Increased Cardiovascular Mortality in Hemodialysis: The Role of Chronic Inflammation, Complement Activation, and Non-Biocompatibility

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
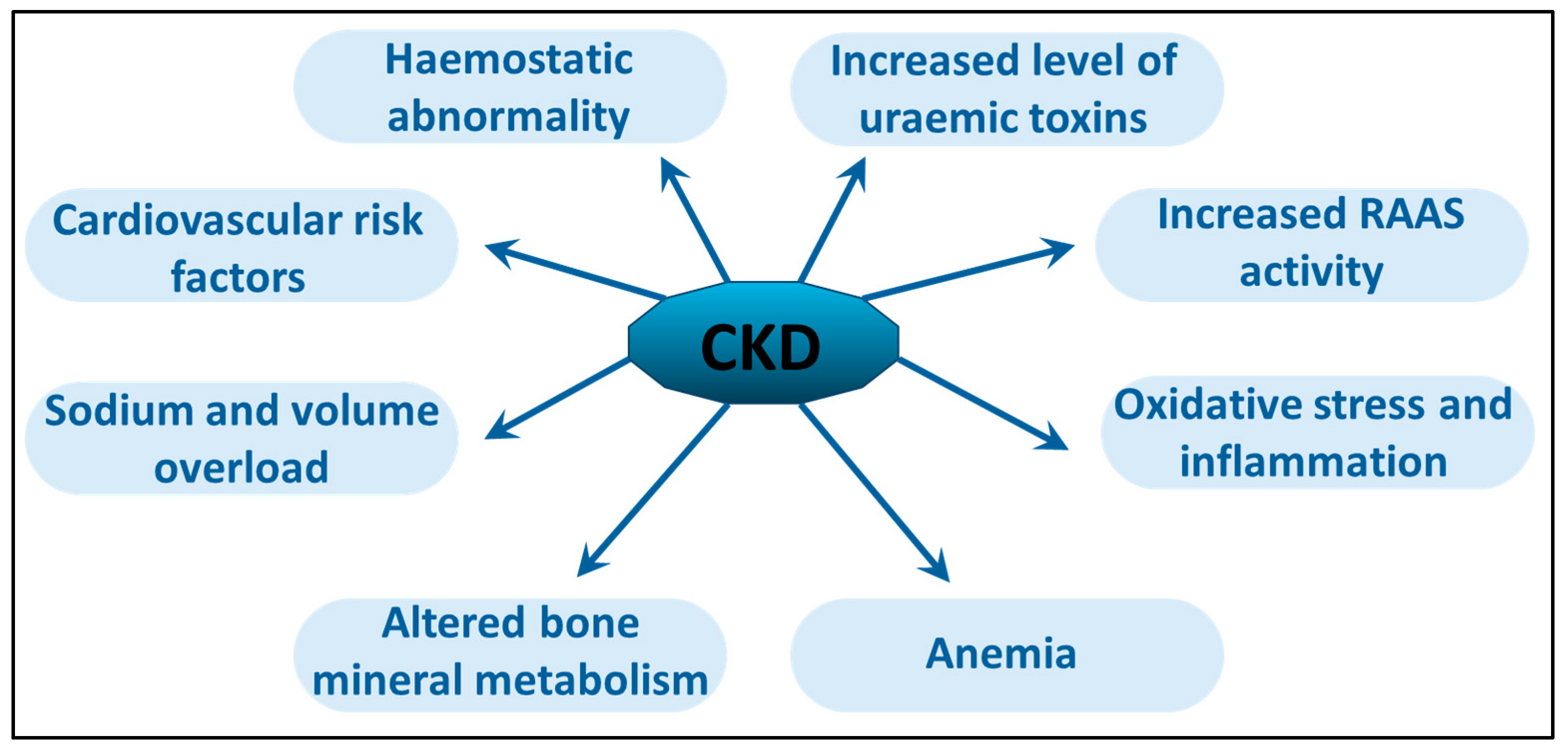
4. Chronic Uremic Inflammation
5. Cardiovascular Mortality in HD Patients
6. Hemodialysis Treatment-Related Inflammation
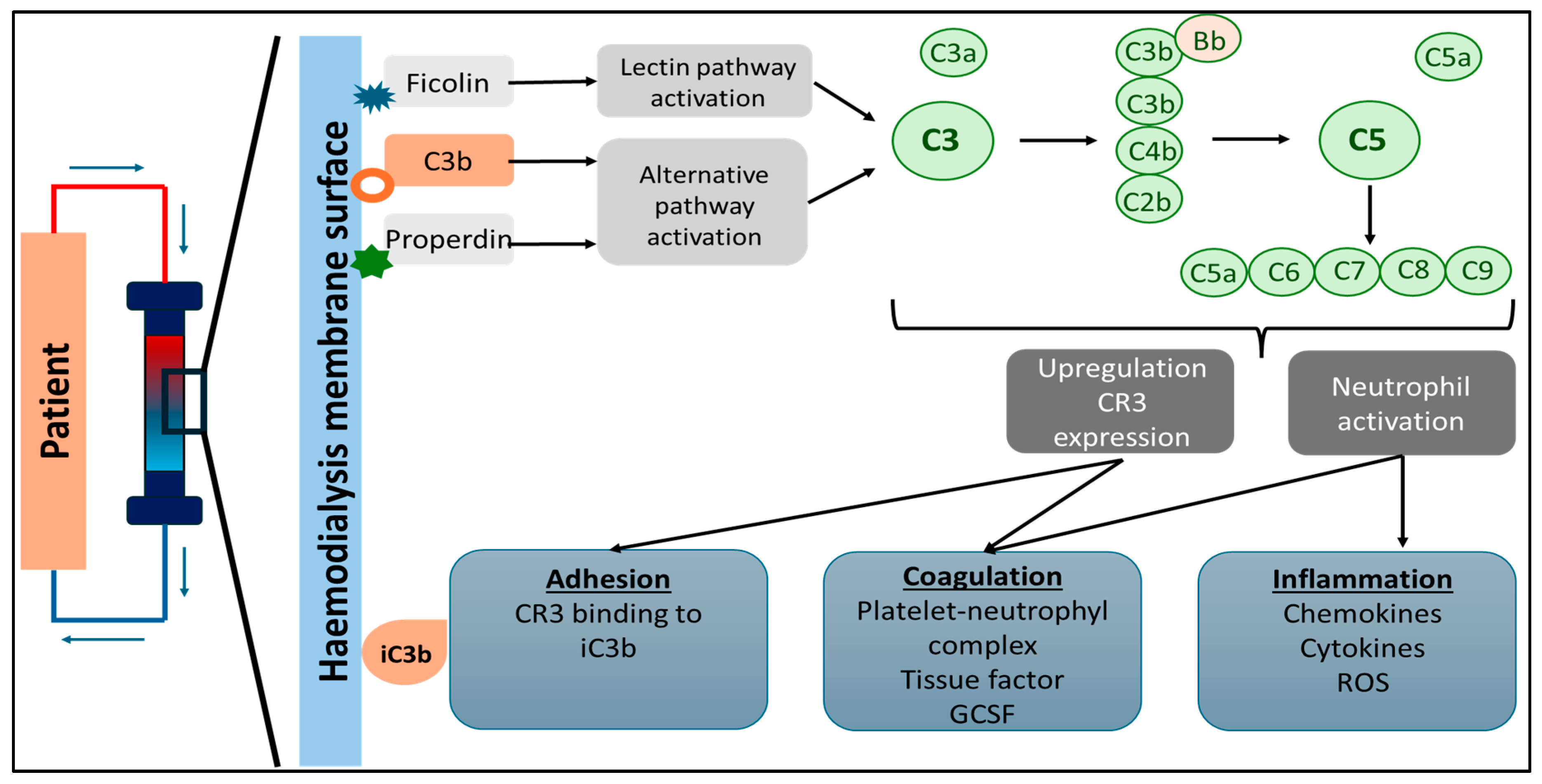
7. Complement System Activation in Hemodialysis
8. Hemodialysis Procedure-Related Reactions
9. Discussion
10. Conclusions
11. Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, J.; Bodegard, J.; Bollmann, A.; Vervloet, M.G.; Mark, P.B.; Karasik, A.; Taveira-Gomes, T.; Botana, M.; Birkeland, K.I.; Thuresson, M.; et al. Prevalence, outcomes, and cost of chronic kidney disease in a contemporary population of 2.4 million patients from 11 countries: The CaReMe CKD study. Lancet Reg. Health Eur. 2022, 20, 100438. [Google Scholar] [CrossRef]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32, S112–S119. [Google Scholar] [CrossRef]
- Neovius, M.; Jacobson, S.H.; Eriksson, J.K.; Elinder, C.G.; Hylander, B. Mortality in chronic kidney disease and renal replacement therapy: A population-based cohort study. BMJ Open 2014, 4, e004251. [Google Scholar] [CrossRef]
- Shahbazi, F.; Doosti-Irani, A.; Soltanian, A.; Poorolajal, J. Global forecasting of chronic kidney disease mortality rates and numbers with the generalized additive model. BMC Nephrol. 2024, 25, 286. [Google Scholar] [CrossRef]
- Broers, N.J.; Cuijpers, A.C.; van der Sande, F.M.; Leunissen, K.M.; Kooman, J.P. The first year on haemodialysis: A critical transition. Clin. Kidney J. 2015, 8, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lukowsky, L.R.; Kheifets, L.; Arah, O.A.; Nissenson, A.R.; Kalantar-Zadeh, K. Patterns and predictors of early mortality in incident hemodialysis patients: New insights. Am. J. Nephrol. 2012, 35, 548–558. [Google Scholar] [CrossRef]
- Gu, L.; Xia, Z.; Qing, B.; Wang, W.; Chen, H.; Wang, J.; Chen, Y.; Gai, Z.; Hu, R.; Yuan, Y. Systemic Inflammatory Response Index (SIRI) is associated with all-cause mortality and cardiovascular mortality in population with chronic kidney disease: Evidence from NHANES (2001–2018). Front. Immunol. 2024, 15, 1338025. [Google Scholar] [CrossRef] [PubMed]
- Descamps-Latscha, B.; Jungers, P. New molecular aspects of chronic uraemia and dialysis-related immunocompetent cell activation. Nephrol. Dial. Transplant. 1996, 11 (Suppl. 2), 121–124. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Montero, R.; Gomez-Lopez, V.E.; Guerrero-Pavon, F.; Carmona-Munoz, A.; Romero-Saldana, M.; Ranchal-Sanchez, A.; Aljama-Garcia, P. Influence of Tunneled Hemodialysis-Catheters on Inflammation and Mortality in Dialyzed Patients. Int. J. Environ. Res. Public Health 2021, 18, 7605. [Google Scholar] [CrossRef] [PubMed]
- Tapolyai, M.B.; Czirok, S.; Szasz, M.; Petho, A.; Fulop, T. Prolonged use of dialysis catheters is associated with elevated chronic inflammatory markers: A single center case series. Ren. Fail. 2025, 47, 2478486. [Google Scholar] [CrossRef]
- Zsom, L.; Zsom, M.; Fulop, T.; Wells, C.; Flessner, M.F.; Eller, J.; Wollheim, C.; Hegbrant, J.; Strippoli, G.F. Correlation of treatment time and ultrafiltration rate with serum albumin and C-reactive protein levels in patients with end-stage kidney disease receiving chronic maintenance hemodialysis: A cross-sectional study. Blood Purif. 2010, 30, 8–15. [Google Scholar] [CrossRef]
- Li, M.; Xue, W.; Li, X.; Song, Y.; Liu, X.; Qin, L. Axl is related to inflammation in hemodialysis patients. Mol. Immunol. 2021, 133, 146–153. [Google Scholar] [CrossRef]
- Qureshi, A.R.; Alvestrand, A.; Divino-Filho, J.C.; Gutierrez, A.; Heimburger, O.; Lindholm, B.; Bergstrom, J. Inflammation, malnutrition, and cardiac disease as predictors of mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 1), S28–S36. [Google Scholar] [CrossRef]
- Xiang, F.; Cao, X.; Chen, X.; Zhang, Z.; Ding, X.; Zou, J.; Shen, B. Decreased Peripheral Naive T Cell Number and Its Role in Predicting Cardiovascular and Infection Events in Hemodialysis Patients. Front. Immunol. 2021, 12, 644627. [Google Scholar] [CrossRef]
- Badawy, A.; Nigm, D.A.; Ezzat, G.M.; Gamal, Y. Interleukin 18 as a new inflammatory mediator in left ventricular hypertrophy in children with end-stage renal disease. Saudi J. Kidney Dis. Transplant. Off. Publ. Saudi Cent. Organ. Transplant. Saudi Arab. 2020, 31, 1206–1216. [Google Scholar] [CrossRef]
- Templier, M.; Paré, G. Transparency in literature reviews: An assessment of reporting practices across review types and genres in top IS journals. Eur. J. Inf. Syst. 2017, 27, 503–550. [Google Scholar] [CrossRef]
