1. Introduction
Clostridium perfringens is a major human gastrointestinal pathogen [
1,
2,
3,
4,
5,
6]. Annually, this bacterium causes one million and five million food poisoning cases in the USA and the European Union, respectively [
3,
7]. The vast majority of
C. perfringens human foodborne disease cases are caused by type F strains [
8], with type F food poisoning ranking as the second most common bacterial foodborne disease in the USA [
3,
7]. Type F strains also cause 5–15% of all cases of nonfoodborne human gastrointestinal disease, most notably including antibiotic-associated diarrhea [
9,
10,
11].
The production of
C. perfringens enterotoxin, a single polypeptide of 35 kDa, is essential for type F strain enteric virulence [
9,
12]. In human enterocyte-like Caco-2 cells, CPE action starts when this toxin binds to receptors, which include certain members of the claudin family of ~20–27 kDa proteins located in epithelial or endothelial tight junctions [
13,
14,
15]. The binding of CPE to these claudin receptors forms an ~90 kDa small complex that contains CPE plus both a receptor and a nonreceptor claudin [
9]. Approximately 6 CPE small complexes oligomerize into an ~425–500 kDa large complex that initially corresponds to a prepore [
16]. Each CPE molecule sequestered in this prepore then extends a β-hairpin to create a β-barrel pore that is selectively permeable to cations, including calcium [
9,
17]. With prolonged treatment times, a secondary large CPE complex of ~550–660 kDa containing CPE, receptor and nonreceptor claudins, and the tight junction protein occludin can sometimes be detected in Caco-2 cells [
9]. Calcium influx into CPE pore-containing Caco-2 cells then activates calpain [
9], which kills these cells via either classical caspase-3 mediated apoptosis [when a lower CPE dose treatment causes limited calpain activation] or necroptosis [when a higher CPE dose treatment causes strong calpain activation] [
9].
In the small intestine, CPE also binds to claudin receptors on villus tip cells, followed by the formation of a large CPE complex [
9,
18]. The ability to form this large CPE complex appears to be important for CPE-induced intestinal damage based on results from studies using several CPE variants blocked at various steps in CPE action [
9]. These experiments revealed that only CPE variants capable of forming large complexes in Caco-2 cells can produce intestinal histologic damage in mouse small intestinal loops. Other studies have strongly suggested that this CPE-induced intestinal histologic damage is responsible for intestinal fluid and electrolyte losses that clinically manifest as diarrhea [
9,
12].
During GI disease, CPE is produced when type F isolates sporulate in the intestines [
1,
9,
19]. Once made, CPE is not secreted but, instead, accumulates in the cytoplasm of the mother cell [
12,
19]. When sporulation is completed, the mother cell lyses to free its mature spore, concomitantly releasing CPE into the intestinal lumen [
12]. After discharge into the lumen, CPE encounters a complex environment replete with proteases. These include pancreatic proteases such as trypsin, chymotrypsin, and elastase but also intestinal cell proteases such as mesotrypsin and microbial proteases produced by the intestinal microbiota [
20,
21,
22,
23,
24,
25]. Adding to the complexity of intestinal protease activity, there are also host dietary and microbial protease inhibitors present and some normal microbiota producing protease-degrading enzymes [
21].
Many bacterial protein toxins produced in the intestines, including several
C. perfringens toxins [
1], are cleaved by intestinal proteases. A number of previous studies have reported that, in vitro, CPE can also be processed using purified trypsin or chymotrypsin [
26,
27,
28,
29]. Specifically, purified trypsin was shown to remove the first 25 amino acids [~3 kDa] from the N-terminus of CPE to generate an ~32 kDa CPE fragment. Results with purified chymotrypsin were less definitive. Initial studies reported chymotrypsin cleaves CPE, but only if the toxin is reduced and S-carboxymethylated [
30,
31]. Under these conditions, a later study [
26] reported that chymotrypsin cleaves CPE at multiple points. Paradoxically, that study concluded that purified chymotrypsin removes the first 36 N-terminal amino acids from CPE to generate a CPE fragment of ~31 kDa [
26], although the diversity and relative abundance of CPE fragments generated by chymotrypsin treatment were never addressed.
