Impact of pH on the Stability and the Cross-Reactivity of Ochratoxin A and Citrinin

 ,
, 
Abstract
:1. Introduction
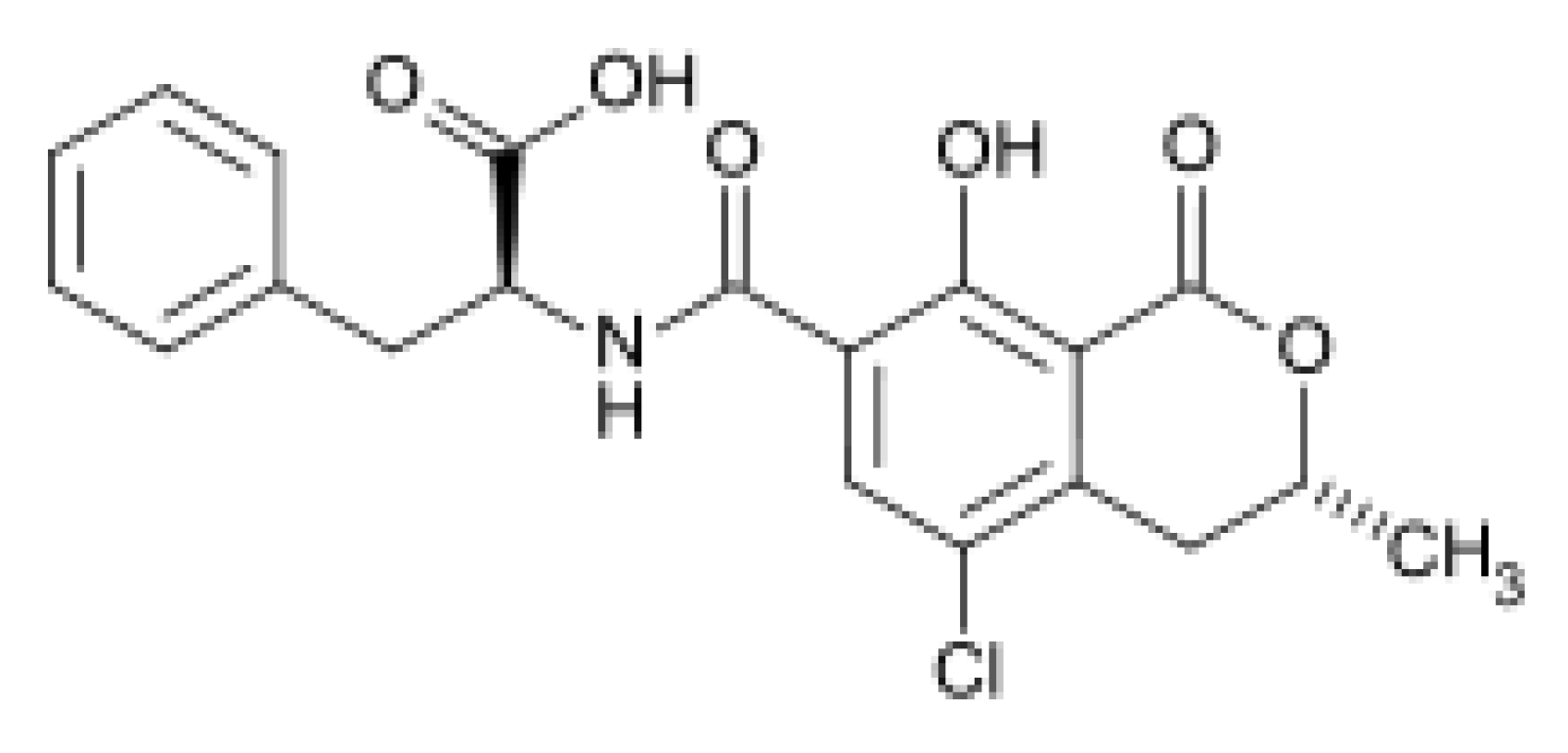
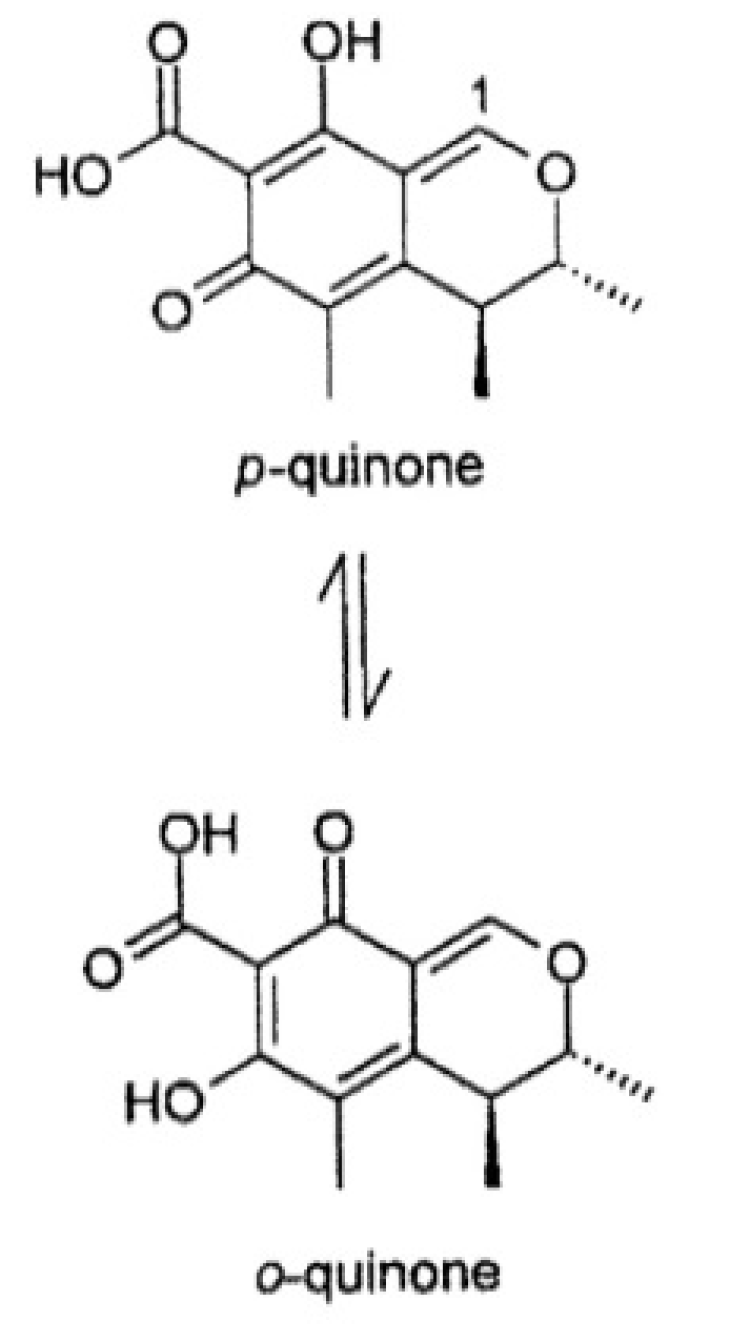
2. Results and Discussion
2.1. Underestimation of OTA Following PEG Clean-Up of Wine
2.2. Interference of CIT with OTA on IAC during Analysis of Wheat Samples
2.2.1. Comparison of OTA/CIT Occurrence in Wheat Using Different Methods of Extraction and Clean-Up

| Samples | OTA ppb (c) | CIT ppb | |||
|---|---|---|---|---|---|
| IAC a/bicarbonate [35] | IAC b/PBS [36] | Liquid-Liquid/acid c [4] | Liquid-Liquid/acid d [4] | ELISA e | |
| F1 | <LOD | trace | 5.58 ± 0.28 | <LOD | <LOD |
| F2 | <LOD | trace | 6.45 ± 0.33 | <LOD | <LOD |
| F4 | <LOD | <LOD | 3.14 ± 0.15 | <LOD | <LOD |
| F6 | <LOD | <LOD | 0.84 ± 0.10 | <LOD | <LOD |
| F7 | <LOD | <LOD | 0.91 ± 0.11 | <LOD | <LOD |
| F8 | <LOD | <LOD | <LOD | <LOD | NA |
| F9 | <LOD | <LOD | Trace (<LOQ) | trace | NA |
| F10 | 128.49 ± 30.10 | 74.40 ± 8.20 | 6.26 ± 0.31 | 512.38 ± 61.5 | 1468.8 |
| F11 | <LOD | <LOD | <LOD | 0.84 ± 0.10 | <LOD |
| F12 | <LOD | <LOD | <LOD | <LOD | NA |
| F13 | <LOD | <LOD | <LOD | <LOD | NA |
| F14 | <LOD | <LOD | 1.07 ± 0.06 | <LOD | <LOD |
| F15 | <LOD | 1.5 ± 1 | 6.73 ± 0.35 | 0.86 ± 0.09 | <LOD |
| F16 | <LOD | <LOD | <LOD | 0.86 ± 0.1 | <LOD |
| F17 | <LOD | <LOD | <LOD | 0.76 ± 0.1 | <LOD |
| F18 | 67.23 ± 8.04 | 67.50 ± 9.50 | 30.77 ± 1.54 | 24.57 ± 2.7 | NA |
| F19 | <LOD | 3.20 ± 0.50 | <LOD | 7.24 ± 0.58 | NA |
| F20 | <LOD | trace | 5.20 ± 0.27 | <LOD | NA |
| S2 | 6.63 ± 2.33 | 5.07 ± 0.60 | 1.74 ± 0.09 | 44.60 ± 4.90 | 151.5 |
| S3 | <LOD | <LOD | <LOD | <LOD | NA |
| S4 | <LOD | <LOD | <LOD | 0.86 ± 0.10 | NA |
| S5 | <LOD | <LOD | <LOD | <LOD | NA |
| S6 | <LOD | <LOD | <LOD | 0.88 ± 0.09 | NA |
| S7 | <LOD | <LOD | <LOD | <LOD | NA |
| S8 | <LOD | <LOD | <LOD | 1.10 ± 0.09 | NA |
| S9 | <LOD | 3.06 | <LOD | 7.46 ± 0.89 | <LOD |
| S10 | <LOD | <LOD | <LOD | 1.04 ± 0.09 | <LOD |
| S11 | <LOD | <LOD | 5.3 ± 0.27 | <LOD | <LOD |
| S12 | <LOD | <LOD | 2.42 ± 0.3 | 1.57 ± 0.126 | <LOD |
| S13 | <LOD | <LOD | <LOD | 1.04 ± 0.09 | <LOD |
| S14 | 27.96 ± 3.01 | 25.20 ± 2.50 | 7.59 ± 0.38 | 3.79 ± 0.30 | <LOD |
| S15 | <LOD | <LOD | <LOD | <LOD | NA |
| S16 | <LOD | 1.00 ± 1.00 | <LOD | 15.00 ± 1.80 | 41.8 |
2.2.2. Recoveries and Confirmation of OTA Metabolites Formed
| Mycotoxin in aqueous solution | Mycotoxin in wheat | |||||
|---|---|---|---|---|---|---|
