Y.; Luik, S.; Laufer, S.; Lang, F. Participation of leukotriene c(4) in the regulation of suicidal erythrocyte death. J. Physiol. Pharmacol. 2009, 60, 135–143. [Google Scholar]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of ctab- and peg-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; de Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef]
- Foller, M.; Sopjani, M.; Koka, S.; Gu, S.; Mahmud, H.; Wang, K.; Floride, E.; Schleicher, E.; Schulz, E.; Munzel, T.; et al. Regulation of erythrocyte survival by amp-activated protein kinase. FASEB J. 2009, 23, 1072–1080. [Google Scholar] [CrossRef]
- Kucherenko, Y.; Zelenak, C.; Eberhard, M.; Qadri, S.M.; Lang, F. Effect of casein kinase 1alpha activator pyrvinium pamoate on erythrocyte ion channels. Cell Physiol. Biochem. 2012, 30, 407–417. [Google Scholar] [CrossRef]
- Zelenak, C.; Eberhard, M.; Jilani, K.; Qadri, S.M.; Macek, B.; Lang, F. Protein kinase ck1alpha regulates erythrocyte survival. Cell Physiol. Biochem. 2012, 29, 171–180. [Google Scholar] [CrossRef]
- Foller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cgki-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef]
- Bhavsar, S.K.; Gu, S.; Bobbala, D.; Lang, F. Janus kinase 3 is expressed in erythrocytes, phosphorylated upon energy depletion and involved in the regulation of suicidal erythrocyte death. Cell Physiol. Biochem. 2011, 27, 547–556. [Google Scholar] [CrossRef]
- Klarl, B.A.; Lang, P.A.; Kempe, D.S.; Niemoeller, O.M.; Akel, A.; Sobiesiak, M.; Eisele, K.; Podolski, M.; Huber, S.M.; Wieder, T.; et al. Protein kinase c mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Physiol. Cell Physiol. 2006, 290, C244–C253. [Google Scholar]
- Gatidis, S.; Zelenak, C.; Fajol, A.; Lang, E.; Jilani, K.; Michael, D.; Qadri, S.M.; Lang, F. P38 mapk activation and function following osmotic shock of erythrocytes. Cell. Physiol. Biochem. 2011, 28, 1279–1286. [Google Scholar] [CrossRef]
- Zelenak, C.; Foller, M.; Velic, A.; Krug, K.; Qadri, S.M.; Viollet, B.; Lang, F.; Macek, B. Proteome analysis of erythrocytes lacking amp-activated protein kinase reveals a role of pak2 kinase in eryptosis. J. Proteome Res. 2011, 10, 1690–1697. [Google Scholar] [CrossRef]
- Lupescu, A.; Shaik, N.; Jilani, K.; Zelenak, C.; Lang, E.; Pasham, V.; Zbidah, M.; Plate, A.; Bitzer, M.; Foller, M.; et al. Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: An in vivo and in vitro study. Cell Physiol. Biochem. 2012, 30, 876–888. [Google Scholar] [CrossRef]
- Shaik, N.; Lupescu, A.; Lang, F. Sunitinib-sensitive suicidal erythrocyte death. Cell Physiol. Biochem. 2012, 30, 512–522. [Google Scholar] [CrossRef]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via cxcl16/sr-psox. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef]
- Pandolfi, A.; di Pietro, N.; Sirolli, V.; Giardinelli, A.; di Silvestre, S.; Amoroso, L.; di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler Thromb Vasc. Biol. 2007, 27, 414–421. [Google Scholar]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef]
- Abed, M.; Towhid, S.T.; Shaik, N.; Lang, F. Stimulation of suicidal death of erythrocytes by rifampicin. Toxicology 2012, 302, 123–128. [Google Scholar] [CrossRef]
- Bottger, E.; Multhoff, G.; Kun, J.F.; Esen, M. Plasmodium falciparum-infected erythrocytes induce granzyme b by nk cells through expression of host-hsp70. PLoS ONE 2012, 7, e33774. [Google Scholar] [CrossRef]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in stz diabetic rats. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef]
- Gao, M.; Cheung, K.L.; Lau, I.P.; Yu, W.S.; Fung, K.P.; Yu, B.; Loo, J.F.; Kong, S.K. Polyphyllin d induces apoptosis in human erythrocytes through Ca2+ rise and membrane permeabilization. Arch. Toxicol. 2012, 86, 741–752. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Abed, M.; Shaik, N.; Lang, F. Enhanced apoptotic death of erythrocytes induced by the mycotoxin ochratoxin a. Kidney Blood Press Res. 2012, 36, 107–118. [Google Scholar] [CrossRef]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Shaik, N.; Lang, F. Withaferin a-stimulated Ca2+ entry, ceramide formation and suicidal death of erythrocytes. Toxicol. In Vitro 2013, 27, 52–58. [Google Scholar] [CrossRef]
- Kucherenko, Y.V.; Lang, F. Inhibitory effect of furosemide on non-selective voltage-independent cation channels in human erythrocytes. Cell. Physiol. Biochem. 2012, 30, 863–875. [Google Scholar] [CrossRef]