The extent of CPE processing, if any, in the complicated protease environment of the intestines has not yet been examined, despite its potential disease relevance given that proteolytic cleavage affects the toxicity of other
C. perfringens toxins [
1]. Therefore, this study evaluated the effects of mouse small intestinal contents on CPE proteolytic processing and compared these ex vivo results to the in vitro effects of purified trypsin or chymotrypsin on CPE to discern the involvement of these two proteases in CPE proteolytic processing by intestinal contents. Studies were then extended to evaluate CPE proteolytic processing and toxicity in the intestines using mouse small intestinal loops; these analyses also provided new insights regarding the formation kinetics and stability of the large CPE complex in vivo.
3. Discussion
Proteases in the intestines process several of the
C. perfringens toxins produced in the intestines, e.g., beta, iota, and epsilon toxins [
1,
33,
34,
35,
36]. Previous studies [
26,
27,
28,
29] have examined the in vitro effects of purified trypsin or chymotrypsin on CPE and reported that these proteases cleave CPE. Using amino acid sequencing, mass spectrometry and Western blot approaches, several studies [
27,
28,
29,
31] reported that purified trypsin removes the first 25 N-terminal amino acids [~3 kDa] from CPE. The current SDS-PAGE results using purified trypsin are consistent with this conclusion. The current study also determined that trypsin treatment decreased Western blot immunoreactivity using a polyclonal CPE antiserum. This result indicates that the first 25 amino acids of CPE contribute to one or more epitopes, which is consistent with a previous epitope mapping study [
28] that showed that a CPE
25-319 fragment lost a non-neutralizing epitope present in native CPE.
Previous studies [
26,
30,
31] concluded that CPE is insensitive to purified chymotrypsin unless the toxin is reduced and S-carboxymethylated. However, when CPE is reduced and S-carboxymethylated, it was reported [
26] that purified chymotrypsin removes the first 36 N-terminal amino acids from CPE. This conclusion has been confusing since, (i) after the S-carboxymethylation and reduction of CPE, chymotrypsin cleavage was also detected at several other N-terminal amino acids preceding CPE amino acid 38 [
26], while (ii) the size or relative abundance of CPE fragment[s] produced by these chymotrypsin cleavage conditions was not evaluated in this study.
In response, the current study evaluated purified chymotrypsin cleavage of CPE, in the absence of S-carboxymethylation, by using Western blotting and Coomassie blue staining after SDS-PAGE. A predominant CPE fragment was identified that is only ~1 kDa smaller than CPE and larger than the CPE fragment produced by trypsin. Since trypsin removes only the first 25 N-terminal amino acids from CPE, this result indicates that, even without S-carboxymethylation, i.e., under relatively physiological conditions, purified chymotrypsin does affect CPE but not by the removal of the 36 N-terminal amino acids.
Results using purified trypsin or chymotrypsin individually are informative, but the intestinal lumen contains multiple other host and microbial proteases and some of these proteases have been shown to process other
C. perfringens toxins. For example, serine proteases initiate the processing of
C. perfringens epsilon toxin, but carboxypeptidase then further processes that toxin [
34]. Consequently, it was unclear whether [i] chymotrypsin or trypsin effects on CPE predominate in the intestines, [ii] these two proteases act synergistically to process CPE beyond their individual cleavage effects, or [iii] other host or microbial proteases in the intestines can, alone or in combination with trypsin or chymotrypsin, cleave CPE beyond the individual effects observed for trypsin or chymotrypsin. Therefore, to better understand CPE processing during type F intestinal disease, the current study evaluated CPE processing by MICs ex vivo and in the small intestine.
The current results indicate that, when incubated ex vivo with MICs or added to the small intestine, CPE is mainly cleaved to a fragment matching the CPE fragment size generated by purified trypsin and ~3 kDa less than native CPE. With some MIC preparations, and in the tested mouse small intestinal loops, a minor CPE band ~7 kDa smaller than CPE was also observed, suggesting there may be some animal-to-animal variability in CPE processing. It should also be noted that this minor ~28 kDa CPE fragment is too small to be cytotoxic based upon results of previous CPE deletion analysis studies [
9].