| IAC/bicarbonate [35] | IAC/PBS [36] | Classic [4] | IAC/bicarbonate [35] | IAC/PBS [36] | Classic | |
| OTA alone | ||||||
| 100 µg/kg c | NA | NA | NA | 25.80 ± 2.50 (25.8%) | 60.10 ± 3.50 (60.1%) | 83.50 ± 4.20 (83.5%) |
| 40 µg/kg | 17.60 ± 1.40 a (44%) b | 30.40 ± 1.40 a (66%) b | 41.00 ± 1.20 (102%) | 11.80 ± 1.87 (29.5%) | 24.40 ± 0.87 (61%) | 28.80 ± 0.90 (72%) |
| 7 µg/kg | 2.03 ± 0.17 (29%) | 4.2 ± 0.27 (60%) | 6.3 ± 0.18 (90%) | 2.24 ± 0.35 (32%) | 3.85 ± 0.35 (55%) | 5.20 ± 0.16 (74%) |
| 3 µg/kg | <LOD (0%) | 1.85 + 0.17 (61.6%) | 2.79 ± 0.08 (93%) | <LOD (0%) | 1.80 + 0.41 (60%) | 2.30 ± 0.07 (76.6%) |
| CIT alone | ||||||
| 5 µg/kg | peak eluting at the OTA retention time | peak eluting at the OTA retention time | 4 ± 0.2 (80%) | peak eluting at the OTA retention time | peak eluting at the OTA retention time | 3.90 ± 0.12 (78%) |
| 40 µg/kg | peak eluting at the OTA retention time | peak eluting at the OTA retention time | 32 ± 1.6 (80%) | peak eluting at the OTA retention time | peak eluting at the OTA retention time | 30.00 ± 1.50 (75%) |
| OTA 40 µg/kg | 20.80 ± 3.11 (52%) | 62.80 ± 3.11 (157%) | 36.00 ± 1.10 (90%) | 42.40 ± 0.90 (106%) | 28.60 ± 1.43 (71.5%) | |
| CIT 40 µg/kg | - | - | 31.20 ± 1.92 (78%) | - | 32.00 ± 1.60 (80%) | |
| OTA 5 µg/kg | 9.10 ± 0.70 (182%) | 7.30 ± 0.70 (146%) | 4.50 ± 0.22 (90%) | 5.75 ± 0.50 (115%) | 3.75 ± 0.11 (75%) | |
| CIT 50 µg/kg | - | - | 39.50 ± 3.16 (79%) | - | 37.50 + 3.00 (75%) | |
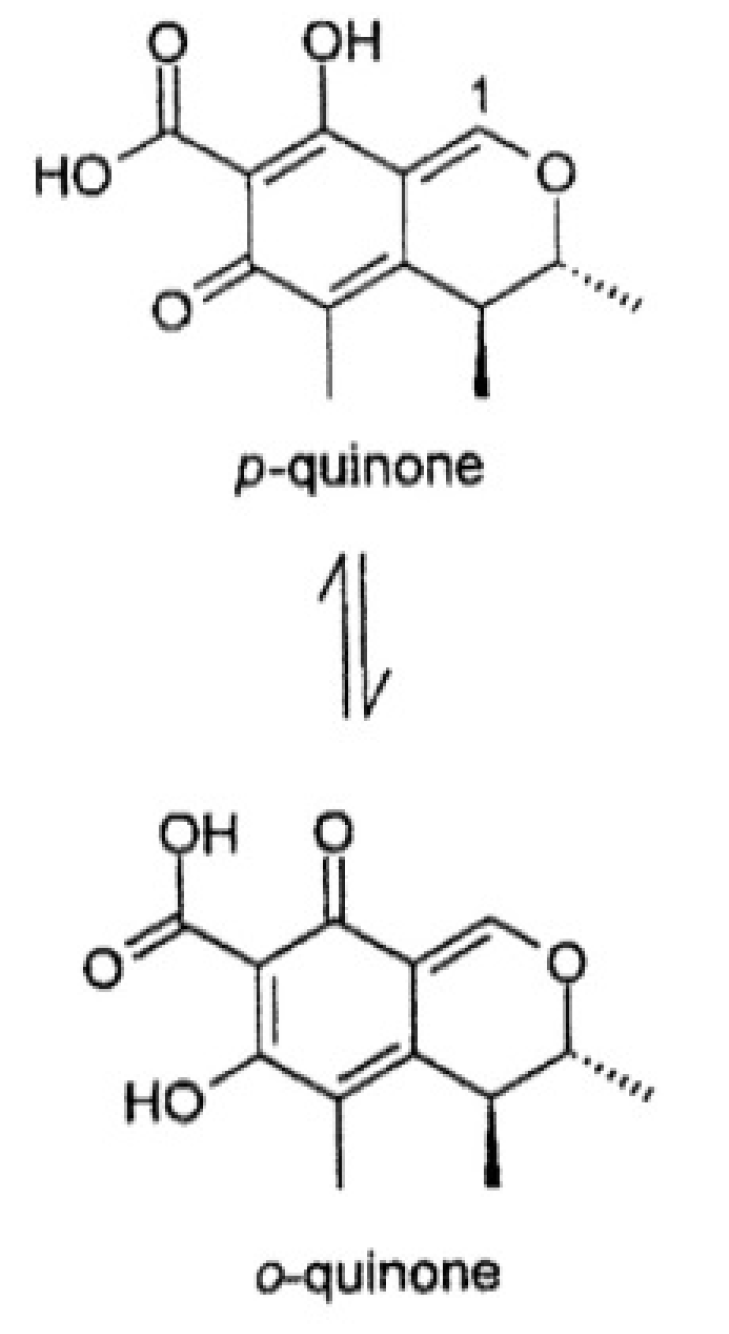
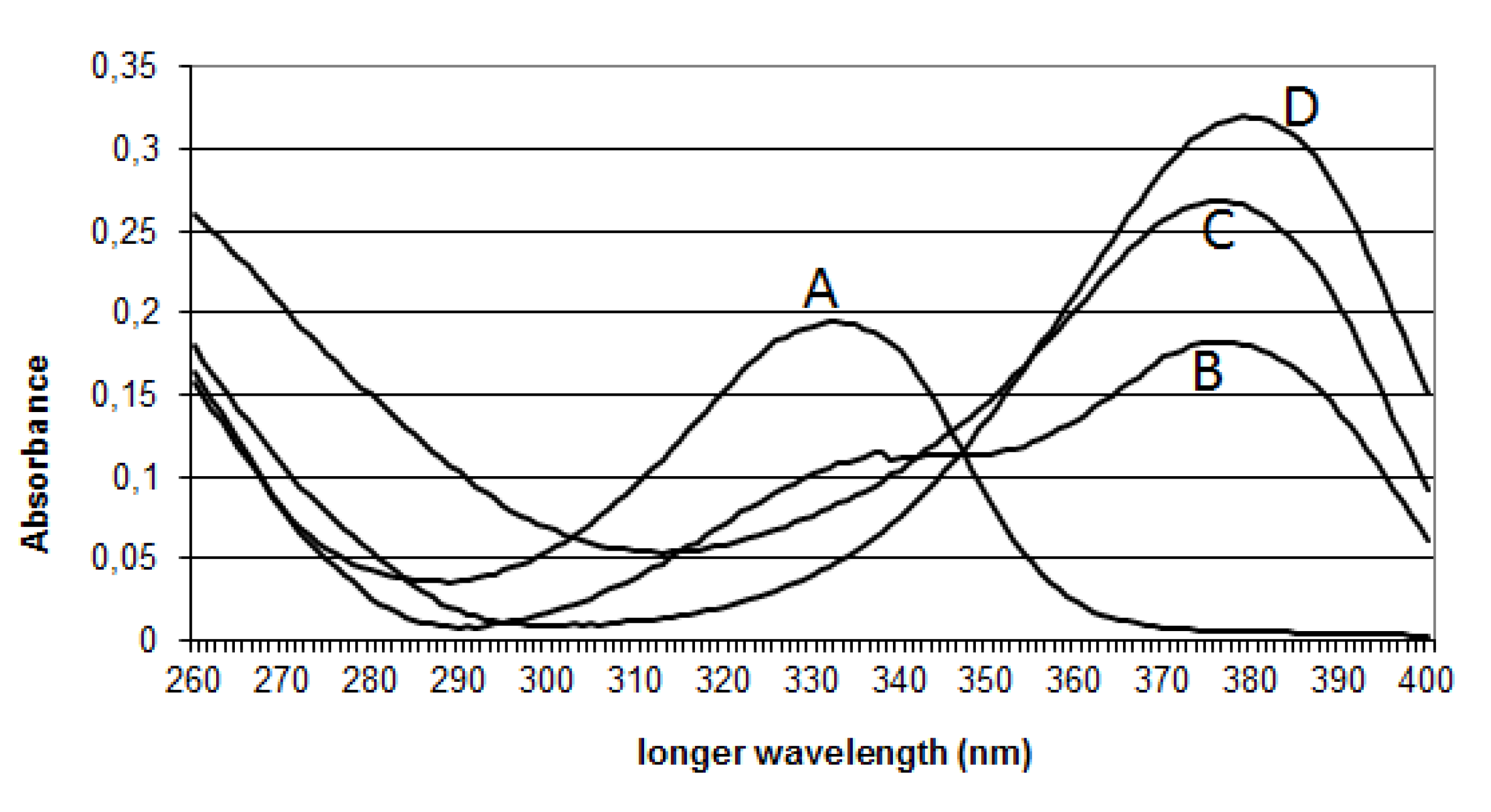
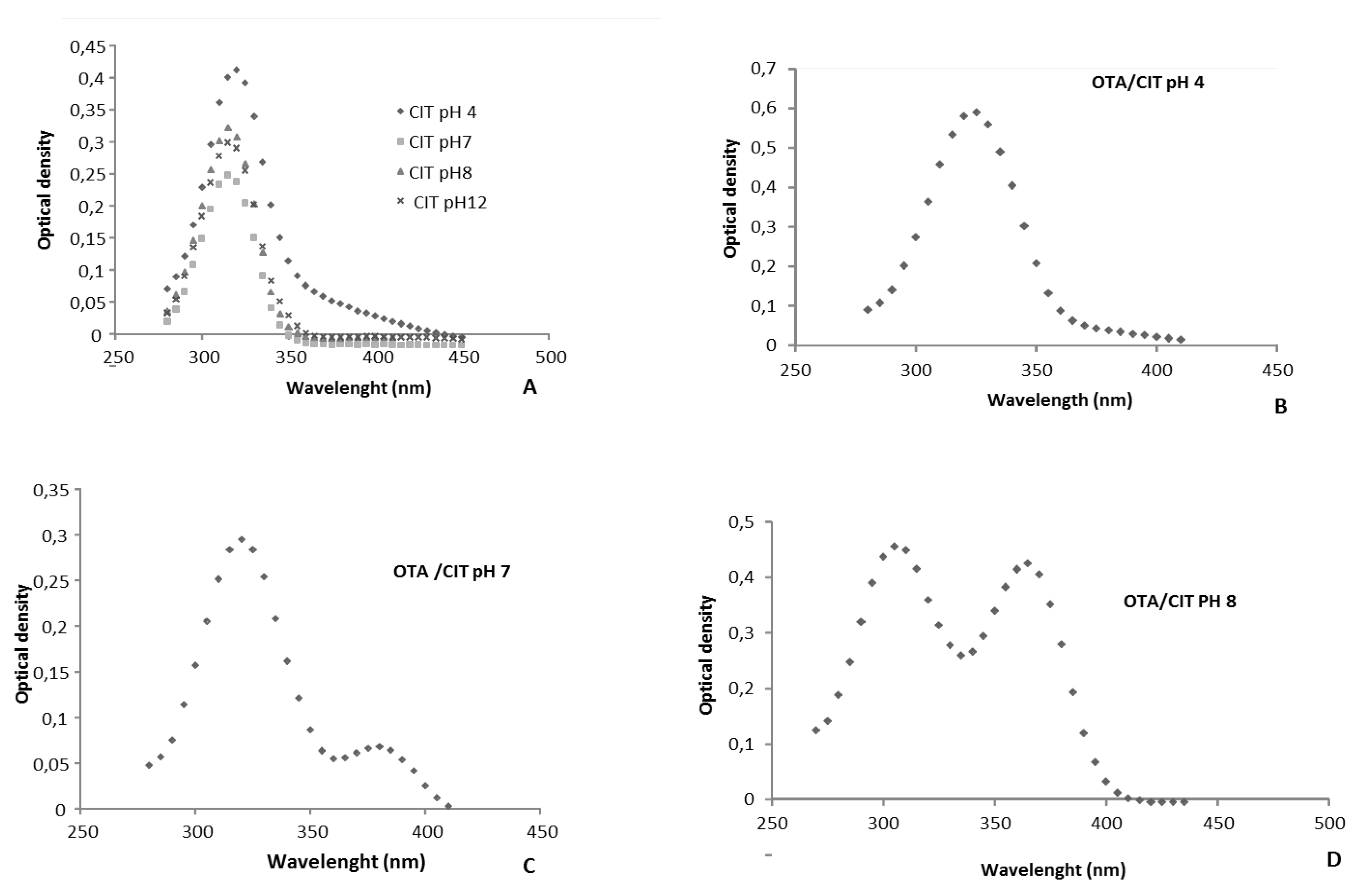
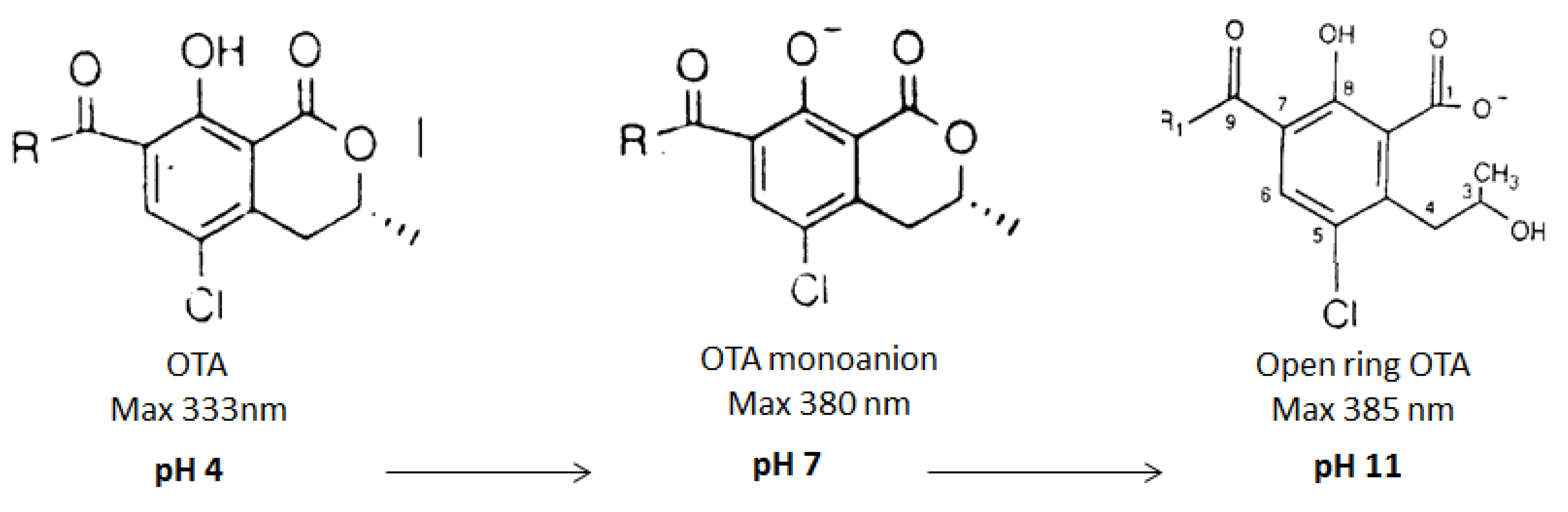
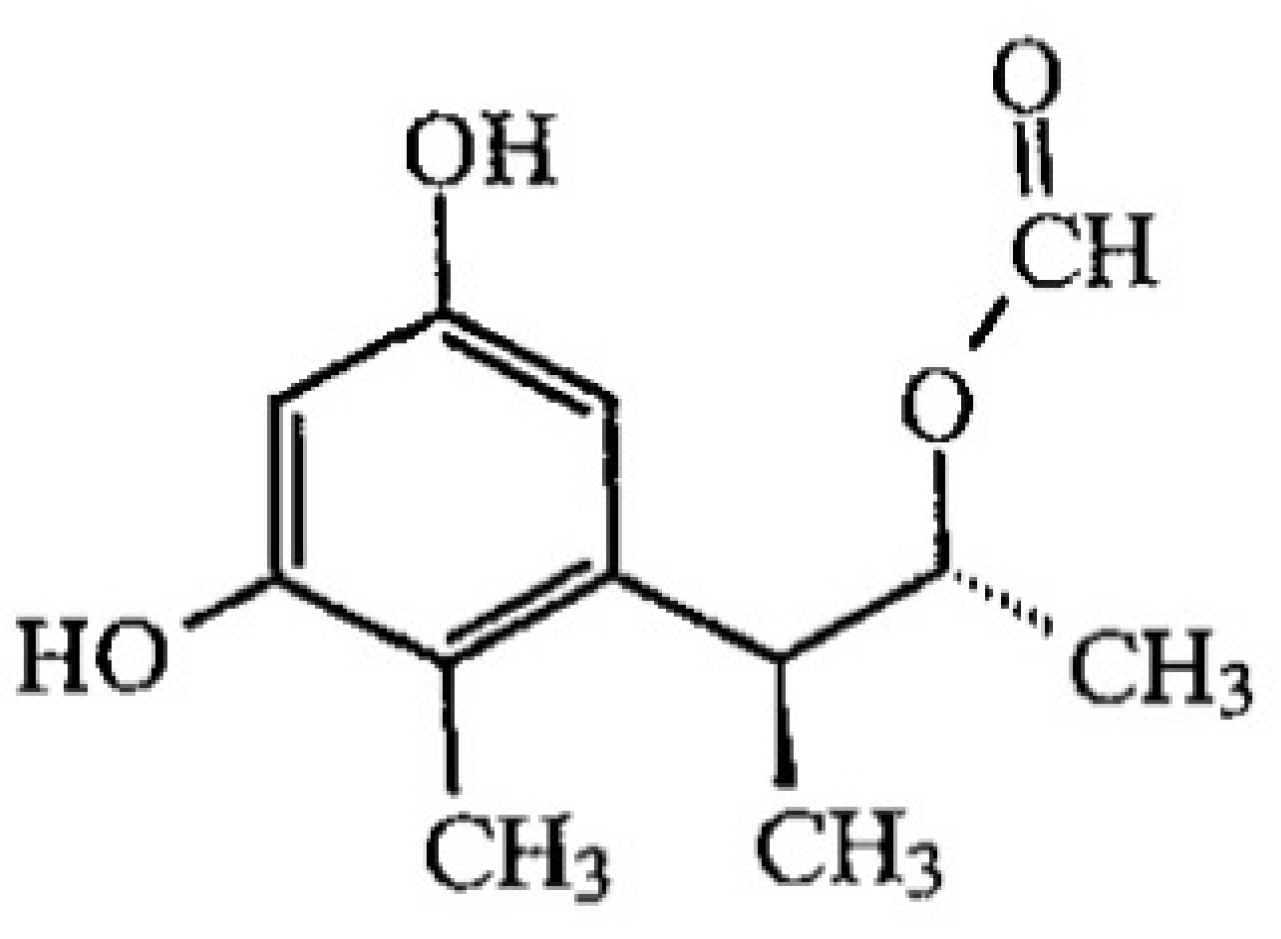
2.2.3. Effect of the pH on OTA Quantification and UV Spectra in Presence or Absence of CIT
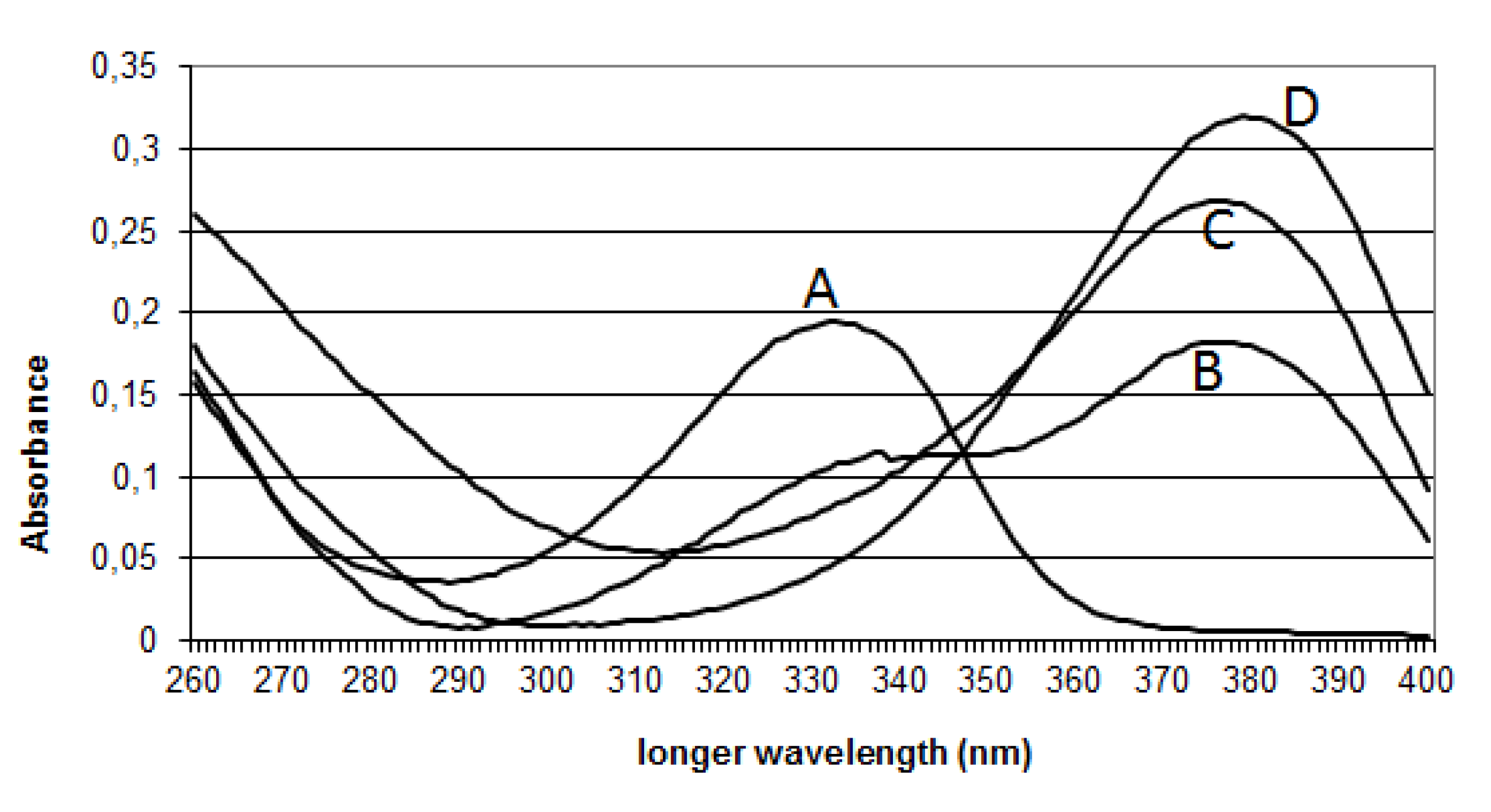
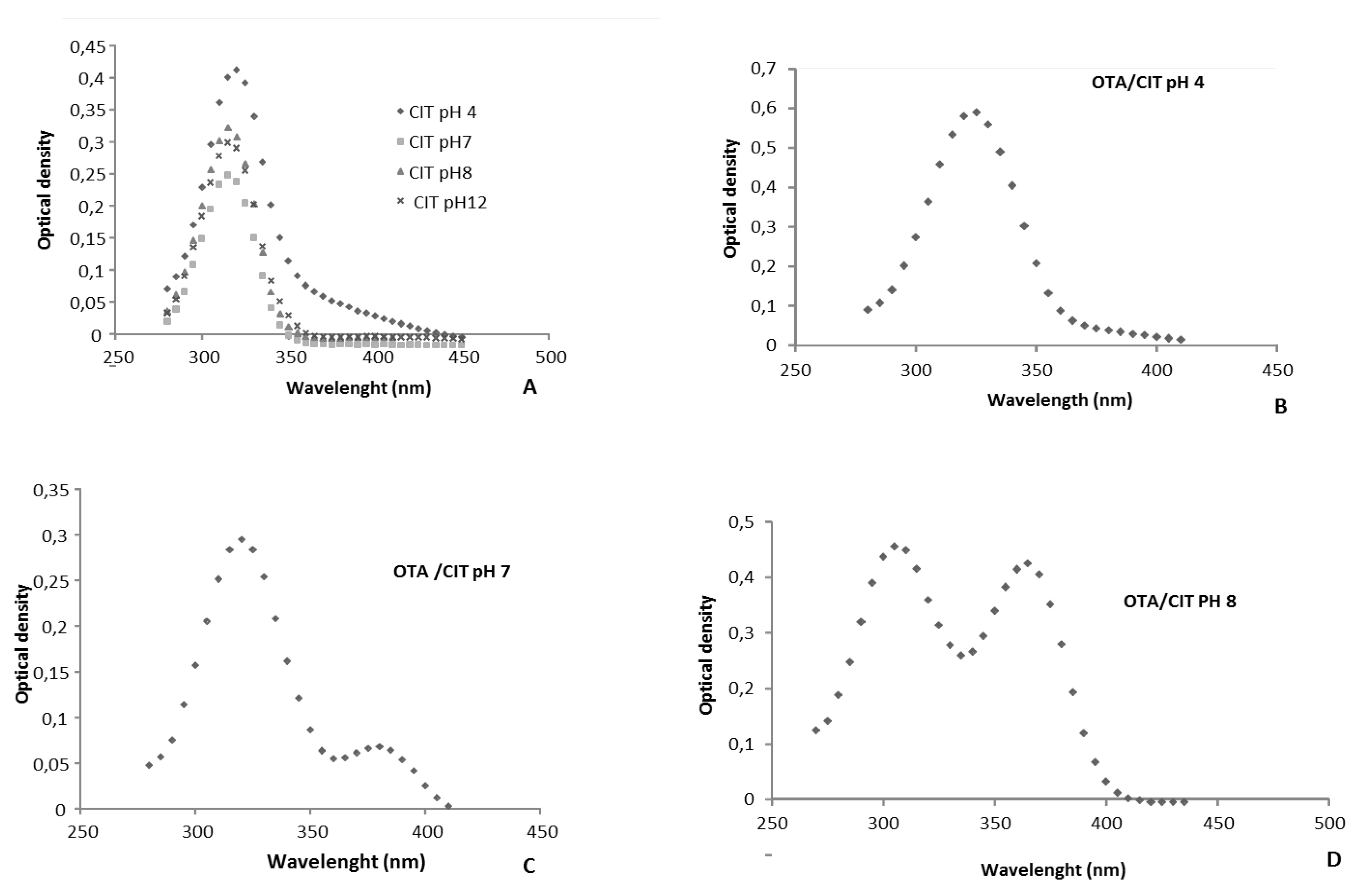
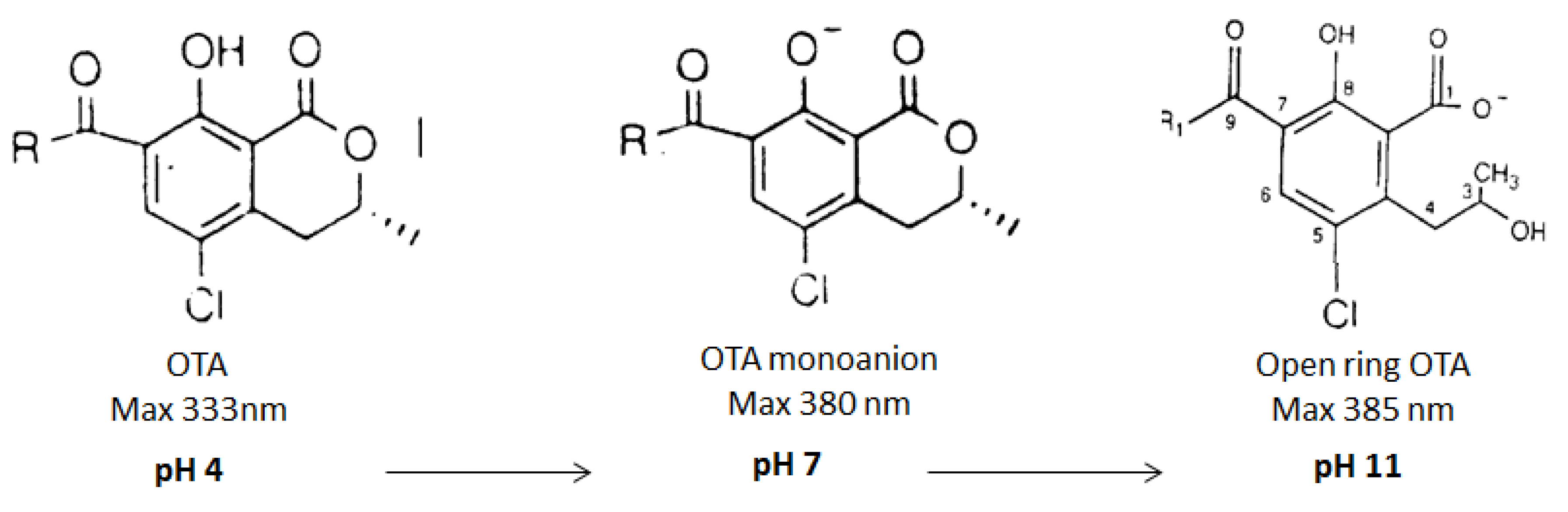
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
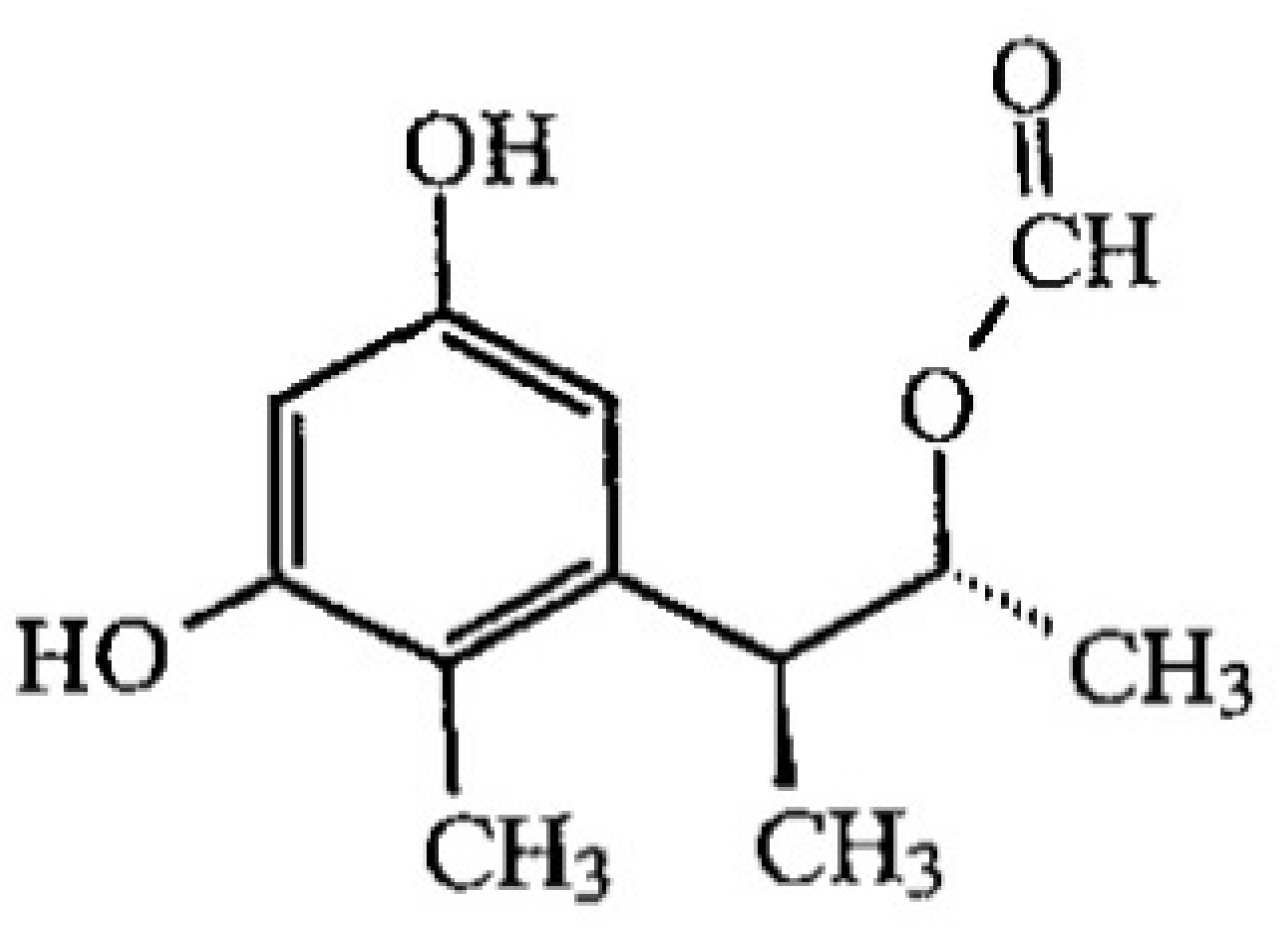
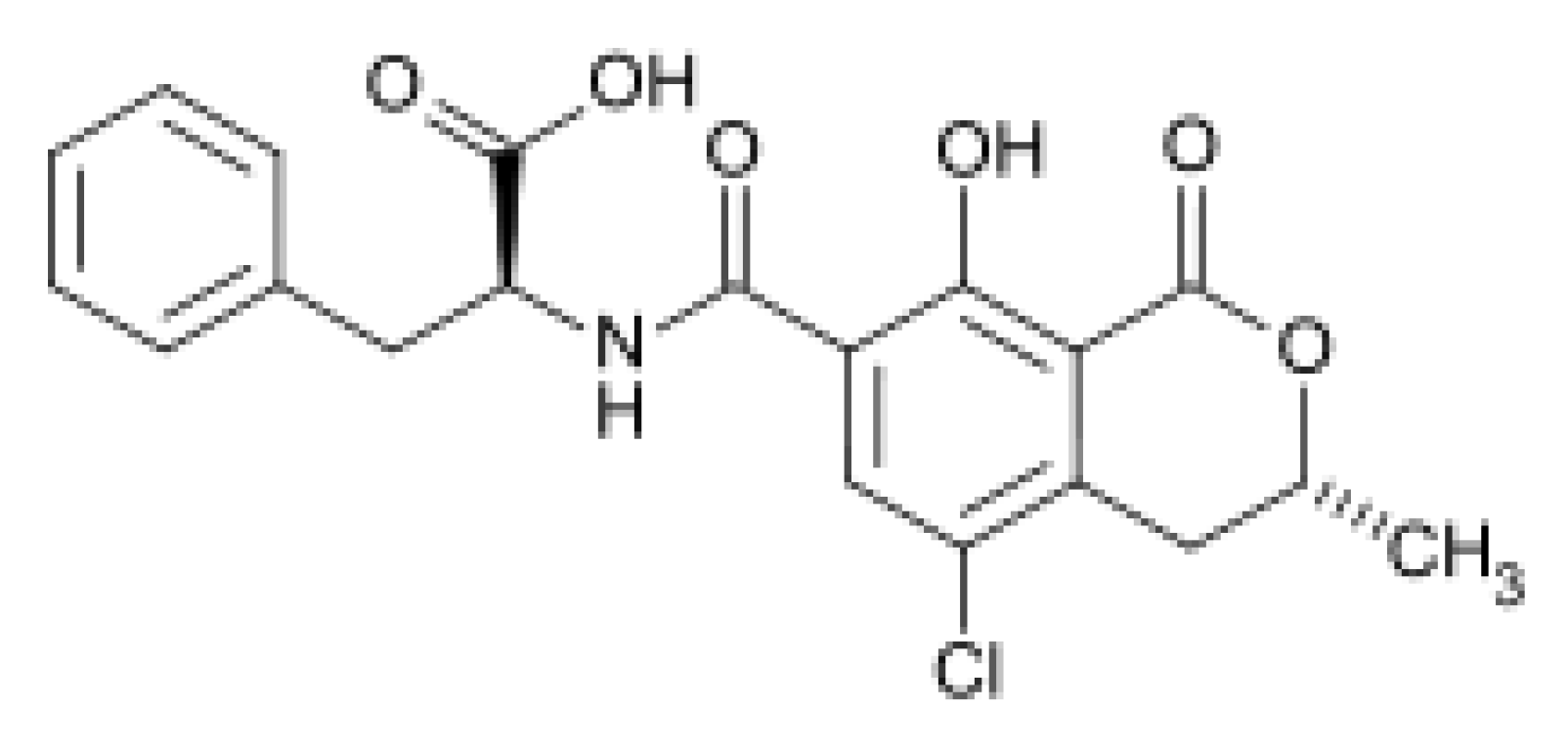
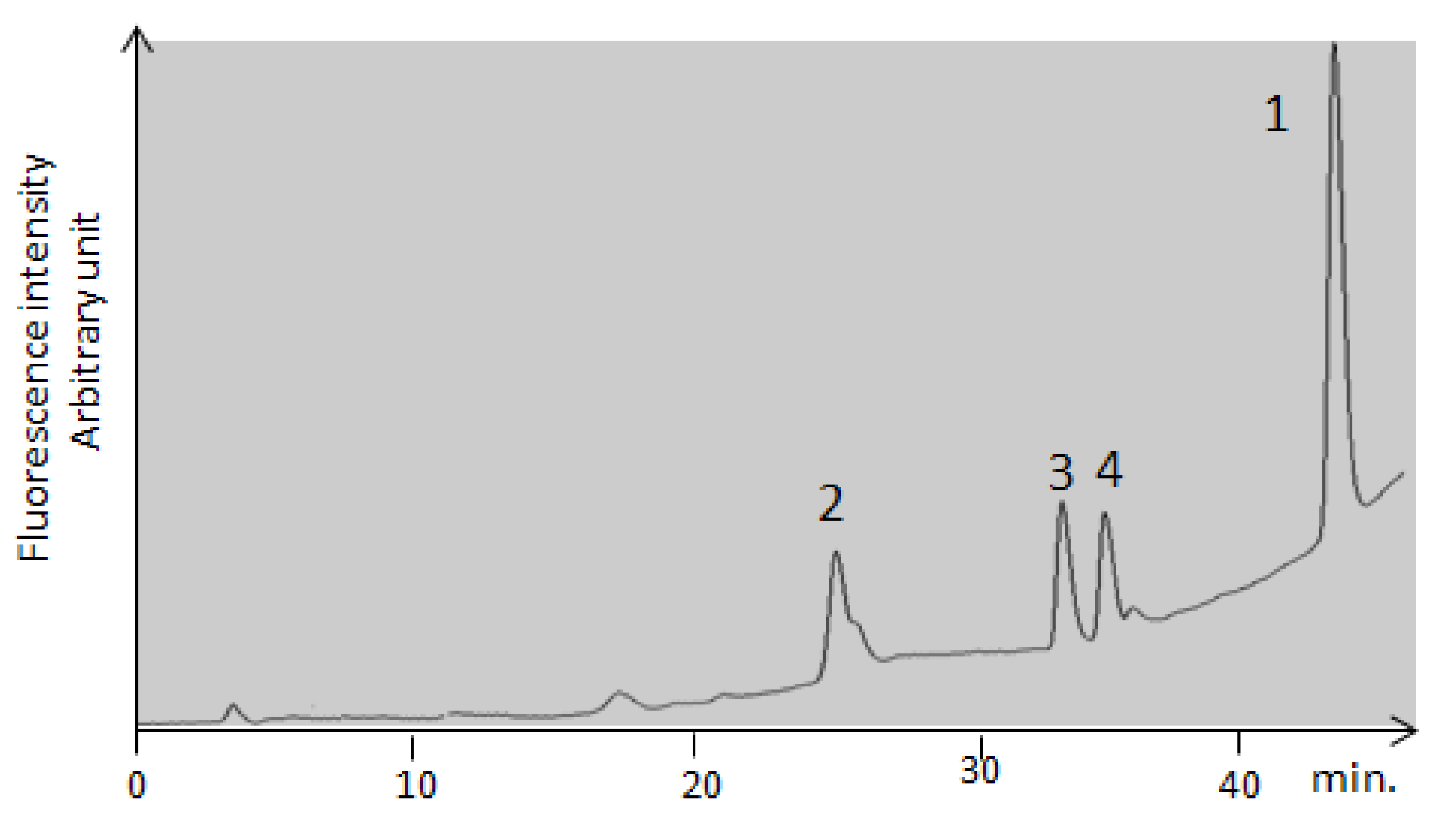
2.2.4. Confirmation by HPLC MS/MS of the Formation of OP-OA; OTHQ and OTB
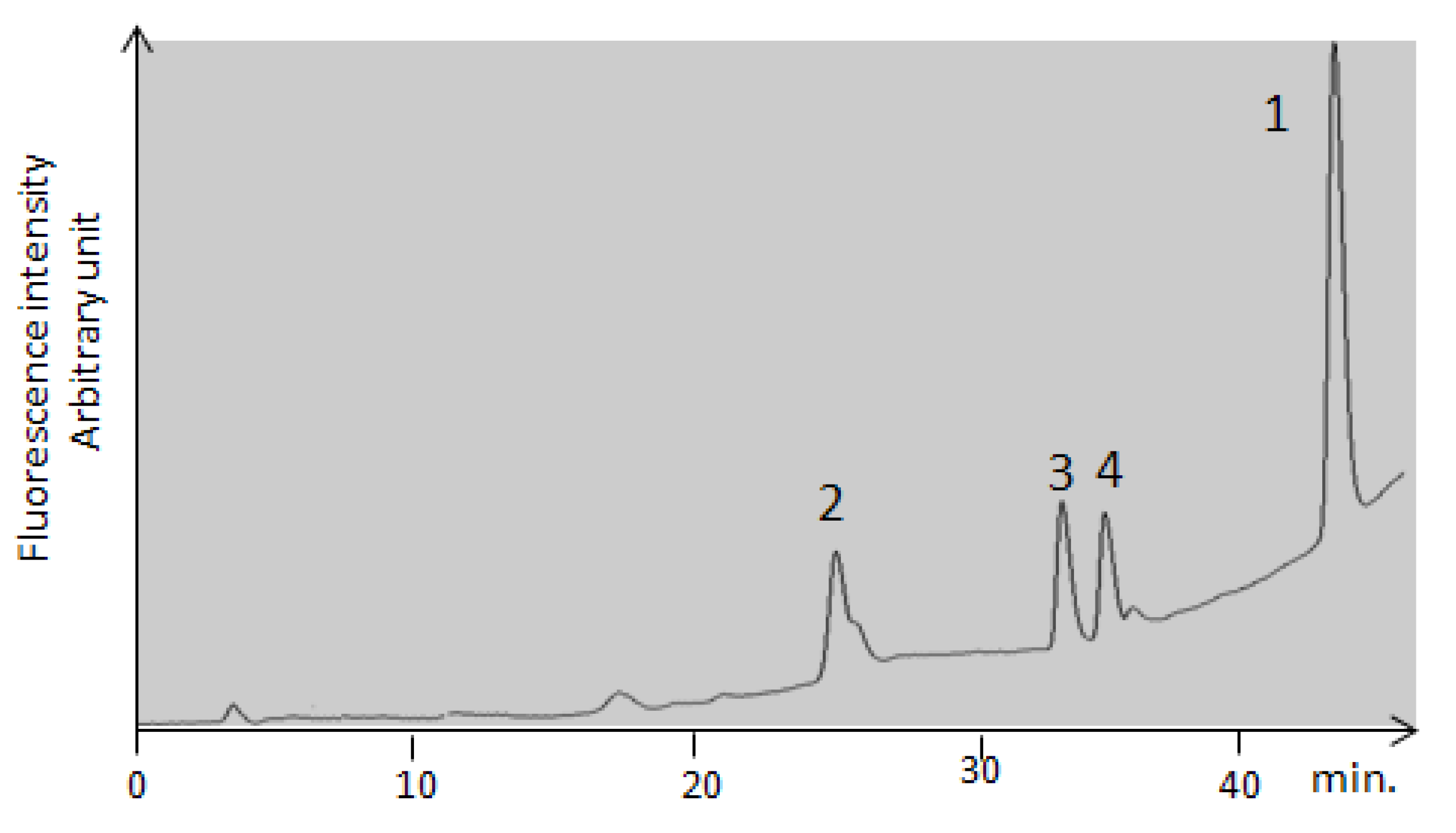
| Peak numbering | Species | RT (min) | λmax | [M−H]− | Fragment ions m/z |
|---|---|---|---|---|---|
| 1 | OTA | 42 | 333 | 402 | 358; 314 |
| 2 | OP-OA | 28 | 380 | 420 | 376; 332 |
| 3 | OTHQ | 34 | 350 | 384 | 340; 296 |
| 4 | OTB | 36 | 316 | 368 | 324; 280 |
| peak | 1 (OTA) | 2 (OP-OA) | 3 (OTHQ) | 4 (OTB) |
|---|---|---|---|---|
