Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Western Blotting
2.4. RT-PCR Assay to Detect XBP1 Splicing
2.5. Quantitative(q) RT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5' to 3') | Reverse Primer (5' to 3') |
|---|---|---|
| Human spliced XBP-1 | TGCTGAGTCCGCAGCAGG | CGCCAGAATCCATGGGGAGA |
| Human total XBP-1 | GCAAGCGACAGCGCCT | TTTTCAGTTTCCTCCTCAGCG |
| Human β-actin | AATGTGGCCGAGGACTTTGATTGC | AGGATGGCAAGGGACTTCCTGTAA |
| Bovine spliced XBP-1 | TGCTGAGTCCGCAGCAGG | CATCAGAGTCCATGGGGAGA |
| Bovine total XBP-1 | GCAAGCGACAGCGCCT | TTTTCAGTTTCCTCCGCAGCG |
| Bovine cyclophilin | GAGCACTGGACAGAAAGGATTTGG | TGAAGTCACCAGCCTGGCACATAA |
2.6. Protein Synthesis Inhibition
2.7. Statistical Analysis
3. Results
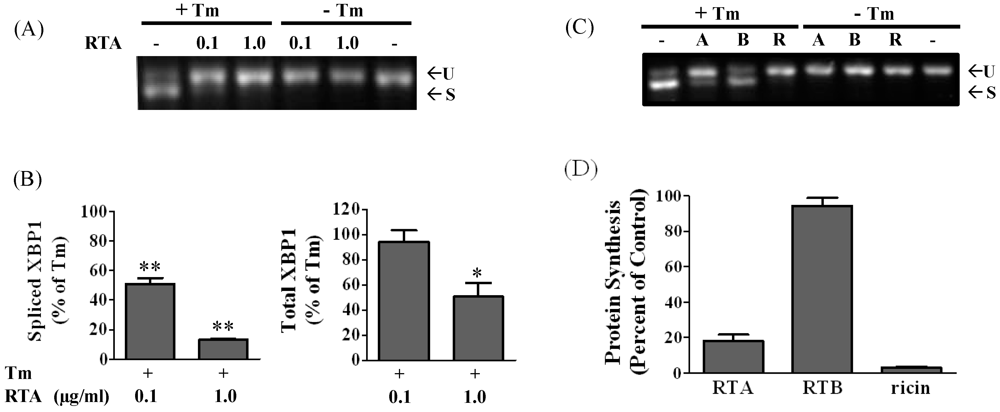
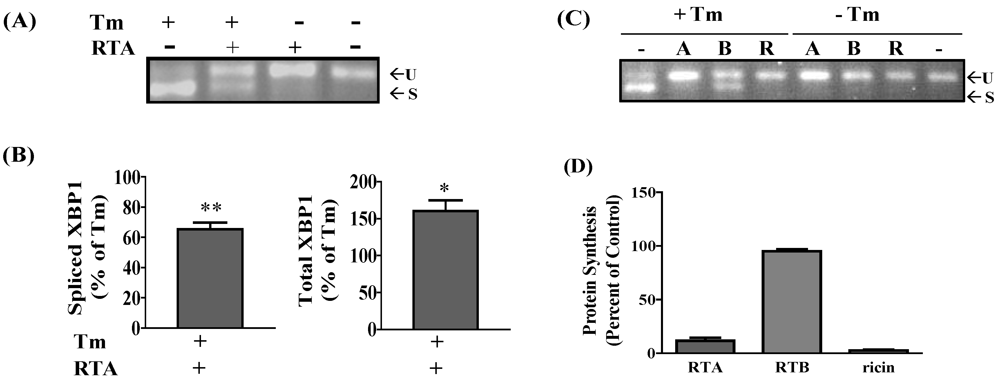
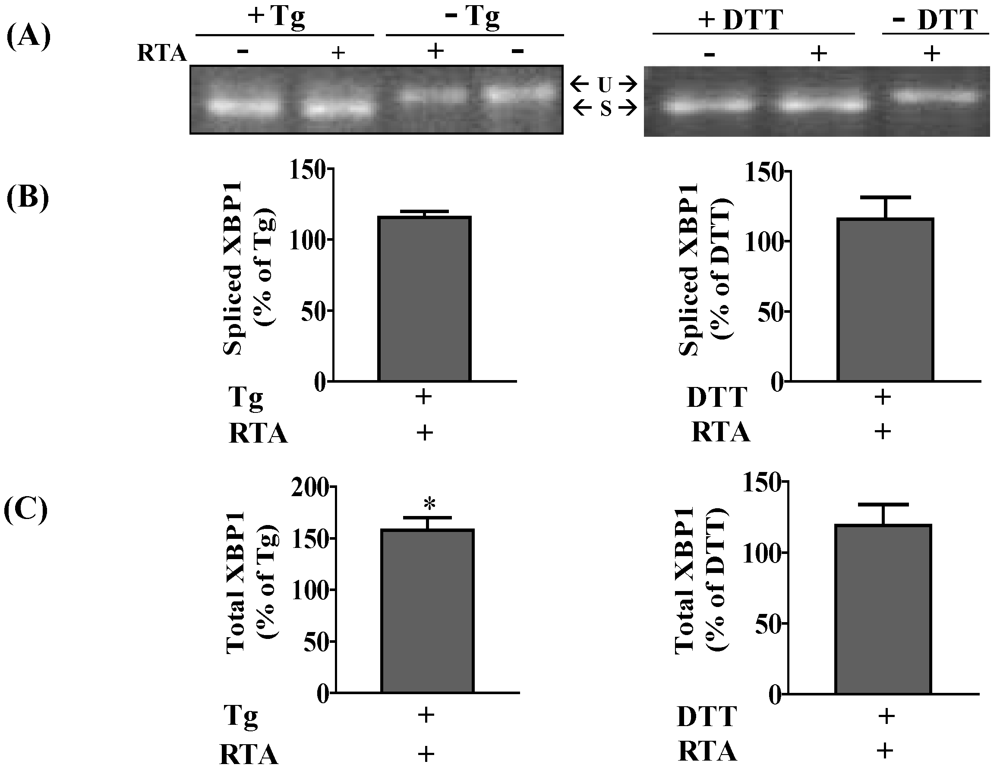
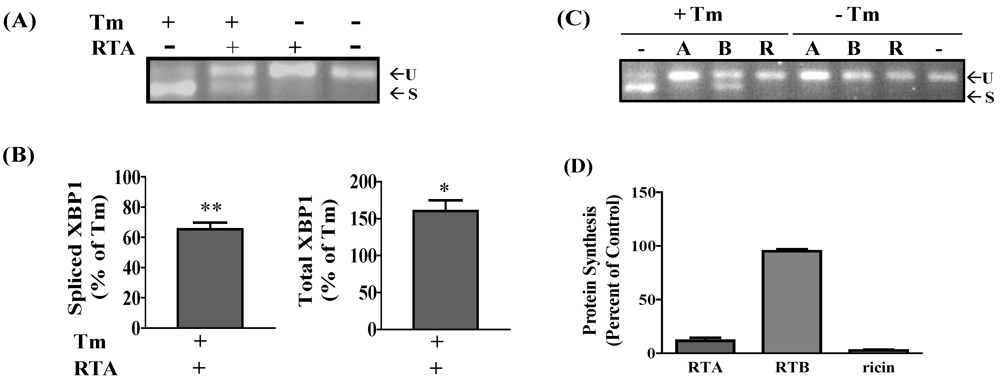