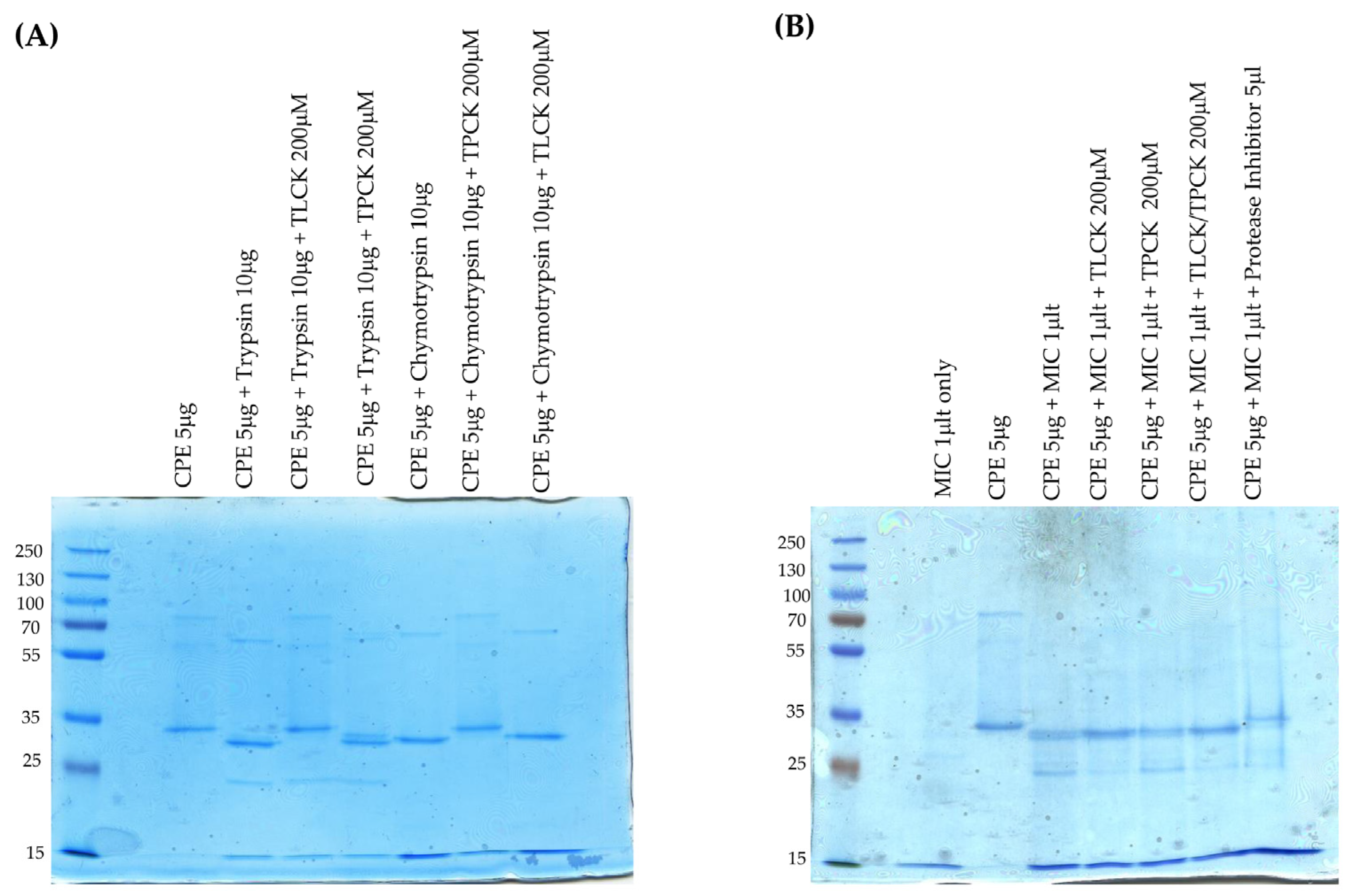
An important role for trypsin in CPE processing ex vivo received support from the ability of the trypsin inhibitor TLCK to affect this CPE processing by MICs. When MICs were preincubated with TLCK, there was a small increase in the size and staining intensity of the main CPE fragment, and the staining intensity of the minor CPE also visibly decreased. Preincubation of MICs with a chymotrypsin inhibitor did not affect normal CPE processing by MICs. When both trypsin and chymotrypsin inhibitors were present, CPE processing occurred similarly as in the presence of TLCK alone. Combining the pan-protease and trypsin-specific inhibitor results indicates that other host or microbial proteases in the intestines can affect CPE processing in the absence of trypsin, although, as mentioned, trypsin appears necessary and sufficient for processing CPE into the major fragment of ~32 kDa. These inhibitor results also indicate that the minor processed CPE band of ~28 kDa observed with some MIC preparations, and produced in the intestine, also involves trypsin activity. Of note, CPE structural biology studies [
9] showed that the N-terminal 34 amino acids of CPE [which includes the N-terminal trypsin cleavage site] have no detectable electron density via X-ray crystallography. Since this CPE region is disordered, it may be more accessible to intestinal proteases, including trypsin, when the toxin is free in solution.