| OTA alone | ||||
| pH 4 | +++ | - | - | - |
| pH 7 | ++ | ++ | - | - |
| pH 8 | + | ++ | - | - |
| pH 12 | - | +++ | - | - |
| OTA + CIT | ||||
| pH 4 | + | - | - | - |
| pH 7 | + | + | + | |
| pH 8 | + | ++ | + | + |
3. Experimental Section
3.1. Chemicals
3.2. Preparation Standard Solution
3.3. OTA extraction in Red Wine Samples
3.3.1. PEG Treatment
3.3.2. PVPP Treatment
3.4. Wheat Analysis
3.4.1. Sampling
3.4.2. OTA Extraction from Wheat Samples
3.4.2.1. Solvent Extraction Clean-Up and Partioning Purification
3.4.2.2. IAC Clean-Up
3.4.2.2.1. OTA Extraction by Bicarbonate
3.4.2.2.2. OTA Extraction by Methanol/Water
3.4.3. OTA/CIT HPLC Conditions
3.4.3.1. Conditions after Extraction by Liquid/Liquid Extraction [4]
3.4.3.2. Condition after IAC Extraction [35,36]
3.4.3.3. Condition for Separation of OTA Metabolites
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rupollo, G.; Gutkoski, L.C.; Martins, I.R.; Elias, M.C. Effects of grain moisture and hermetic storage on fungi contamination and mycotoxin production in oats. Ciênc. Agrotec. 2006, 30, 118. [Google Scholar]
- Food and Agriculture Organization (FAO). Worldwide Regulations for Mycotoxins. In A Compendium, FAO Food and Nutrition Paper No. 64; FAO: Rome, Italy, 1995. [Google Scholar]
- Nguyen, M.T.; Tozlovanu, M.; Tran, T.L.; Pfohl-Leszkowicz, A. Occurrence of aflatoxin B1, citrinin and ochratoxin a in rice in five provinces of central region in Vietnam. Food Chem. 2007, 105, 42–47. [Google Scholar]
- Molinié, A.; Faucet, M.; Castegnaro, M.; Pfohl-Leszkowicz, A. Analysis of some breakfast cereals on the French market for their contents of ochratoxin A, citrinin and fumonisin B1: Development of a method for simultaneous extraction of ochratoxin A and citrinin. Food Chem. 2005, 92, 391. [Google Scholar] [CrossRef]
- Jorgensen, K. Occurrence of ochratoxin A in commodities and processes food—A review of EU occurence dat. Food Addit. Contam. 2005, 22, 26–30. [Google Scholar] [CrossRef]
- Duarte, S.C.; Pena, A.; Lino, C.M. A review on ochratoxin A occurrence and effects of processing of cereal and cereal derived food products. Food Microbiol. 2010, 27, 187–198. [Google Scholar] [CrossRef]
- Duarte, S.C.; Lino, C.; Pena, A. Ochratoxin A in feed of food-producing animals: An undesirable mycotoxin with health and performance effects. Veterinary Microbiol. 2011, 154, 1–13. [Google Scholar] [CrossRef]