- Paré, G.; Kitsiou, S. Methods for Literature Reviews. In Handbook of eHealth Evaluation: An Evidence-Based Approach; Lau, F., Kuziemsky, C., Eds.; University of Victoria: Victoria, BC, Canada, 2017; pp. 157–179. [Google Scholar]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.W.; Salerno, F.R. 47—Avoidance and Treatment of Cardiovascular Disease in Dialysis. In Handbook of Dialysis Therapy, 6th ed.; Nissenson, A.R., Fine, R.N., Mehrotra, R., Zaritsky, J., Eds.; Elsevier: New Delhi, India, 2023; pp. 421–429. [Google Scholar]
- Eloueyk, A.; Osta, B.; Alameldinne, R.; Awad, D. Uremic Serum Induces Inflammation in Cultured Human Endothelial Cells and Triggers Vascular Repair Mechanisms. Inflammation 2019, 42, 2003–2010. [Google Scholar] [CrossRef]
- Sun, Y.; Johnson, C.; Zhou, J.; Wang, L.; Li, Y.F.; Lu, Y.; Nanayakkara, G.; Fu, H.; Shao, Y.; Sanchez, C.; et al. Uremic toxins are conditional danger- or homeostasis-associated molecular patterns. Front. Biosci. (Landmark Ed.) 2018, 23, 348–387. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Jovanovich, A.; Farmer-Bailey, H.; Bispham, N.; Struemph, T.; Malaczewski, M.; Wang, W.; Chonchol, M. Vascular Dysfunction, Oxidative Stress, and Inflammation in Chronic Kidney Disease. Kidney360 2020, 1, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Jerotic, D.; Suvakov, S.; Matic, M.; Alqudah, A.; Grieve, D.J.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Damjanovic, T.; Dimkovic, N.; McClements, L.; et al. GSTM1 Modulates Expression of Endothelial Adhesion Molecules in Uremic Milieu. Oxid. Med. Cell. Longev. 2021, 2021, 6678924. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef]
- Zsom, L.; Faludi, M.; Fulop, T.; Dossabhoy, N.R.; Rosivall, L.; Tapolyai, M.B. The association of overhydration with chronic inflammation in chronic maintenance hemodiafiltration patients. Hemodial. Int. 2019, 23, 384–391. [Google Scholar] [CrossRef]
- Weiner, D.E.; Tighiouart, H.; Elsayed, E.F.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. The Framingham predictive instrument in chronic kidney disease. J. Am. Coll. Cardiol. 2007, 50, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Roehm, B.; Weiner, D.E. Blood pressure targets and kidney and cardiovascular disease: Same data but discordant guidelines. Curr. Opin. Nephrol. Hypertens. 2019, 28, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Group, A.C.; Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Zewinger, S.; Kleber, M.E.; Rohrer, L.; Lehmann, M.; Triem, S.; Jennings, R.T.; Petrakis, I.; Dressel, A.; Lepper, P.M.; Scharnagl, H.; et al. Symmetric dimethylarginine, high-density lipoproteins and cardiovascular disease. Eur. Heart J. 2017, 38, 1597–1607. [Google Scholar] [CrossRef]
- Ganesh, S.K.; Hulbert-Shearon, T.; Port, F.K.; Eagle, K.; Stack, A.G. Mortality differences by dialysis modality among incident ESRD patients with and without coronary artery disease. J. Am. Soc. Nephrol. 2003, 14, 415–424. [Google Scholar] [CrossRef]
- Aeddula, A.S.M.F.H.N.R. Chronic Kidney Disease-Mineral Bone Disorder (CKD-MBD). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Shetty, M.; Chowdhury, Y.; Yegneswaran, B. Calcific uremic arteriolopathy. Cleve Clin. J. Med. 2018, 85, 584–585. [Google Scholar] [CrossRef]
- Wang, N.; Angioi, A.; Hanset, N.; Ye, X.; Lu, S.; Zhu, Y. Individualizing the lifesaving journey for calciphylaxis: Addressing rapidly progressive attacks with multidimensional and AI research for regenerative medicine. Ren. Fail. 2024, 46, 2392846. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Dominic, E.A.; Anderson, A.H.; Beddhu, S.; Rahman, M.; Wolf, M.; Reilly, M.; Ojo, A.; Townsend, R.R.; et al. Use of Measures of Inflammation and Kidney Function for Prediction of Atherosclerotic Vascular Disease Events and Death in Patients With CKD: Findings from the CRIC Study. Am. J. Kidney Dis. 2019, 73, 344–353. [Google Scholar] [CrossRef]
- Marshall, M.R. The benefit of early survival on PD versus HD-Why this is (still) very important. Perit. Dial. Int. 2020, 40, 405–418. [Google Scholar] [CrossRef]
- Fulop, T.; Zsom, L.; Tapolyai, M.B.; Molnar, M.Z.; Abdul Salim, S.; Arany, I.; Hamrahian, M.; Rosivall, L. Peritoneal dialysis: The unique features by compartmental delivery of renal replacement therapy. Med. Hypotheses 2017, 108, 128–132. [Google Scholar] [CrossRef]
- Velloso, M.S.; Otoni, A.; de Paula Sabino, A.; de Castro, W.V.; Pinto, S.W.; Marinho, M.A.; Rios, D.R. Peritoneal dialysis and inflammation. Clin. Chim. Acta 2014, 430, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Canaud, B.; Kooman, J.P.; Selby, N.M.; Taal, M.W.; Francis, S.; Maierhofer, A.; Kopperschmidt, P.; Collins, A.; Kotanko, P. Dialysis-Induced Cardiovascular and Multiorgan Morbidity. Kidney Int. Rep. 2020, 5, 1856–1869. [Google Scholar] [CrossRef]