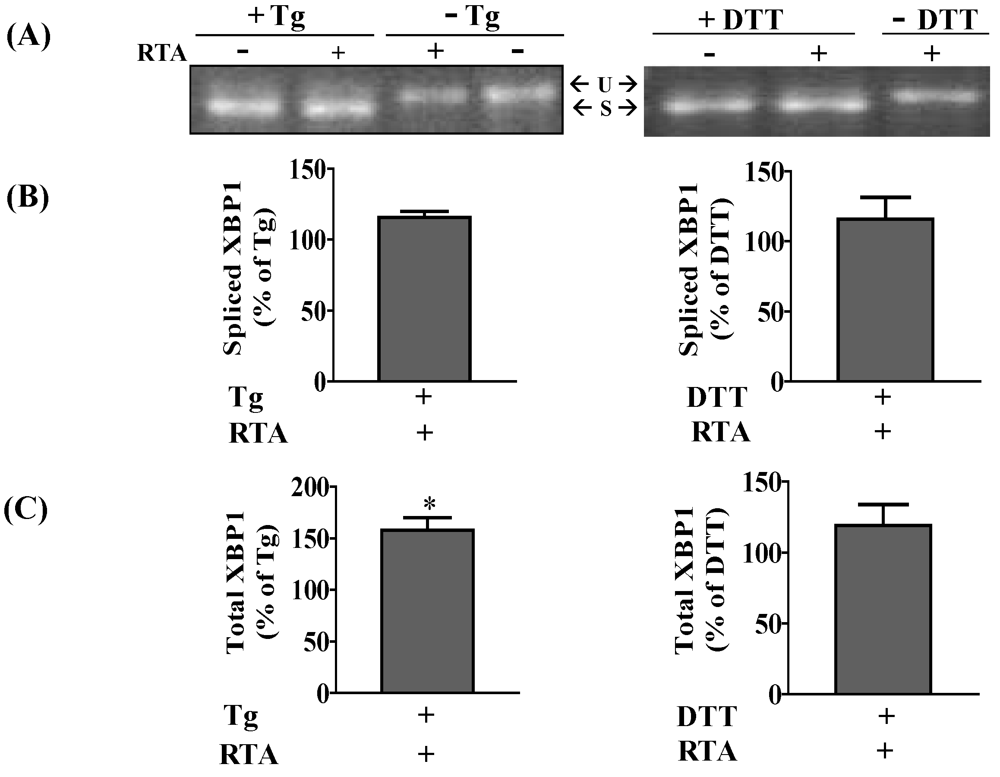
3.1. RTA Inhibits XBP1 Splicing in Mammalian Cells
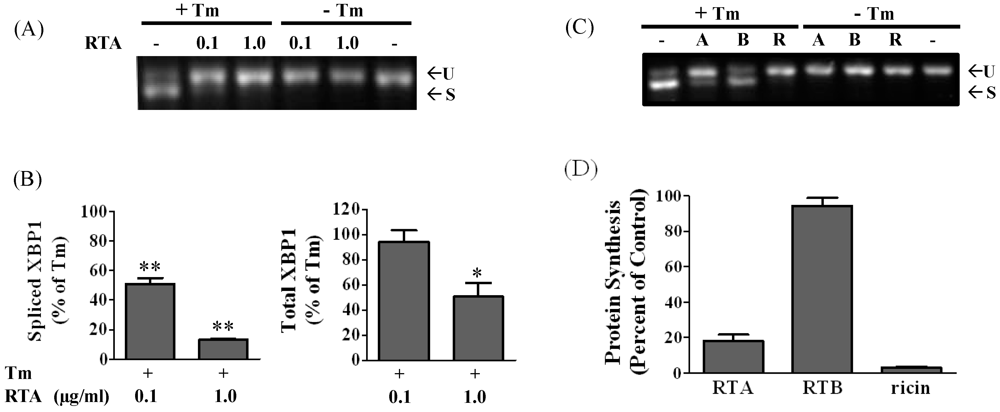
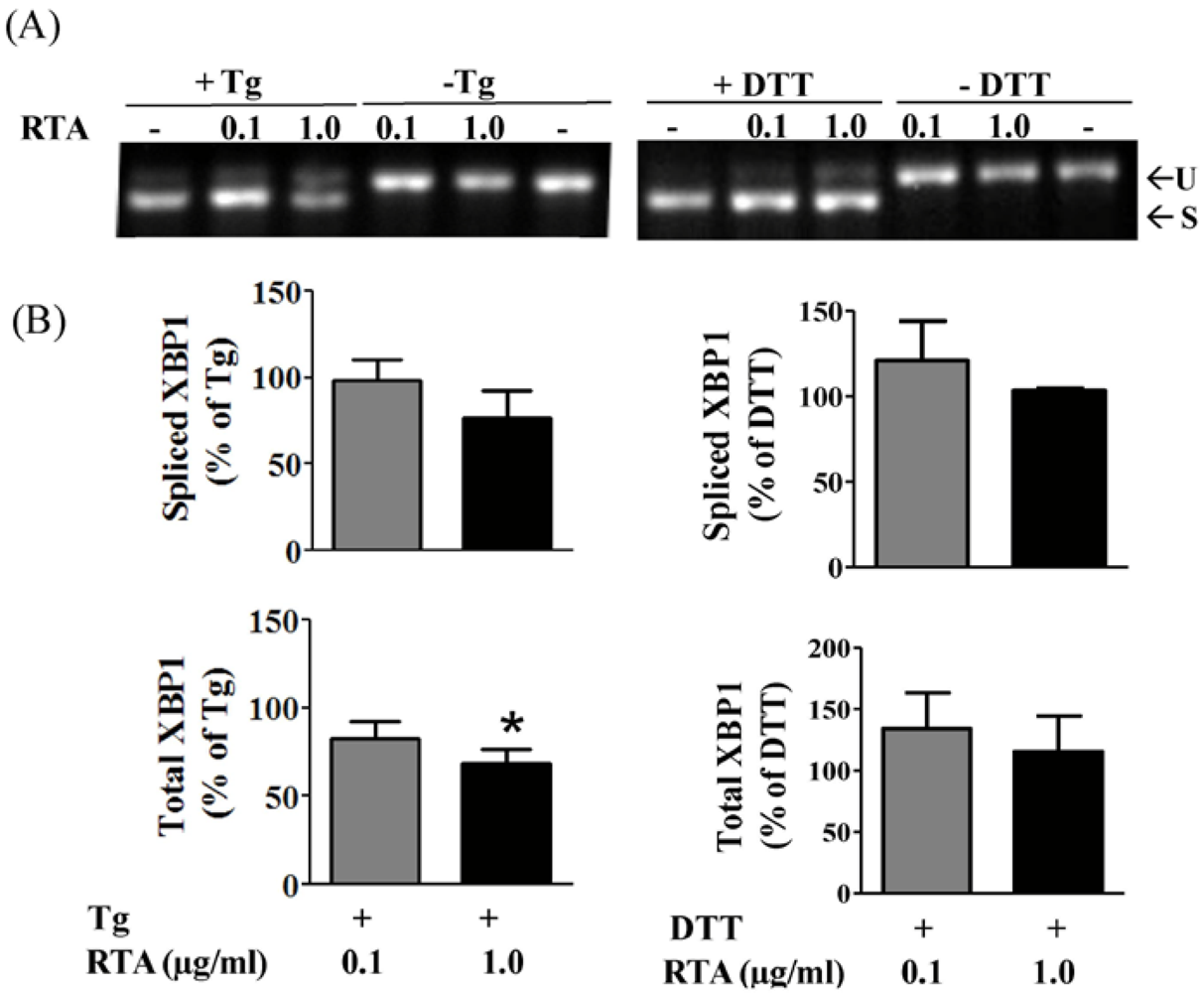
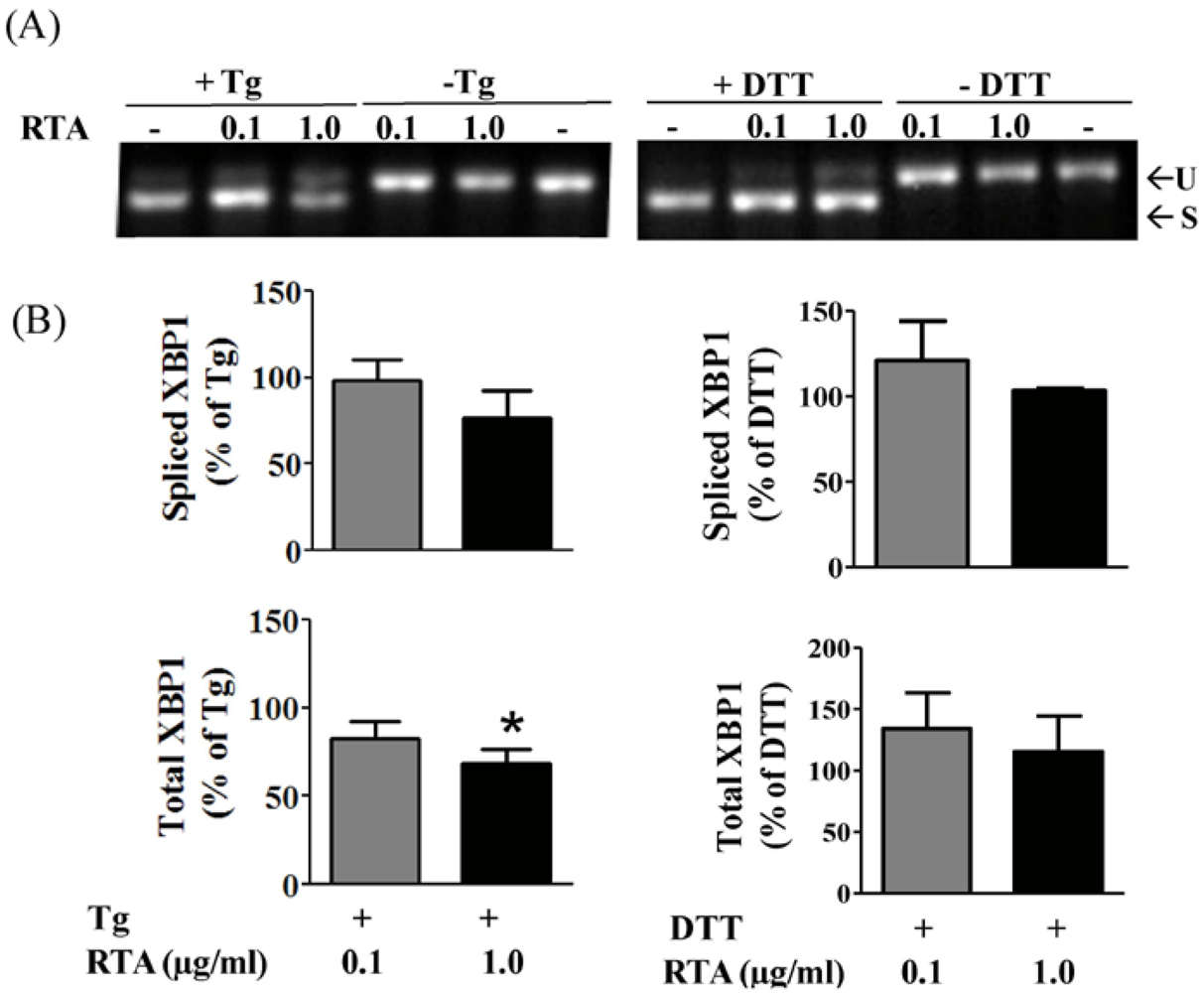
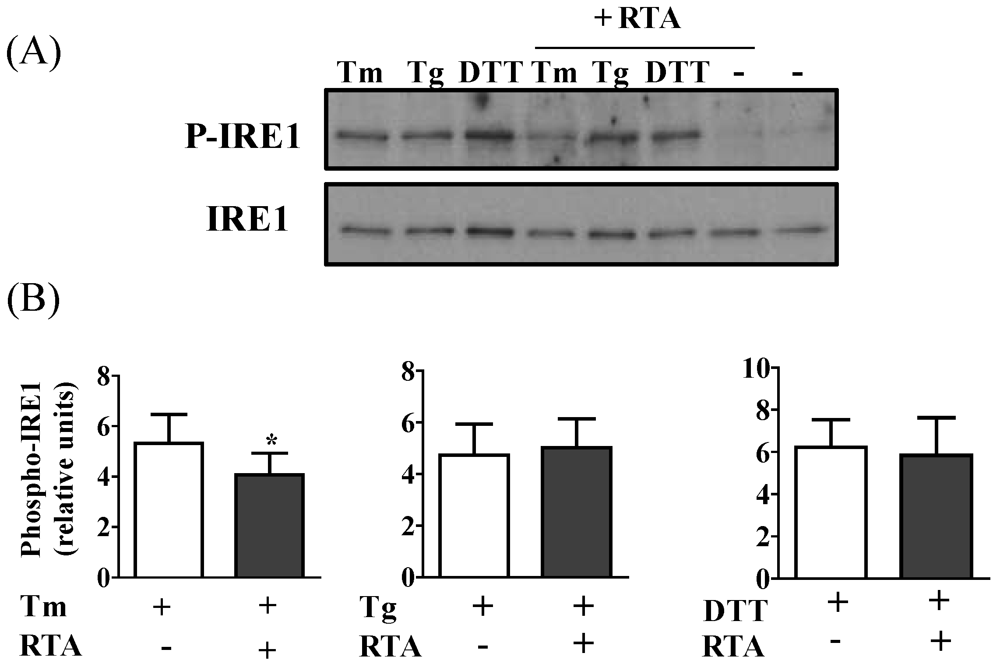
3.2. RTA Inhibits Tm-Induced Phosphorylation of IRE1
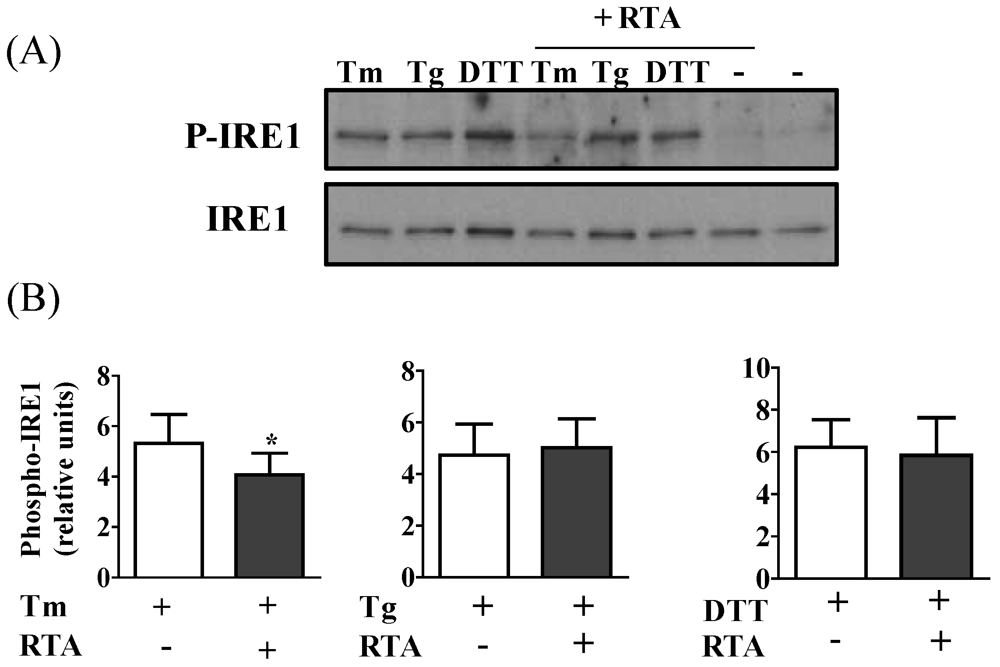
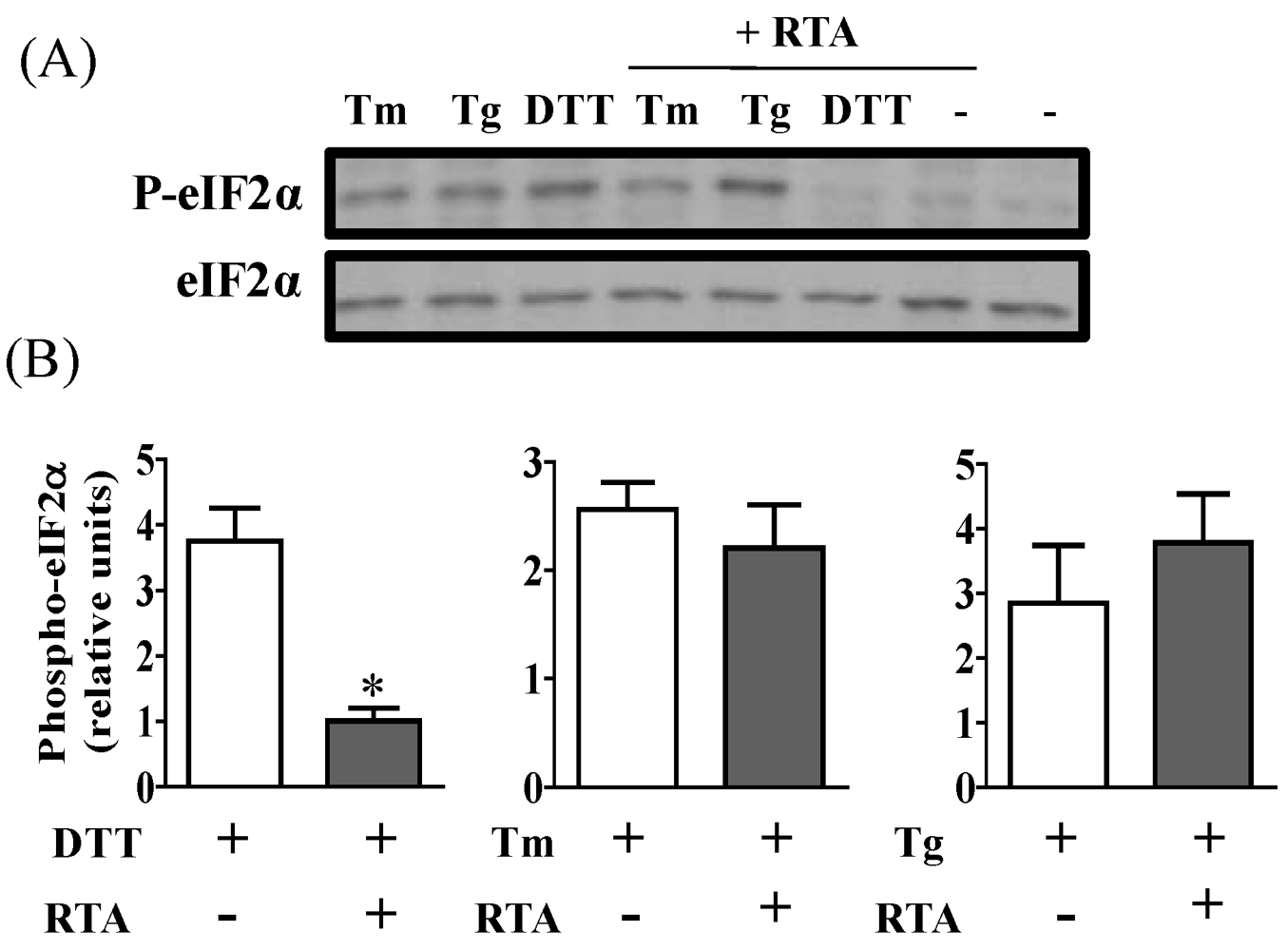
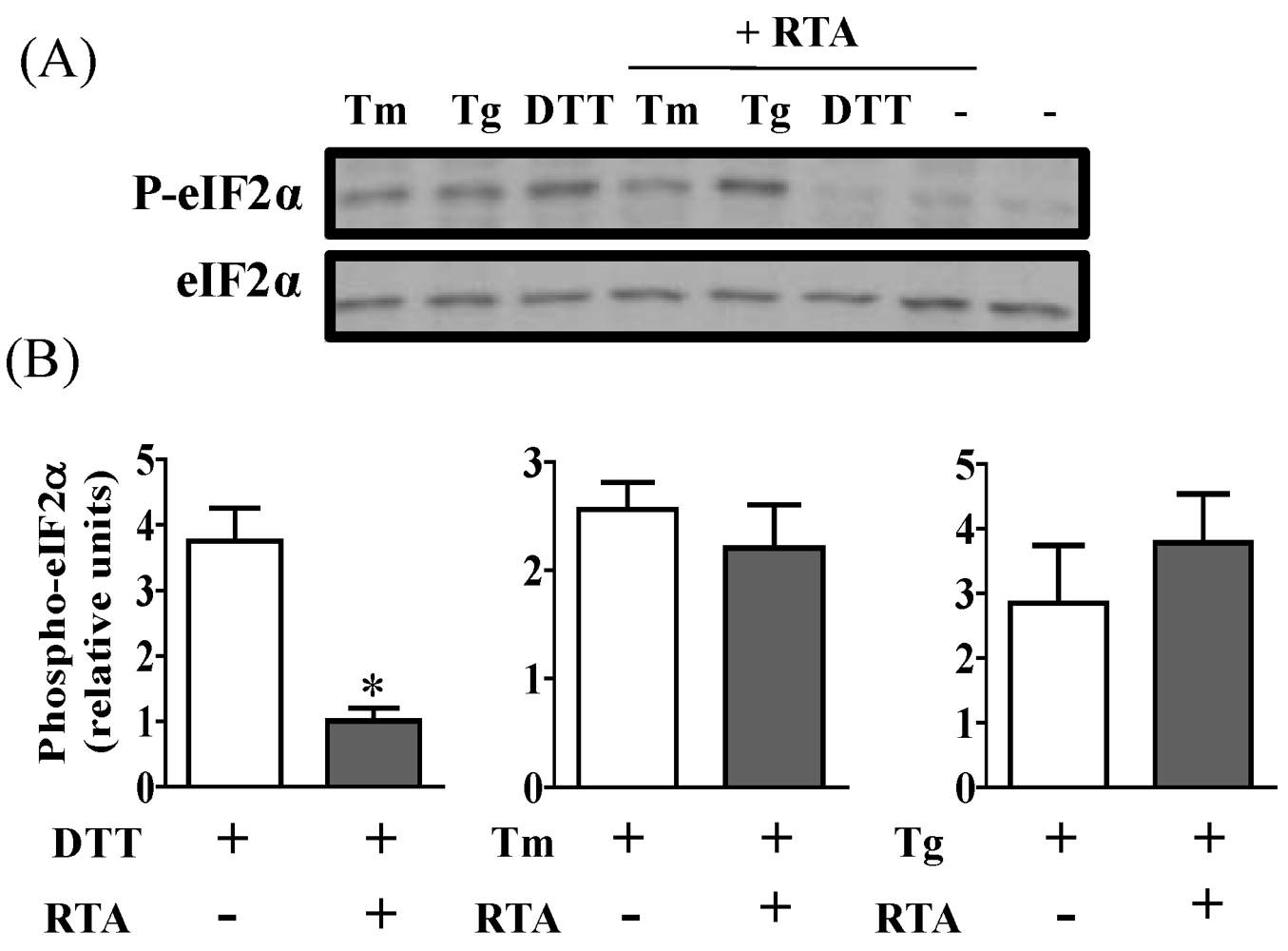
3.3. RTA Inhibits DTT-Induced Phosphorylation of eIF2α
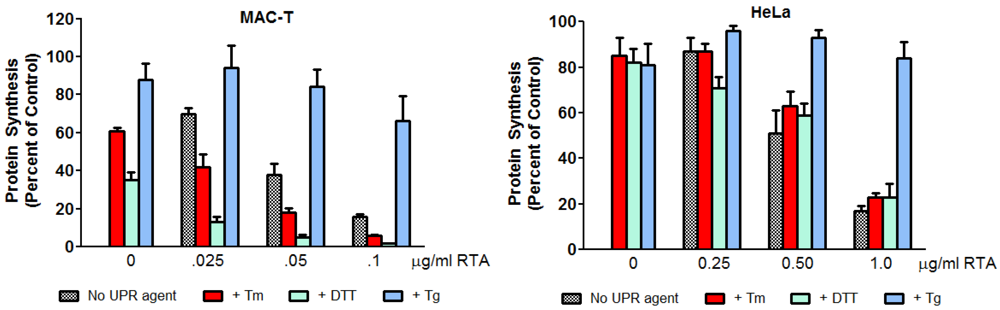
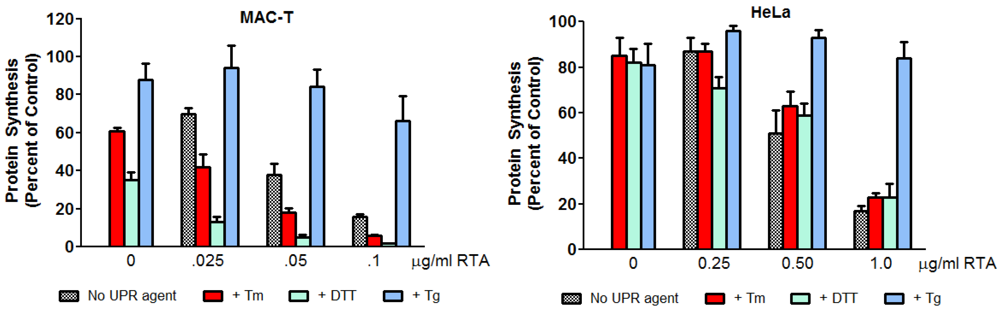
3.4. Inhibition of ER Stress by RTA Differentially Affects Protein Synthesis Inhibition in MAC-T and HeLa Cells
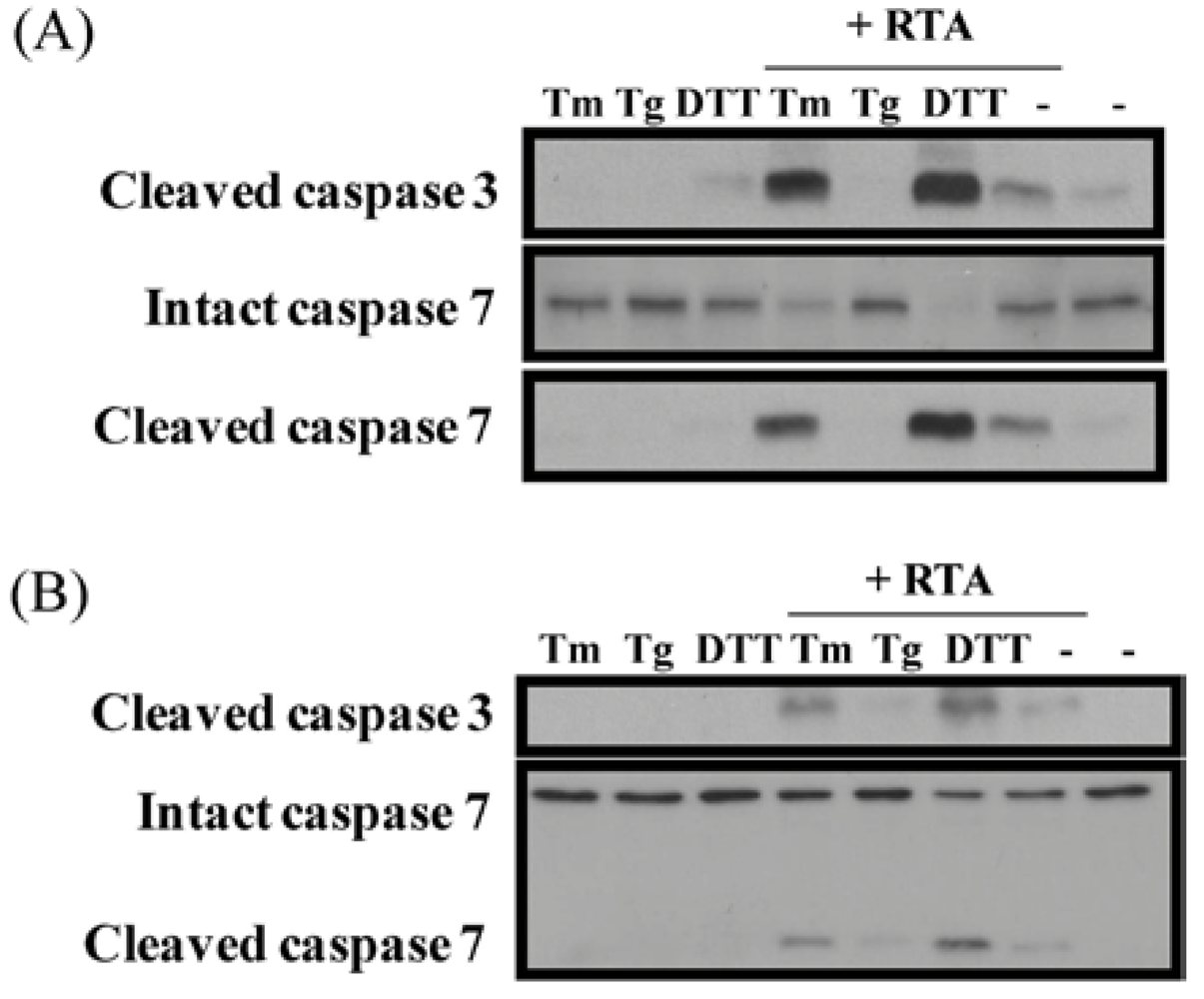
3.5. Inhibition of ER Stress Sensitizes Cells to RTA-Induced Caspase Activation
4. Discussion
5. Conclusions
Acknowledgements
References