- Navas, S.A.; Sabino, M.; Rodriguez-Amaya, D.B. Aflatoxin M1 and ochratoxin A in a human milk bank in the city of Sao Paulo, Brazil. Food Addit. Contam. 2005, 22, 457–462. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R. Review on Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar]
- European Commission. Commission Regulation (EC) No. 1881/2006 of 19th December 2006. Offi. J. Eur. Union 2006, 364, 5–24. [Google Scholar]
- Hetherington, C.; Raistrick, H. Studies in the Biochemistry of Micro-Organisms. Part XIV. In On the Production and Chemical Constitution of the New Yellow Colouring Matter, Citrinin, Produced from Glucose by Penicillium citrinum; The Royal Society: London, UK, 1931; Volume 220, pp. 269–295. [Google Scholar]
- Krogh, P.; Hald, B.; Pedersen, E.J. Occurrence of ochratoxin A and citrinin in cereals associated with 905 mycotoxic porcine nephropathy. Acta Pathol. Microbiol. Scand 1973, 816, 689–695. [Google Scholar]
- Pfohl-Leszkowicz, A.; Petkova-Bocharova, T.; Chernozemsky, I.N.; Castegnaro, M. Balkan endemic nephropathy and the associated urinary tract tumours: Review on etiological causes, potential role of mycotoxins. Food Addit. Contam. 2002, 19, 282–302. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Tozlovanu, M.; Manderville, R.; Peraica, M.; Castegnaro, M.; Stefanovic, V. New molecular and field evidences for the implication of mycotoxins but not aristolochic acid in Human Nephropathy and Urinary tract tumor. Mol. Nutr. Food Res. 2007, 51, 131–146. [Google Scholar]
- Pfohl-Leszkowicz, A. Ochratoxin A and aristolochic acid in the Nephropathies and Associated Urothelial Tract Tumours. Arh. Hig. Rada Toksikol. 2009, 60, 465–483. [Google Scholar] [CrossRef]
- Sabater-Vilar, M.; Maas, R.F.; Fink-Gremmels, J. Mutagenicity of commercial Monascus fermentation products and the role of citrinin contamination. Mutat. Res. 1999, 444, 7–16. [Google Scholar]
- Segvic-Klaric, M.; Zeljezic, D.; Domijan, A.M.; Peraica, M.; Pepeljnjak, S. Cytotoxicity, genotoxicity and apoptosis induced by ochratoxin A and citrinin in porcine kidney PK15 cells: Effects of single and combined mycotoxins. Toxicol. Lett. 2007, 172, 56. [Google Scholar]
- Iwahashi, H.; Kitagawa, E.; Suzuki, Y.; Ueda, Y.; Ishizawa, Y.; Nobumasa, H.; Kuboki, Y.; Hosoda, H.; Iwahashi, Y. Evaluation of toxicity of the mycotoxin citrinin using yeast ORF DNA microarray and oligo DNA microarray. BMC Genom. 2007, 8, 95. [Google Scholar]