- Butani, L.; Calogiuri, G. Hypersensitivity reactions in patients receiving hemodialysis. Ann. Allergy Asthma Immunol. 2017, 118, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Allon, M. Diagnosis, Treatment, and Prevention of Hemodialysis Emergencies. Clin. J. Am. Soc. Nephrol. 2017, 12, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Maurice, F.; Moneret-Vautrin, D.A. Anaphylactic reactions during hemodialysis: Responsibility of the dialysis membrane (cuprophan). Nephrologie 1984, 5, 119–122. [Google Scholar]
- Foley, R.J.; Reeves, W.B. Acute Anaphylactoid Reactions in Hemodialysis. Am. J. Kidney Dis. 1985, 5, 132–135. [Google Scholar] [CrossRef]
- Schaefer, R.M.; Schaefer, L.; Horl, W.H. Anaphylactoid reactions during hemodialysis. Clin. Nephrol. 1994, 42, S44–S47. [Google Scholar]
- Fulop, T.; Elliott, A.B.; Jenssen, F.; Ullian, M.E.; Herberth, J. Fatal anaphylactic shock in a patient undergoing hemodialysis via a polyurethane catheter. Clin. Nephrol. 2019, 92, 218–219. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Mukaya, J.E.; Jacobson, M.S.; Esprit, D.; Ajayi, T. Allergic reaction to polysulphone membrane dialyser masquerading as infection. BMJ Case Rep. 2015, 2015, bcr2014208591. [Google Scholar] [CrossRef]
- Yang, R.C.; Lindsay, R.M. Dialyzer reactions in a patient switching from peritoneal dialysis to hemodialysis. Hemodial. Int. 2005, 9, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Montagnac, R.; Schillinger, F.; Milcent, T.; Croix, J.C. Hypersensitivity reactions during hemodialysis. Roles of high permeability, backfiltration and bacterial contamination of dialysis fluid. Nephrologie 1988, 9, 29–32. [Google Scholar]
- Galan, A.; Perez Garcia, R.; Garcia Vinuesa, M.; Anaya, F. Anaphylactoid reactions in patients on hemodialysis with the AN69 membrane: The ACE inhibitors. Nefrologia 1991, 11, 556–557. [Google Scholar]
- Davies, S.J.; Phillips, L.; Naish, P.F.; Russell, G.I. Peritoneal glucose exposure and changes in membrane solute transport with time on peritoneal dialysis. J. Am. Soc. Nephrol. 2001, 12, 1046–1051. [Google Scholar] [CrossRef]
- Krediet, R.T.; Van Esch, S.; Smit, W.; Michels, W.M.; Zweers, M.M.; Ho-Dac-Pannekeet, M.M.; Struijk, D.G. Peritoneal membrane failure in peritoneal dialysis patients. Blood Purif. 2002, 20, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Witowski, J.; Kamhieh-Milz, J.; Kawka, E.; Catar, R.; Jorres, A. IL-17 in Peritoneal Dialysis-Associated Inflammation and Angiogenesis: Conclusions and Perspectives. Front. Physiol. 2018, 9, 1694. [Google Scholar] [CrossRef] [PubMed]
- Craddock, P.R.; Fehr, J.; Dalmasso, A.P.; Brighan, K.L.; Jacob, H.S. Hemodialysis leukopenia. Pulmonary vascular leukostasis resulting from complement activation by dialyzer cellophane membranes. J. Clin. Investig. 1977, 59, 879–888. [Google Scholar] [CrossRef]
- Craddock, P.R.; Fehr, J.; Brigham, K.L.; Kronenberg, R.S.; Jacob, H.S. Complement and leukocyte-mediated pulmonary dysfunction in hemodialysis. N. Engl. J. Med. 1977, 296, 769–774. [Google Scholar] [CrossRef]
- Stepniewska, J.; Dolegowska, B.; Golembiewska, E.; Marchelek-Mysliwiec, M.; Domanski, M.; Ciechanowski, K.; Zair, L. The activation of complement system in different types of renal replacement therapy. J. Physiol. Pharmacol. 2020, 71, 275–281. [Google Scholar] [CrossRef]
- Suzuki, Y.; Uchida, J.; Tsuji, H.; Kuzuhara, K.; Hara, S.; Nihei, H.; Ogura, Y.; Otsubo, O.; Mimura, N. Acute changes in C3a and C5a in an anaphylactoid reaction in hemodialysis patients. Tohoku J. Exp. Med. 1987, 152, 35–45. [Google Scholar] [CrossRef]
- Hakim, R.M.; Fearon, D.T.; Lazarus, J.M. Biocompatibility of dialysis membranes: Effects of chronic complement activation. Kidney Int. 1984, 26, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Vienken, J.; Diamantoglou, M.; Hahn, C.; Kamusewitz, H.; Paul, D. Considerations on developmental aspects of biocompatible dialysis membranes. Artif. Organs 1995, 19, 398–406. [Google Scholar] [CrossRef]
- Vanholder, R. Biocompatibility issues in hemodialysis. Clin. Mater. 1992, 10, 87–133. [Google Scholar] [CrossRef]
- Rodríguez-Sanz, A.; Sánchez-Villanueva, R.; Domínguez-Ortega, J.; Fiandor, A.M.; Ruiz, M.P.; Trocoli, F.; Díaz-Tejeiro, R.; Cadenillas, C.; González, E.; Martínez, V.; et al. Mechanisms Involved in Hypersensitivity Reactions to Polysulfone Hemodialysis Membranes. Artif. Organs 2017, 41, E285–E295. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, A.A.; Horoz, M.; Ozcan, T.; Yildiz, I.; Sari, S.; Genctoy, G. Pulmonary hypertension in hemodialysis patients without arteriovenous fistula: The effect of dialyzer composition. Ren. Fail. 2010, 32, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Arenas Jiménez, M.D.; Gil, M.T.; Carretón, M.A.; Moledous, A.; Albiach, B. Adverse reactions to polisulphone membrane dialyzers durind hemodialysis abstract. Nefrologia 2007, 27, 638–642. [Google Scholar]