- Lang, E.; Jilani, K.; Zelenak, C.; Pasham, V.; Bobbala, D.; Qadri, S.M.; Lang, F. Stimulation of suicidal erythrocyte death by benzethonium. Cell Physiol. Biochem. 2011, 28, 347–354. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Jilani, K.; Zelenak, C.; Lupescu, A.; Schleicher, E.; Lang, F. Carbon monoxide-sensitive apoptotic death of erythrocytes. Basic Clin. Pharmacol. Toxicol. 2012, 111, 348–355. [Google Scholar]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, E.; Lang, F. Enhanced Ca(2+) entry, ceramide formation, and apoptotic death of erythrocytes triggered by plumbagin. J. Nat. Prod. 2012, 75, 1956–1961. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of apoptotic erythrocyte death by rotenone. Toxicology 2012, 300, 132–137. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zelenak, C.; Zbidah, M.; Qadri, S.M.; Lang, F. Hexavalent chromium-induced erythrocyte membrane phospholipid asymmetry. Biometals 2012, 25, 309–318. [Google Scholar] [CrossRef]
- Polak-Jonkisz, D.; Purzyc, L. Ca influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef]
- Qian, E.W.; Ge, D.T.; Kong, S.K. Salidroside protects human erythrocytes against hydrogen peroxide-induced apoptosis. J. Nat. Prod. 2012, 75, 531–537. [Google Scholar] [CrossRef]
- Shaik, N.; Zbidah, M.; Lang, F. Inhibition of Ca(2+) entry and suicidal erythrocyte death by naringin. Cell Physiol. Biochem. 2012, 30, 678–686. [Google Scholar] [CrossRef]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca(2+) dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell. Calcium 2012, 51, 51–56. [Google Scholar]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? J. Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef]
- Zbidah, M.; Lupescu, A.; Jilani, K.; Lang, F. Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin. Pharmacol. Toxicol. 2013, 112, 346–351. [Google Scholar] [CrossRef]
- Zbidah, M.; Lupescu, A.; Shaik, N.; Lang, F. Gossypol-induced suicidal erythrocyte death. Toxicology 2012, 302, 101–105. [Google Scholar] [CrossRef]
- Zelenak, C.; Pasham, V.; Jilani, K.; Tripodi, P.M.; Rosaclerio, L.; Pathare, G.; Lupescu, A.; Faggio, C.; Qadri, S.M.; Lang, F. Tanshinone iia stimulates erythrocyte phosphatidylserine exposure. Cell Physiol. Biochem. 2012, 30, 282–294. [Google Scholar] [CrossRef]
- Abed, M.; Herrmann, T.; Alzoubi, K.; Pakladok, T.; Lang, F. Tannic acid induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1106–1116. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res. 2013, 37, 158–167. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of nfkappab abundance and programmed cell death in erythrocytes induced by nfkappab inhibitors. Cell Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of erythrocyte cation channels by erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef]
- Lang, P.A.; Beringer, O.; Nicolay, J.P.; Amon, O.; Kempe, D.S.; Hermle, T.; Attanasio, P.; Akel, A.; Schafer, R.; Friedrich, B.; et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. Berl. 2006, 84, 378–388. [Google Scholar] [CrossRef]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 269–277. [Google Scholar]
- Lang, P.A.; Kasinathan, R.S.; Brand, V.B.; Duranton, C.; Lang, C.; Koka, S.; Shumilina, E.; Kempe, D.S.; Tanneur, V.; Akel, A.; et al. Accelerated clearance of plasmodium-infected erythrocytes in sickle cell trait and annexin-a7 deficiency. Cell Physiol. Biochem. 2009, 24, 415–428. [Google Scholar] [CrossRef]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006, 20, 368–370. [Google Scholar]
- Qadri, S.M.; Mahmud, H.; Lang, E.; Gu, S.; Bobbala, D.; Zelenak, C.; Jilani, K.; Siegfried, A.; Foller, M.; Lang, F. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J. Cell Mol. Med. 2012, 16, 1085–1093. [Google Scholar] [CrossRef]
- Birka, C.; Lang, P.A.; Kempe, D.S.; Hoefling, L.; Tanneur, V.; Duranton, C.; Nammi, S.; Henke, G.; Myssina, S.; Krikov, M.; et al. Enhanced susceptibility to erythrocyte “apoptosis” following phosphate depletion. Pflugers Arch. 2004, 448, 471–477. [Google Scholar]
- Sinha, A.; Singh, A.; Satchidanandam, V.; Natarajan, K. Impaired generation of reactive oxygen species during differentiation of dendritic cells (dcs) by mycobacterium tuberculosis secretory antigen (mtsa) and subsequent activation of mtsa-dcs by mycobacteria results in increased intracellular survival. J. Immunol. 2006, 177, 468–478. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alzoubi, K.; Alktifan, B.; Oswald, G.; Fezai, M.; Abed, M.; Lang, F. Breakdown of Phosphatidylserine Asymmetry Following Treatment of Erythrocytes with Lumefantrine. Toxins 2014, 6, 650-664. https://doi.org/10.3390/toxins6020650
Alzoubi K, Alktifan B, Oswald G, Fezai M, Abed M, Lang F. Breakdown of Phosphatidylserine Asymmetry Following Treatment of Erythrocytes with Lumefantrine. Toxins. 2014; 6(2):650-664. https://doi.org/10.3390/toxins6020650
Chicago/Turabian StyleAlzoubi, Kousi, Bassel Alktifan, Gergely Oswald, Myriam Fezai, Majed Abed, and Florian Lang. 2014. "Breakdown of Phosphatidylserine Asymmetry Following Treatment of Erythrocytes with Lumefantrine" Toxins 6, no. 2: 650-664. https://doi.org/10.3390/toxins6020650
APA StyleAlzoubi, K., Alktifan, B., Oswald, G., Fezai, M., Abed, M., & Lang, F. (2014). Breakdown of Phosphatidylserine Asymmetry Following Treatment of Erythrocytes with Lumefantrine. Toxins, 6(2), 650-664. https://doi.org/10.3390/toxins6020650