- Pfohl-Leszkowicz, A.; Molinié, A.; Tozlovanu, M. Manderville R.A. Combined Toxic Effects of Ochratoxin A and Citrinin, in Vitro and in Vivo. In Food Contaminants, Mycotoxins & Food Allergen; Siantar, D.P., Trucksess, M.W., Scott, P.M., Herman, E.M., Eds.; ACS Symposium: Washington, DC, USA, 2008; Volume 1001, pp. 56–60. [Google Scholar]
- Sweeney, M.J.; Dobson, A.D. Mycotoxin production by Aspergillus, Fusarium and Penicillium species. Int. J. Food Microbiol. 1998, 43, 141–158. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Lund, F.; Elmholt, S. Ochratoxin A producing Penicillium verrucosum isolates from cereals reveal large AFLP fingerprinting variability. J. Appl. Microbiol. 2005, 98, 684–692. [Google Scholar] [CrossRef]
- Hackbart, H.C.S.; Prietto, L.; Primel, E.G.; Garda-Buffon, J.; Badiale-Furlong, E. Simultaneous extraction and detection of ochratoxin A and citrinin in rice. J. Braz. Chem. Soc. 2012, 23, 103–109. [Google Scholar] [CrossRef]
- Tokusoglu, Ö.; Bozoglu, F. Citrinin risk in black and green table olives: Simultaneous determination with ochratoxin-a by optimized extraction and IAC-HPLC-FD. Ital. J. Food Sci. 2010, 22, 284–291. [Google Scholar]
- El Adlouni, C.; Tozlovanu, M.; Natman, F.; Faid, M.; Pfohl-Leszkowicz, A. Preliminary data on the presence of mycotoxins (ochratoxin A, citrinin, aflatoxin B1) in black olives “greek styles” of Moroccan origin. Mol. Nutr. Food Res. 2006, 50, 507–512. [Google Scholar] [CrossRef]
- Vrabcheva, T.; Usleber, E.; Dietrich, R.; Märtlbauer, E. Co-occurrence of ochratoxin A and citrinin in cereals from bulgarian villages with a history of Balkan endemic nephropathy. J. Agric. Food Chem. 2000, 48, 2483–2488. [Google Scholar]
- Polisenska, I.; Pfohl-Leszkowicz, A.; Hadjeba, K.; Dohnal, V.; Jirsa, O.; Denesova, O.; Jezkova, A.; Macharackova, P. Occurrence of ochratoxin A and citrinin in Czech cereals and comparison of two HPLC methods for ochratoxin A detection. Food Addit. Contam. 2010, 27, 1545–1557. [Google Scholar] [CrossRef]
- Manderville, R.; Pfohl-Leszkowicz, A. Bioactivation and DNA Adduction as a Rationale for Ochratoxin A Carcinogenesis. World Mycot. J. 2008, 1, 357–367. [Google Scholar]
- Trucksess, M.W. Official Methods of Analysis of AOAC International; Horwitz, W., Ed.; AOAC International: Gaithersburg, MD, USA, 2003; p. 56. [Google Scholar]
- Tabata, S.; Iida, K.; Kimura, K.; Iwasaki, Y.; Nakazato, M.; Kamata, K.; Hirokado, M. Simultaneous determination of ochratoxin A, B and citrinin in foods by HPLC-FL and LC/MS/MS. J. Food Hyg. Soc. Jpn. 2008, 49, 100–105. [Google Scholar] [CrossRef]