- Coombs, R.R.A.; Gell, P.G.H. Classification of allergic reactions responsible for drug hypersensitivity reactions. In Clinical Aspects of Immunology; Davis, B., Ed.; Davis: Philadelphia, PA, USA, 1968; pp. 575–596. [Google Scholar]
- Dezsi, L.; Fulop, T.; Meszaros, T.; Szenasi, G.; Urbanics, R.; Vazsonyi, C.; Orfi, E.; Rosivall, L.; Nemes, R.; Kok, R.J.; et al. Features of complement activation-related pseudoallergy to liposomes with different surface charge and PEGylation: Comparison of the porcine and rat responses. J. Control. Release 2014, 195, 2–10. [Google Scholar] [CrossRef]
- Kozma, G.T.; Meszaros, T.; Vashegyi, I.; Fulop, T.; Orfi, E.; Dezsi, L.; Rosivall, L.; Bavli, Y.; Urbanics, R.; Mollnes, T.E.; et al. Pseudo-anaphylaxis to Polyethylene Glycol (PEG)-Coated Liposomes: Roles of Anti-PEG IgM and Complement Activation in a Porcine Model of Human Infusion Reactions. ACS Nano 2019, 13, 9315–9324. [Google Scholar] [CrossRef]
- Szebeni, J.; Fontana, J.L.; Wassef, N.M.; Mongan, P.D.; Morse, D.S.; Dobbins, D.E.; Stahl, G.L.; Bunger, R.; Alving, C.R. Hemodynamic changes induced by liposomes and liposome-encapsulated hemoglobin in pigs: A model for pseudoallergic cardiopulmonary reactions to liposomes. Role of complement and inhibition by soluble CR1 and anti-C5a antibody. Circulation 1999, 99, 2302–2309. [Google Scholar] [CrossRef]
- Szebeni, J.; Baranyi, L.; Savay, S.; Bodo, M.; Milosevits, J.; Alving, C.R.; Bunger, R. Complement activation-related cardiac anaphylaxis in pigs: Role of C5a anaphylatoxin and adenosine in liposome-induced abnormalities in ECG and heart function. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1050–H1058. [Google Scholar] [CrossRef]
- Szebeni, J.; Bedocs, P.; Csukas, D.; Rosivall, L.; Bunger, R.; Urbanics, R. A porcine model of complement-mediated infusion reactions to drug carrier nanosystems and other medicines. Adv. Drug Deliv. Rev. 2012, 64, 1706–1716. [Google Scholar] [CrossRef]
- Szebeni, J.; Bedocs, P.; Rozsnyay, Z.; Weiszhar, Z.; Urbanics, R.; Rosivall, L.; Cohen, R.; Garbuzenko, O.; Bathori, G.; Toth, M.; et al. Liposome-induced complement activation and related cardiopulmonary distress in pigs: Factors promoting reactogenicity of Doxil and AmBisome. Nanomedicine 2012, 8, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Bedocs, P.; Urbanics, R.; Bunger, R.; Rosivall, L.; Toth, M.; Barenholz, Y. Prevention of infusion reactions to PEGylated liposomal doxorubicin via tachyphylaxis induction by placebo vesicles: A porcine model. J. Control. Release 2012, 160, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.A.; Meszaros, T.; Fulop, T.; Urbanics, R.; Szebeni, J.; Cho, N.J. Comparison of complement activation-related pseudoallergy in miniature and domestic pigs: Foundation of a validatable immune toxicity model. Nanomedicine 2016, 12, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Mechanism of nanoparticle-induced hypersensitivity in pigs: Complement or not complement? Drug Discov. Today 2018, 23, 487–492. [Google Scholar] [CrossRef]
- Meszaros, T.; Kozma, G.T.; Shimizu, T.; Miyahara, K.; Turjeman, K.; Ishida, T.; Barenholz, Y.; Urbanics, R.; Szebeni, J. Involvement of complement activation in the pulmonary vasoactivity of polystyrene nanoparticles in pigs: Unique surface properties underlying alternative pathway activation and instant opsonization. Int. J. Nanomed. 2018, 13, 6345–6357. [Google Scholar] [CrossRef]
- Dezsi, L.; Meszaros, T.; Kozma, G.; Mária, H.-V.; Olah, C.Z.; Szabo, M.; Patko, Z.; Fulop, T.; Hennies, M.; Szebeni, M.; et al. A naturally hypersensitive porcine model may help understand the mechanism of COVID-19 mRNA vaccine-induced rare (pseudo) allergic reactions: Complement activation as a possible contributing factor. Geroscience 2022, 44, 597–618. [Google Scholar] [CrossRef]
- Bakos, T.; Meszaros, T.; Kozma, G.T.; Berenyi, P.; Facsko, R.; Farkas, H.; Dezsi, L.; Heirman, C.; de Koker, S.; Schiffelers, R.; et al. mRNA-LNP COVID-19 Vaccine Lipids Induce Complement Activation and Production of Proinflammatory Cytokines: Mechanisms, Effects of Complement Inhibitors, and Relevance to Adverse Reactions. Int. J. Mol. Sci. 2024, 25, 3595. [Google Scholar] [CrossRef]
- Milosevits, G.; Meszaros, T.; Orfi, E.; Bakos, T.; Garami, M.; Kovacs, G.; Dezsi, L.; Hamar, P.; Gyorffy, B.; Szabo, A.; et al. Complement-mediated hypersensitivity reactions to an amphotericin B-containing lipid complex (Abelcet) in pediatric patients and anesthetized rats: Benefits of slow infusion. Nanomedicine 2021, 34, 102366. [Google Scholar] [CrossRef]