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. J. Am. Med. Assoc. 2005, 294, 2342–2351. [Google Scholar]
- Xu, F.; Leadon, S.A.; Yu, Y.; Boyer, C.M.; O’briant, K.; Ward, K.; Mcwatters, A.; Zhao, X.; Bae, D.S.; Desombre, K.; et al. Synergistic interaction between anti-p185HER-2 ricin A chain immunotoxins and radionuclide conjugates for inhibiting growth of ovarian and breast cancer cells that overexpress HER-2. Clin. Cancer Res. 2000, 6, 3334–3341. [Google Scholar] [PubMed]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar] [PubMed]
- Olsnes, S.; Kozlov, J.V. Ricin. Toxicon 2001, 39, 1723–1728. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosen, A.; Solda, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004, 67, 1721–1731. [Google Scholar]
- Day, P.J.; Pinheiro, T.J.; Roberts, L.M.; Lord, J.M. Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 2002, 41, 2836–2843. [Google Scholar]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 274, 34443–34449. [Google Scholar]
- Slominska-Wojewodzka, M.; Gregers, T.F.; Walchli, S.; Sandvig, K. EDEM is involved in retrotranslocation of ricin from the endoplasmic reticulum to the cytosol. Mol. Biol. Cell 2006, 17, 1664–1675. [Google Scholar]
- Li, S.; Spooner, R.A.; Allen, S.C.; Guise, C.P.; Ladds, G.; Schnoder, T.; Schmitt, M.J.; Lord, J.M.; Roberts, L.M. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol. Biol. Cell 2010, 21, 2543–2554. [Google Scholar]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Hohfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar]
- Moazed, D.; Robertson, J.M.; Noller, H.F. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 1988, 334, 362–364. [Google Scholar]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Holmberg, L.; Nygard, O. Depurination of A4256 in 28 S rRNA