- Monaci, L.; Palmisano, F. Determination of ochratoxin A in pig tissues by liquid-liquid extraction and clean-up and high-performance liquid chromatography. Anal. Bioanal. Chem. 2004, 378, 1777. [Google Scholar] [CrossRef]
- González-Peñas, E.; Leache, C.; Bizcarte, M.; Obanos, A.P. Determination of ochratoxin A in wine using liquid-phase microextraction combined with liquid chromatography with fluorescence detection. J. Chromatogr. A 2004, 1025, 163. [Google Scholar]
- Senyuva, H.Z.; Gilbert, J.; Ozcan, S.; Ulken, U. Survey for co-occurrence of ochratoxin A and aflatoxin B1 in dried figs in Turkey by using a single laboratory-validated alkaline extraction method for ochratoxin A. J. Food Prot. 2005, 68, 1512. [Google Scholar]
- Prieto-Simón, B.; Campàs, M.; Marty, J.-L.; Noguer, T. Novel highly-performing immunosensor-based strategy for ochratoxin A detection in wine samples. Biosens. Bioelectron. 2008, 23, 995–1002. [Google Scholar] [CrossRef]
- Rhone Diagnostics technologies. Cereal Ochratoxin A Extraction Method, Application Note for Analysis of Ochratoxin A in Cereal Using Sodium Bicarbonate Extraction in Conjunction with Ochraprep®; Application note Ref No. A9-P14.V1; Rhone Diagnostics Technologies: Saint-Didier Mont d’Or, France, 1999. [Google Scholar]
- Entwilse, A.C.; Williams, A.C.; Man, P.J.; Slack, P.T.; Gilbert, J. Liquid chromatography method with immunoaffinity column clean-up for determination of ochratoxin A in barley: Collaborative study. J. Assoc. Off. Anal. Chem. 2000, 83, 1377–138. [Google Scholar]
- Fernandes, P.J.; Barros, N.; Camara, J.S. A Survey of the occurrence of ochratoxin A in Madeira wines based on a modified QuEChERS extraction procedure combined with Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry. Food Res. Int. 2013, 54, 293–301. [Google Scholar] [CrossRef]
- Cao, J.; Zhou, S.; Kong, W.; Wan, L.; Yang, M. Molecular imprinted polymer-based solid phase clean-up for analysis of ochratoxin A in ginger and LC-MS/MS confirmation. Food Control 2013, 33, 337–343. [Google Scholar]
- Frenich, A.G.; Romero-González, R.; Gómez-Pérez, M.L.; Vidal, J.L.M. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A. 2011, 1218, 4349–4356. [Google Scholar]
- Radi, A.-E.; Muñoz-Berbel, X.; Lates, V.; Marty, J.-L. Label free impedimetric immunosensor for sensitive detection of ochratoxin A. Biosens. Bioelectron. 2008, 24, 1888–1892. [Google Scholar]