- Nilsson, B.; Ekdahl, K.N.; Mollnes, T.E.; Lambris, J.D. The role of complement in biomaterial-induced inflammation. Mol. Immunol. 2007, 44, 82–94. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, S. Risk factors for mortality in patients undergoing hemodialysis: A systematic review and meta-analysis. Int. J. Cardiol. 2017, 238, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, J.; Lu, H.; Shi, Z.; Wang, X. Effect of hemodiafiltration and hemodialysis on mortality of patients with end-stage kidney disease: A meta-analysis. BMC Nephrol. 2024, 25, 372. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V. Neutrophil Extracellular Traps in the Second Decade. J. Innate Immun. 2018, 10, 414–421. [Google Scholar] [CrossRef]
- Yang, Y.; Jiao, Y.Y.; Zhang, Z.; Zhuo, L.; Li, W.G. Neutrophil extracellular trap is an important connection between hemodialysis and acute myocardial infarction. Ren. Fail. 2023, 45, 2216307. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Aziz, M.; Wang, P. The vitals of NETs. J. Leukoc. Biol. 2021, 110, 797–808. [Google Scholar] [CrossRef]
- Korabecna, M.; Tesar, V. NETosis provides the link between activation of neutrophils on hemodialysis membrane and comorbidities in dialyzed patients. Inflamm. Res. 2017, 66, 369–378. [Google Scholar] [CrossRef]
- Lee, H.W.; Nizet, V.; An, J.N.; Lee, H.S.; Song, Y.R.; Kim, S.G.; Kim, J.K. Uremic serum damages endothelium by provoking excessive neutrophil extracellular trap formation. Sci. Rep. 2021, 11, 21439. [Google Scholar] [CrossRef]
- Bieber, S.; Muczynski, K.A.; Lood, C. Neutrophil Activation and Neutrophil Extracellular Trap Formation in Dialysis Patients. Kidney Med. 2020, 2, 692–698.e1. [Google Scholar] [CrossRef]
- Cheung, A.K.; LeWinter, M.; Chenoweth, D.E.; Lew, W.Y.; Henderson, L.W. Cardiopulmonary effects of cuprophane-activated plasma in the swine. Kidney Int. 1986, 29, 799–806. [Google Scholar] [CrossRef]
- Petho, A.; Piecha, D.; Meszaros, T.; Urbanics, R.; Moore, C.; Canaud, B.; Rosivall, L.; Mollnes, T.E.; Steppan, S.; Szenasi, G.; et al. A porcine model of hemodialyzer reactions: Roles of complement activation and rinsing back of extracorporeal blood. Ren. Fail. 2021, 43, 1609–1620. [Google Scholar] [CrossRef]
- Petho, A.; Szebeni, J. Pulmonary hypertension in a hemodialysis porcine model: Possible unforeseen causes? Ren. Fail. 2022, 44, 693. [Google Scholar] [CrossRef]
- Tang, M.; Batty, J.A.; Lin, C.; Fan, X.; Chan, K.E.; Kalim, S. Pulmonary Hypertension, Mortality, and Cardiovascular Disease in CKD and ESRD Patients: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2018, 72, 75–83. [Google Scholar] [CrossRef]
- Edmonston, D.L.; Parikh, K.S.; Rajagopal, S.; Shaw, L.K.; Abraham, D.; Grabner, A.; Sparks, M.A.; Wolf, M. Pulmonary Hypertension Subtypes and Mortality in CKD. Am. J. Kidney Dis. 2020, 75, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, N.C.; Argula, R.G.; Klings, E.S.; Wilson, K.C.; Farber, H.W. Prevalence and Mortality of Pulmonary Hypertension in ESRD: A Systematic Review and Meta-analysis. Lung 2020, 198, 535–545. [Google Scholar] [CrossRef]
- Utley, J.R. Pathophysiology of cardiopulmonary bypass: Current issues. J. Card. Surg. 1990, 5, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Boylan, J.F.; Cheng, D.C. The systemic inflammatory response to cardiac surgery: Implications for the anesthesiologist. Anesthesiology 2002, 97, 215–252. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.G.; Hagen, P.O. Systemic inflammatory response syndrome. Br. J. Surg. 1997, 84, 920–935. [Google Scholar] [CrossRef]
- Paparella, D.; Yau, T.M.; Young, E. Cardiopulmonary bypass induced inflammation: Pathophysiology and treatment. An update. Eur. J. Cardiothorac. Surg. 2002, 21, 232–244. [Google Scholar] [CrossRef]
- Finn, A.; Naik, S.; Klein, N.; Levinsky, R.J.; Strobel, S.; Elliott, M. Interleukin-8 release and neutrophil degranulation after pediatric cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 1993, 105, 234–241. [Google Scholar] [CrossRef]
- Edmunds, L.H., Jr. Why cardiopulmonary bypass makes patients sick: Strategies to control the blood-synthetic surface interface. Adv. Card. Surg. 1995, 6, 131–167. [Google Scholar]
- Li, T.; Luo, N.; Du, L.; Liu, J.; Gong, L.; Zhou, J. Early and marked up-regulation of TNF-alpha in acute respiratory distress syndrome after cardiopulmonary bypass. Front. Med. 2012, 6, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Manché, A.; Derias, N.W.; Hearse, D.J. Thromboxane A2 mediates pulmonary hypertension after cardiopulmonary bypass in the rabbit. J. Thorac. Cardiovasc. Surg. 1993, 106, 959–967. [Google Scholar] [CrossRef]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, hypertension, and cardiovascular disease: Clinical insights and vascular mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, B.; Chen, X.; Duan, J.; Liu, X.; Zhang, R.; Li, G. Fragmented QRS complex on a 12-lead electrocardiogram predicts cardiovascular and all-cause mortality in dialysis patients. Semin. Dial. 2023, 36, 43–52. [Google Scholar] [CrossRef]