by the ribosome-inactivating proteins from barley and ricin results in different ribosome conformations. J. Mol. Biol. 1996, 259, 81–94. [Google Scholar]
- Li, X.P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar]
- Alford, S.C.; Pearson, J.D.; Carette, A.; Ingham, R.J.; Howard, P.L. Alpha-sarcin catalytic activity is not required for cytotoxicity. BMC Biochem. 2009, 10, 9. [Google Scholar]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell. Dev. Biol. 2007, 18, 716–731. [Google Scholar]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Meth. Mol. Biol. 2009, 559, 191–204. [Google Scholar]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar]
- Horrix, C.; Raviv, Z.; Flescher, E.; Voss, C.; Berger, M.R. Plant ribosome-inactivating proteins type II induce the unfolded protein response in human cancer cells. Cell. Mol. Life Sci. 2010, 68, 1269–1281. [Google Scholar]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar]
- Parikh, B.A.; Tortora, A.; Li, X.P.; Tumer, N.E. Ricin inhibits activation of the unfolded protein response by preventing splicing of the HAC1 mRNA. J. Biol. Chem. 2008, 283, 6145–6153. [Google Scholar]
- Huynh, H.T.; Robitaille, G.; Turner, J.D. Establishment of bovine mammary epithelial cells (MAC-T): an in vitro model for bovine lactation. Exp. Cell Res. 1991, 197, 191–199. [Google Scholar] [CrossRef]
- Jetzt, A.E.; Cheng, J.S.; Tumer, N.E.; Cohick, W.S. Ricin A-chain requires c-Jun N-terminal kinase to induce apoptosis in nontransformed epithelial cells. Int. J. Biochem. Cell Biol. 2009, 41, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, K.; Kaufman, R.J. The unfolded protein response-a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar]