- Li, Y.; Wu, H.; Guo, L.; Zheng, Y.; Guo, Y. Microsphere-based flow cytometric immunoassay for the determination of citrinin in red yeast rice. Food Chem. 2012, 134, 2540–2545. [Google Scholar] [CrossRef]
- Yuan, J.; Deng, D.; Lauren, D.R.; Aguilar, M.; Wu, Y. Surface plasmon resonance biosensor for the detection of ochratoxin A in cereals and beverages. Anal. Chim. Acta 2009, 656, 63–71. [Google Scholar] [CrossRef]
- Arévalo, F.J.; Granero, A.M.; Fernández, H.; Raba, J.; Zón, M.A. Citrinin (CIT) determination in rice samples using a micro fluidic electrochemical immunosensor. Talanta 2011, 83, 966–973. [Google Scholar] [CrossRef]
- Liu, X.-P.; Deng, Y.-J.; Jin, X.-Y.; Chen, L.-G.; Juang, J.-H.; Shen, G.-L.; Yu, R.-Q. Ultrasensitive electrochemical immunosensor for ochratoxin A using gold colloid-mediated hapten immobilization. Anal. Biochem. 2009, 389, 63–68. [Google Scholar]
- Zachetti, V.G.; Granero, A.M.; Robledo, S.N.; Zon, M.A.; Fernández, H. Development of an amperometric biosensor based on peroxidases to quantify citrinin in rice samples. Bioelectrochemistry 2013, 91, 37–43. [Google Scholar]
- Klarić, M.S.; Cvetnić, Z.; Pepeljnjak, S.; Kosalec, I. Co-occurrence of aflatoxins, ochratoxin A, fumonisins, and zearalenone in cereals and feed, determined by competitive direct enzyme-linked immunosorbent assay and thin-layer chromatography. Arh. Hig. Rada Toksikol. 2009, 60, 427–434. [Google Scholar]
- Li, Y.; Wang, Y.; Guo, Y. Preparation of synthetic antigen and monoclonal antibody for indirect competitive ELISA of citrinin. Food Agric. Immunol. 2012, 23, 145–156. [Google Scholar]
- Bazin, I.; Nabais, E.; Lopez-Ferber, M. Rapid Visual Tests: Fast and Reliable Detection of Ochratoxin A. Toxins 2010, 2, 2230–2241. [Google Scholar] [CrossRef]
- Visconti, A.; Pascale, M.; Centonze, G. Determination of ochratoxin A in wine by means of immunoaffinity column clean-up and high-performance liquid chromatography. J. Chromatogr. A 1999, 864, 89–101. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Molinie, A.; Castegnaro, M. Underestimation of Fumonisin B1 and Ochratoxin A, from Complex Matrices by Use of Immunoaffinity Columns. In Mycotoxins and Phycotoxins; Njapau, H., Trujillio, S., van Egmond, H.P., Park, D.L., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2006; pp. 83–89. [Google Scholar]
- Castegnaro, M.; Tozlovanu, M.; Wild, C.; Molinie, A.; Sylla, A.; Pfohl-Leszkowicz, A. Advantages and drawbacks of immunoaffinity columns in analysis of mycotoxins in food. Mol. Nutr. Food Res. 2006, 50, 480–481. [Google Scholar] [CrossRef]