- Franklin, S.S.; Wong, N.D. Hypertension and Cardiovascular Disease: Contributions of the Framingham Heart Study. Glob. Heart 2013, 8, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yuan, Y.; Zheng, M.; Pan, A.; Wang, M.; Zhao, M.; Li, Y.; Yao, S.; Chen, S.; Wu, S. Association of age of onset of hypertension with cardiovascular diseases and mortality. J. Am. Coll. Cardiol. 2020, 75, 2921–2930. [Google Scholar] [CrossRef]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease: The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Resnick, H.E.; Howard, B.V. Diabetes and cardiovascular disease. Annu. Rev. Med. 2002, 53, 245–267. [Google Scholar] [CrossRef]
- Matheus, A.S.d.M.; Tannus, L.R.M.; Cobas, R.A.; Palma, C.C.S.; Negrato, C.A.; Gomes, M.d.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef]
- Gonna, H.; Ray, K.K. The importance of dyslipidaemia in the pathogenesis of cardiovascular disease in people with diabetes. Diabetes Obes. Metab. 2019, 21, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Lorenzatti, A.J.; Toth, P.P. New perspectives on atherogenic dyslipidaemia and cardiovascular disease. Eur. Cardiol. Rev. 2020, 15, e04. [Google Scholar] [CrossRef]
- Pascual, V.; Serrano, A.; Pedro-Botet, J.; Ascaso, J.; Barrios, V.; Millán, J.; Pintó, X.; Cases, A. Chronic kidney disease and dyslipidaemia. Clínica Investig. Arterioscler. (Engl. Ed.) 2017, 29, 22–35. [Google Scholar] [CrossRef]
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Grams, M.; Ouyang, P.; Szklo, M.; De Boer, I.H. Coronary heart disease risk associated with the dyslipidaemia of chronic kidney disease. Heart 2018, 104, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewska, A.E.; Adamska, P.; Iwanczyk-Skalska, E.; Ostromecka, K.; Niepolski, L.; Marcinkowski, W.; Mostowska, A.; Warchol, W.; Zaba, C.; Jagodzinski, P.P. Paraoxonase 1 concerning dyslipidaemia, cardiovascular diseases, and mortality in haemodialysis patients. Sci. Rep. 2021, 11, 6773. [Google Scholar] [CrossRef]
- Martin, K.J.; González, E.A. Metabolic bone disease in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 875–885. [Google Scholar] [CrossRef]
- O’Riordan, E.; Foley, R.N. Effects of anaemia on cardiovascular status. Nephrol. Dial. Transplant. 2000, 15, 19–22. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Tighiouart, H.; Manjunath, G.; MacLeod, B.; Griffith, J.; Salem, D.; Levey, A.S. Anemia as a risk factor for cardiovascular disease in The Atherosclerosis Risk in Communities (ARIC) study. J. Am. Coll. Cardiol. 2002, 40, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lanser, L.; Fuchs, D.; Scharnagl, H.; Grammer, T.; Kleber, M.E.; März, W.; Weiss, G.; Kurz, K. Anemia of chronic disease in patients with cardiovascular disease. Front. Cardiovasc. Med. 2021, 8, 666638. [Google Scholar] [CrossRef]
- Mikhail, A.; Brown, C.; Williams, J.A.; Mathrani, V.; Shrivastava, R.; Evans, J.; Isaac, H.; Bhandari, S. Renal association clinical practice guideline on Anaemia of Chronic Kidney Disease. BMC Nephrol. 2017, 18, 345. [Google Scholar] [CrossRef]
- Stauffer, M.E.; Fan, T. Prevalence of anemia in chronic kidney disease in the United States. PLoS ONE 2014, 9, e84943. [Google Scholar] [CrossRef] [PubMed]
- Murdeshwar, H.N.; Anjum, F. Hemodialysis; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Bello, A.K.; Okpechi, I.G.; Osman, M.A.; Cho, Y.; Htay, H.; Jha, V.; Wainstein, M.; Johnson, D.W. Epidemiology of haemodialysis outcomes. Nat. Rev. Nephrol. 2022, 18, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.Y.; Jeon, Y.; Kim, Y.S.; Kang, S.W.; Yang, C.W.; Kim, N.H.; Noh, H.W.; Jeon, S.J.; Lim, J.H.; Choi, J.Y.; et al. Sex disparities in mortality among patients with kidney failure receiving dialysis. Sci. Rep. 2022, 12, 18555. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Ke, G.; Huang, J.; Yang, D.; Pi, M.; Li, L.; Liu, X.; Tao, S.; Chen, L.; Liang, G.; et al. Effects of Carvedilol on Cardiovascular Events and Mortality in Hemodialysis Patients, A Systematic Review and Meta-Analysis. Iran. J. Kidney Dis. 2020, 14, 256–266. [Google Scholar]
- Maki, K.C.; Wilcox, M.L.; Dicklin, M.R.; Kakkar, R.; Davidson, M.H. Left ventricular mass regression, all-cause and cardiovascular mortality in chronic kidney disease: A meta-analysis. BMC Nephrol. 2022, 23, 34. [Google Scholar] [CrossRef]
- Zimmermann, J.; Herrlinger, S.; Pruy, A.; Metzger, T.; Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999, 55, 648–658. [Google Scholar] [CrossRef]
- Ebert, T.; Pawelzik, S.C.; Witasp, A.; Arefin, S.; Hobson, S.; Kublickiene, K.; Shiels, P.G.; Back, M.; Stenvinkel, P. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins 2020, 12, 227. [Google Scholar] [CrossRef]