- Koumenis, C.; Wouters, B.G. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Canc. Res. 2006, 4, 423–436. [Google Scholar]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: Co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar]
- Kimata, Y.; Oikawa, D.; Shimizu, Y.; Ishiwata-Kimata, Y.; Kohno, K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell Biol. 2004, 167, 445–456. [Google Scholar]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar]
- Gill, D.L.; Waldron, R.T.; Rys-Sikora, K.E.; Ufret-Vincenty, C.A.; Graber, M.N.; Favre, C.J.; Alfonso, A. Calcium pools, calcium entry, and cell growth. Biosci. Rep. 1996, 16, 139–157. [Google Scholar] [CrossRef]
- Zhou, X.X.; Ji, F.; Zhao, J.L.; Cheng, L.F.; Xu, C.F. Anti-cancer activity of anti-p185HER-2 ricin A chain immunotoxin on gastric cancer cells. J. Gastroenterol. Hepatol. 2010, 25, 1266–1275. [Google Scholar]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Canc. Res. 2005, 3, 597–605. [Google Scholar]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, C.-T.; Jetzt, A.E.; Cheng, J.-S.; Cohick, W.S. Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells. Toxins 2011, 3, 453-468. https://doi.org/10.3390/toxins3050453
Wang C-T, Jetzt AE, Cheng J-S, Cohick WS. Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells. Toxins. 2011; 3(5):453-468. https://doi.org/10.3390/toxins3050453
Chicago/Turabian StyleWang, Chao-Ting, Amanda E. Jetzt, Ju-Shun Cheng, and Wendie S. Cohick. 2011. "Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells" Toxins 3, no. 5: 453-468. https://doi.org/10.3390/toxins3050453
APA StyleWang, C.-T., Jetzt, A. E., Cheng, J.-S., & Cohick, W. S. (2011). Inhibition of the Unfolded Protein Response by Ricin A-Chain Enhances Its Cytotoxicity in Mammalian Cells. Toxins, 3(5), 453-468. https://doi.org/10.3390/toxins3050453