- Tozlovanu, M.; Pfohl-Leszkowicz, A. Ochratoxin A in roasted coffee purchased in French supermarket-Transfer in coffee beverage/Comparison of several methods of analysis. Toxins 2010, 2, 1928–1942. [Google Scholar] [CrossRef]
- Frenette, C.; Paugh, R.; Tozlovanu, M.; Juzio, M.; Pfohl-Leszkowicz, A.; Manderville, R. Structure-activity relationships for the fluorescence of ochratoxin A: Insight for detection of ochratoxin A metabolites. Anal. Chim. Acta 2008, 617, 153–161. [Google Scholar]
- Valenta, H. Chromatographic methods for the determination of ochratoxin A. J. Chrom. A 1998, 815, 75–92. [Google Scholar] [CrossRef]
- Verrone, R.; Catucci, L.; Cosma, P.; Fini, P.; Agostiano, A.; Lippolis, V.; Pascale, M. Effect of β-cyclodextrin on spectroscopic properties of ochratoxin A in aqueous solution. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 475–479. [Google Scholar]
- Hirota, M.; Mehta, A.; Yoneyama, K.; Kitabatake, N. A major decomposition product, citrinin H2, from citrinin on heating with moisture. Biosci. Biotechnol. Biochem. 2002, 66, 1617. [Google Scholar]
- Faucet-Marquis, V.; Pont, F.; Størmer, F.; Rizk, T.; Castegnaro, M.; Pfohl-Leszkowicz, A. Evidence of a new dechlorinated OTA derivative formed in opossum kidney cell cultures after pre-treatment by modulators of glutathione pathways. Correlation with DNA adducts formation. Mol. Nutr. Food Res. 2006, 50, 531–542. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bazin, I.; Faucet-Marquis, V.; Monje, M.-C.; El Khoury, M.; Marty, J.-L.; Pfohl-Leszkowicz, A. Impact of pH on the Stability and the Cross-Reactivity of Ochratoxin A and Citrinin. Toxins 2013, 5, 2324-2340. https://doi.org/10.3390/toxins5122324
Bazin I, Faucet-Marquis V, Monje M-C, El Khoury M, Marty J-L, Pfohl-Leszkowicz A. Impact of pH on the Stability and the Cross-Reactivity of Ochratoxin A and Citrinin. Toxins. 2013; 5(12):2324-2340. https://doi.org/10.3390/toxins5122324
Chicago/Turabian StyleBazin, Ingrid, Virginie Faucet-Marquis, Marie-Carmen Monje, Micheline El Khoury, Jean-Louis Marty, and Annie Pfohl-Leszkowicz. 2013. "Impact of pH on the Stability and the Cross-Reactivity of Ochratoxin A and Citrinin" Toxins 5, no. 12: 2324-2340. https://doi.org/10.3390/toxins5122324
APA StyleBazin, I., Faucet-Marquis, V., Monje, M.-C., El Khoury, M., Marty, J.-L., & Pfohl-Leszkowicz, A. (2013). Impact of pH on the Stability and the Cross-Reactivity of Ochratoxin A and Citrinin. Toxins, 5(12), 2324-2340. https://doi.org/10.3390/toxins5122324