Kinetically, the current study determined that CPE processing by MICs ex vivo is very rapid, reaching completion within 10 min. This rapid processing likely provides a major explanation for the detection of processed CPE in intestinal loops treated with CPE for only 15 min, i.e., considerable amounts of CPE may be proteolytically processed even before this toxin binds to the intestines or becomes sequestered in the large CPE complex. However, it remains possible that some CPE may be processed once it becomes localized in the large CPE complex given that CPE binding and large CPE complex formation also occur quickly in vivo, becoming detectable by the 15 min treatment time-point in mouse small intestinal loops. Consistent with the possibility that some CPE cleavage might occur after the toxin becomes sequestered in the large complex, a previous study [
37] showed that a portion of CPE in the large complex remains exposed on the membrane surface, i.e., CPE antibodies react with CPE that is present in the large complex in intestinal brush border membranes. If some processing of CPE localized in the large complex does occur, this might involve the unstructured N-terminus of CPE remaining outside the large complex and therefore being accessible to trypsin. It is also possible that CPE remains more susceptible to proteases when transiently located in the small complex or before the prepore to pore shift. Given the absence of structural information concerning CPE complexes, these issues will await future studies.
Intestinal proteases vary in their effects on the toxicity of
C. perfringens toxins produced in the intestines [
1,
33,
34]. For some toxins [e.g., epsilon toxin], this processing has a toxicity-activating effect, but for other toxins [e.g., beta toxin], proteolytic cleavage is detrimental, with a toxin-inactivating effect. The current study first explored this issue in vitro. When CPE and MICs were preincubated together and that mix was then added to Caco-2 cells, this experiment revealed that CPE preincubated with MICs exhibits similar Caco-2 cell cytotoxicity as does CPE preincubated without MICs, i.e., proteolytic processing by MICs did not substantially affect CPE cytotoxicity under the tested conditions. Consistent with the MIC-processed CPE retaining cytotoxic activity in vitro, the current study also demonstrated that, while much of the CPE associated with the large complex formed in the intestines is proteolytically processed, it retains intestinal enterotoxicity, i.e., in the presence of intestinal proteases, CPE still causes the intestinal damage important for type F disease [
9,
12,
38].