- Ulrich, C.; Wildgrube, S.; Fiedler, R.; Seibert, E.; Kneser, L.; Fick, S.; Schafer, C.; Markau, S.; Trojanowicz, B.; Girndt, M. NLRP3 Inflammasome Activation in Hemodialysis and Hypertensive Patients with Intact Kidney Function. Toxins 2020, 12, 675. [Google Scholar] [CrossRef]
- Li, H.; Lu, X.; Xiong, R.; Wang, S. High Neutrophil-to-Lymphocyte Ratio Predicts Cardiovascular Mortality in Chronic Hemodialysis Patients. Mediat. Inflamm. 2017, 2017, 9327136. [Google Scholar] [CrossRef]
- Kumbar, L.; Yee, J. Current Concepts in Hemodialysis Vascular Access Infections. Adv. Chronic Kidney Dis. 2019, 26, 16–22. [Google Scholar] [CrossRef]
- Almeida, B.M.; Moreno, D.H.; Vasconcelos, V.; Cacione, D.G. Interventions for treating catheter-related bloodstream infections in people receiving maintenance haemodialysis. Cochrane Database Syst. Rev. 2022, 4, CD013554. [Google Scholar] [CrossRef] [PubMed]
- Balbino, K.P.; Hermsdorff, H.H.M.; Bressan, J. Polymorphism related to cardiovascular risk in hemodialysis subjects: A systematic review. J. Bras. Nefrol. 2018, 40, 179–192. [Google Scholar] [CrossRef]
- Mineshima, M. Optimal Design of Dialyzers. Contrib. Nephrol. 2017, 189, 204–209. [Google Scholar] [CrossRef]
- Lemke, H.D.; Kuentz, F.; Forêt, M. Mechanisms of hypersensitivity reactions during hemodialysis. Trans.-Am. Soc. Artif. Intern. Organs 1985, 31, 149–152. [Google Scholar]
- Bacelar Marques, I.D.; Pinheiro, K.F.; de Freitas do Carmo, L.P.; Costa, M.C.; Abensur, H. Anaphylactic reaction induced by a polysulfone/polyvinylpyrrolidone membrane in the 10th session of hemodialysis with the same dialyzer. Hemodial. Int. 2011, 15, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Boer, W.; Liem, Y.; De Beus, E.; Abrahams, A. Acute reactions to polysulfone/polyethersulfone dialysers: Literature review and management. Neth. J. Med. 2017, 75, 4–13. [Google Scholar]
- Dahe, G.J.; Teotia, R.S.; Kadam, S.S.; Bellare, J.R. The biocompatibility and separation performance of antioxidative polysulfone/vitamin E TPGS composite hollow fiber membranes. Biomaterials 2011, 32, 352–365. [Google Scholar] [CrossRef]
- Kohlová, M.; Rocha, S.; Gomes Amorim, C.; de Nova Araújo, A.; Santos-Silva, A.; Solich, P.; Branco da Silva Montenegro, M.C. Doping Polysulfone Membrane with Alpha-Tocopherol and Alpha-Lipoic Acid for Suppressing Oxidative Stress Induced by Hemodialysis Treatment. Macromol. Biosci. 2020, 20, 2000046. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.-I.; Matsuda, M.; Okuoka, M.; Yakushiji, T.; Fukuda, M.; Miyasaka, T.; Matsumoto, Y.; Sakai, K. Antioxidation property of vitamin E-coated polysulfone dialysis membrane and recovery of oxidized vitamin E by vitamin C treatment. J. Membr. Sci. 2007, 302, 115–118. [Google Scholar] [CrossRef]
- Panichi, V.; Rosati, A.; Paoletti, S.; Ferrandello, P.; Migliori, M.; Beati, S.; Bernabini, G.; Daini, R.; Casani, A.; Angelini, D. A vitamin E-coated polysulfone membrane reduces serum levels of inflammatory markers and resistance to erythropoietin-stimulating agents in hemodialysis patients: Results of a randomized cross-over multicenter trial. Blood Purif. 2011, 32, 7–14. [Google Scholar] [CrossRef]
- Amorim, C.; Montenegro, M.; Araujo, A.; Kohlová, M.; Solich, P.; Zelena, L. Preparation of Polysulfone Membrane with α-Tocopherol and α Lipoic acid to Reduce Oxidative Stress. In Proceedings of the 9th Meeting of Division of Analytical Chemistry, Porto, Portugal, 26–27 March 2018. [Google Scholar]
- Li, Y.; Luo, X.; Yang, M.; Su, B. Alleviation of Oxidative Stress during Hemodialysis Sessions by Hemodialysis Membrane Innovation: A Multidisciplinary Perspective. Blood Purif. 2023, 52, 905–916. [Google Scholar] [CrossRef] [PubMed]
- de Borst, M.H. The complement system in hemodialysis patients: Getting to the heart of the matter. Nephron 2016, 132, 1–4. [Google Scholar] [CrossRef]
- Skinner, S.C.; Derebail, V.K.; Poulton, C.J.; Bunch, D.O.; Roy-Chaudhury, P.; Key, N.S. Hemodialysis-related complement and contact pathway activation and cardiovascular risk: A narrative review. Kidney Med. 2021, 3, 607–618. [Google Scholar] [CrossRef]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Vonesh, E.F.; Snyder, J.J.; Foley, R.N.; Collins, A.J. The differential impact of risk factors on mortality in hemodialysis and peritoneal dialysis. Kidney Int. 2004, 66, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Luhana, S.; Sadarat, F.; Parkash, O.; Rahaman, Z.; Wang, H.Y.; Kiran, F.; Lohana, A.C.; Sapna, F.; Kumari, R. Mortality and mode of dialysis: Meta-analysis and systematic review. BMC Nephrol. 2024, 25, 1. [Google Scholar] [CrossRef]
- Ethier, I.; Hayat, A.; Pei, J.; Hawley, C.M.; Johnson, D.W.; Francis, R.S.; Wong, G.; Craig, J.C.; Viecelli, A.K.; Htay, H.; et al. Peritoneal dialysis versus haemodialysis for people commencing dialysis. Cochrane Database Syst. Rev. 2024, 6, CD013800. [Google Scholar] [CrossRef]
- Baroni, G.; Schuinski, A.; de Moraes, T.P.; Meyer, F.; Pecoits-Filho, R. Inflammation and the peritoneal membrane: Causes and impact on structure and function during peritoneal dialysis. Mediat. Inflamm. 2012, 2012, 912595. [Google Scholar] [CrossRef]
- Flessner, M.F.; Credit, K.; Henderson, K.; Vanpelt, H.M.; Potter, R.; He, Z.; Henegar, J.; Robert, B. Peritoneal changes after exposure to sterile solutions by catheter. J. Am. Soc. Nephrol. 2007, 18, 2294–2302. [Google Scholar] [CrossRef]
- Razeghi, E.; Omati, H.; Maziar, S.; Khashayar, P.; Mahdavi-Mazdeh, M. Chronic inflammation increases risk in hemodialysis patients. Saudi J. Kidney Dis. Transplant. 2008, 19, 785–789. [Google Scholar]
- Lonnemann, G. Chronic inflammation in hemodialysis: The role of contaminated dialysate. Blood Purif. 2000, 18, 214–223. [Google Scholar] [CrossRef]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Spittle, M.A.; Hoenich, N.A.; Handelman, G.J.; Adhikarla, R.; Homel, P.; Levin, N.W. Oxidative stress and inflammation in hemodialysis patients. Am. J. Kidney Dis. 2001, 38, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L. Inflammation and cardiovascular disease associated with hemodialysis for end-stage renal disease. Front. Pharmacol. 2022, 13, 800950. [Google Scholar] [CrossRef]
- Dheda, S.; Vesey, D.A.; Hawley, C.; Johnson, D.W.; Fahim, M. Effect of a hemodialysis session on markers of inflammation and endotoxin. Int. J. Inflamm. 2022, 2022, 8632245. [Google Scholar] [CrossRef]
- Ebert, T.; Neytchev, O.; Witasp, A.; Kublickiene, K.; Stenvinkel, P.; Shiels, P.G. Inflammation and oxidative stress in chronic kidney disease and dialysis patients. Antioxid. Redox Signal. 2021, 35, 1426–1448. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, Y.; Su, B.; Zhao, W.; Kizhakkedathu, J.N.; Zhao, C. Advances in enhancing hemocompatibility of hemodialysis hollow-fiber membranes. Adv. Fiber Mater. 2023, 5, 1198–1240. [Google Scholar] [CrossRef]
- Motofelea, A.C.; Mihaescu, A.; Olariu, N.; Marc, L.; Chisavu, L.; Pop, G.N.; Crintea, A.; Jura, A.M.C.; Ivan, V.M.; Apostol, A.; et al. Machine Learning Models for Predicting Mortality in Hemodialysis Patients: A Systematic Review. Appl. Sci. 2025, 15, 5776. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Search Keywords | Number of Publications Until 1 May 2025 | First Publication in This Field |
|---|---|---|
| chronic uremic inflammation | 1284 | 1966 |
| hemodialysis treatment- related inflammation | 2845 | 1969 |
| cardiovascular mortality in HD patients | 2389 | 1968 |
| hemodialysis treatment- related inflammation and cardiovascular mortality in HD patients | 100 | 1998 |
| mortality between HD and PD | 4907 | 1968 |
| complement system activation in hemodialysis | 423 | 1964 |
| hemodialysis procedure- related reactions | 1305 | 1964 |
| Small Molecular Weight (<500 Da) | Middle Molecular Weight (≥500 Da) | High Molecular Weight (Mostly < 500 Da) |
|---|---|---|
| Urea | PTH | Indoxyl sulfate |
| Creatinine | β2-microglobulin | Indole acetic acid |
| Uric acid | Endothelin | p-cresylsulfate |
| ADMA | FGF23 | Phenylacetic acid |
| Carbamylated compounds | Ghrelin | Kynurenines |
| SDMA | Immunoglobulin light chains | AGEs |
| TMAO | IL-6, IL-8, IL-18 | Homocysteine |
| Lipids and lipoproteins | ||
| Neuropeptide Y | ||
| ANP | ||
| Retinol binding protein | ||
| TNF-α |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pethő, Á.G.; Fülöp, T.; Orosz, P.; Szénási, G.; Tapolyai, M.; Dézsi, L. Increased Cardiovascular Mortality in Hemodialysis: The Role of Chronic Inflammation, Complement Activation, and Non-Biocompatibility. Toxins 2025, 17, 345. https://doi.org/10.3390/toxins17070345
Pethő ÁG, Fülöp T, Orosz P, Szénási G, Tapolyai M, Dézsi L. Increased Cardiovascular Mortality in Hemodialysis: The Role of Chronic Inflammation, Complement Activation, and Non-Biocompatibility. Toxins. 2025; 17(7):345. https://doi.org/10.3390/toxins17070345
Chicago/Turabian StylePethő, Ákos Géza, Tibor Fülöp, Petronella Orosz, Gábor Szénási, Mihály Tapolyai, and László Dézsi. 2025. "Increased Cardiovascular Mortality in Hemodialysis: The Role of Chronic Inflammation, Complement Activation, and Non-Biocompatibility" Toxins 17, no. 7: 345. https://doi.org/10.3390/toxins17070345
APA StylePethő, Á. G., Fülöp, T., Orosz, P., Szénási, G., Tapolyai, M., & Dézsi, L. (2025). Increased Cardiovascular Mortality in Hemodialysis: The Role of Chronic Inflammation, Complement Activation, and Non-Biocompatibility. Toxins, 17(7), 345. https://doi.org/10.3390/toxins17070345