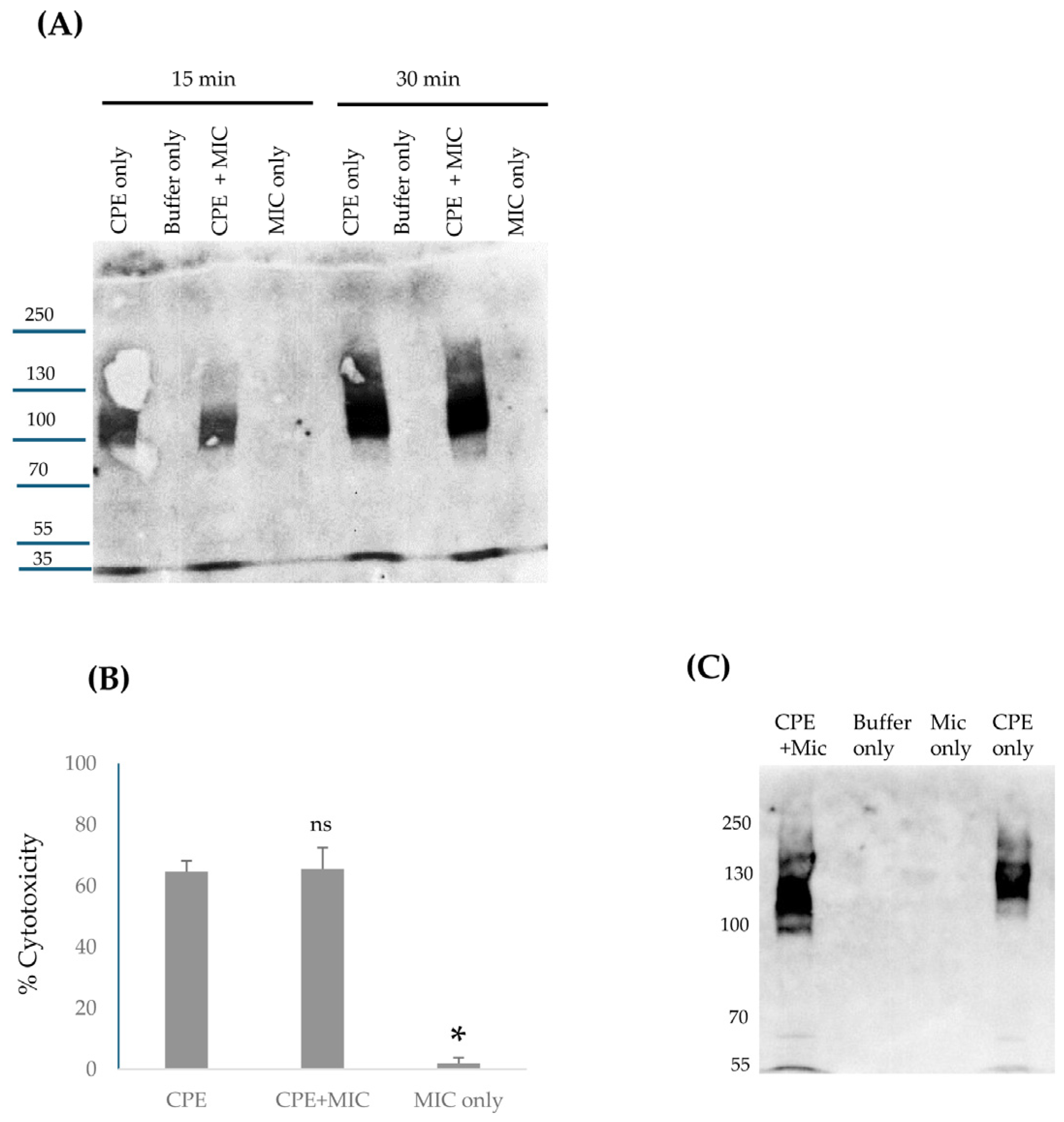
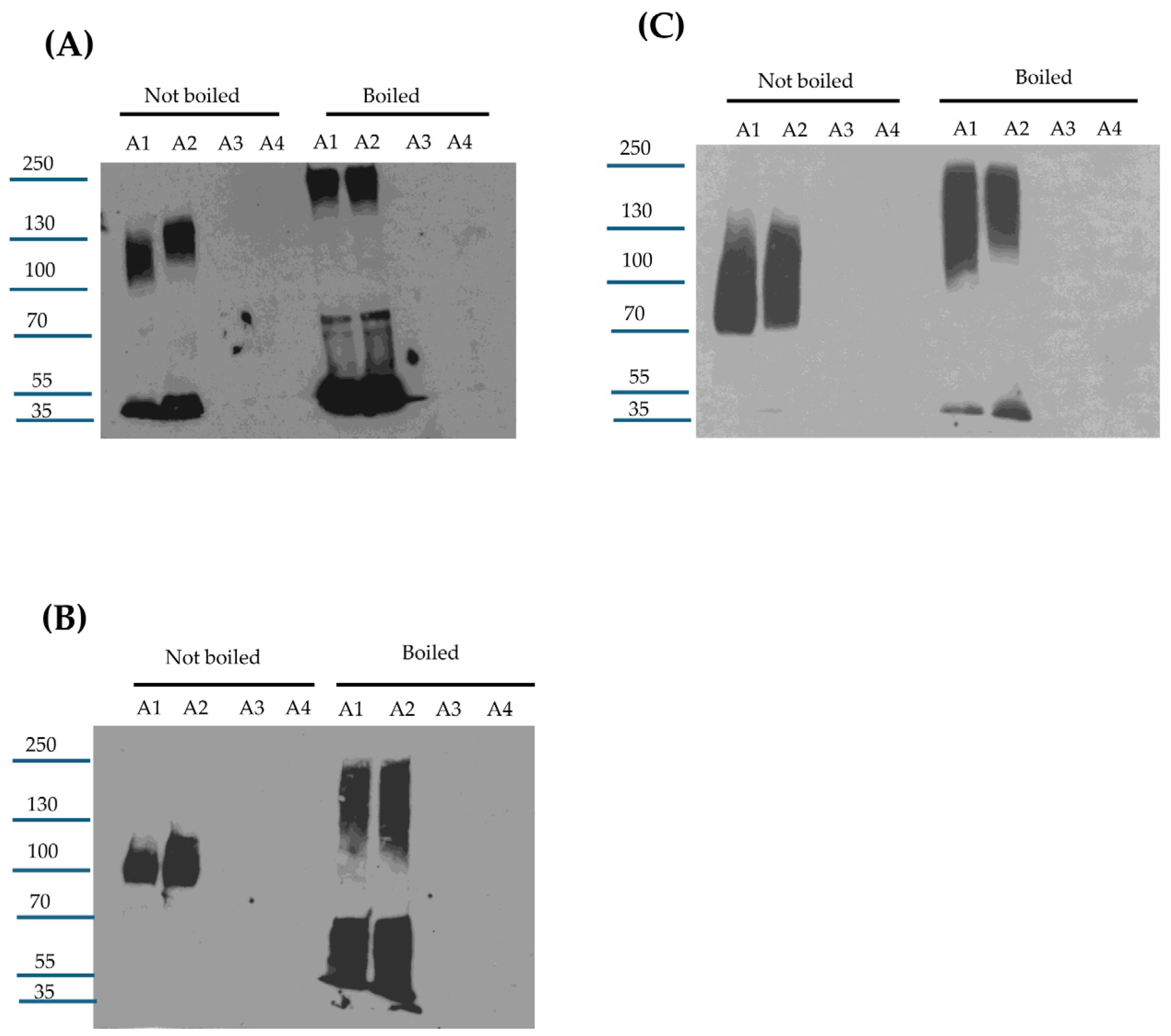
Beyond providing important insights into CPE proteolytic processing under intestinal conditions, the current study also offers new information regarding large CPE complex formation and stability in vivo. The availability of mouse small intestinal loops treated for different times with CPE allowed this study to analyze large CPE complex formation kinetics and large CPE complex stability in vivo. CPE Western blot studies showed that large CPE complex formation and, by extension, CPE binding [since it is required for large CPE complex formation [
9]] occur rapidly in the intestines. These Western blot results further indicate that by 30 min, the vast majority of CPE injected into mouse small intestinal loops had already bound and become sequestered in large CPE complex. However, a significant percentage of the CPE present in large CPE complex after this 30 min treatment was still susceptible to dissociation by sample boiling prior to SDS-PAGE. In contrast, after 4 h of CPE treatment, very little CPE could be released from the large complex by sample boiling. Collectively, these results indicate that the large CPE complex formed in vivo becomes more physically stable with time. Further studies are needed to better analyze how the formation and composition of the large CPE complex influence stability of this complex in vivo. It would also be worthwhile in the future to examine whether the strain, age, or diet of mice influence CPE complex formation or stability in vivo.
4. Materials and Methods
4.1. Proteases and Inhibitors
Purified trypsin [Bovine Pancreas TPCK-treated Trypsin] was purchased from Thermo Scientific (Rockford, IL, USA), while purified chymotrypsin [Alpha Chymotrypsin TLCK-treated] was obtained from Worthington Biochemical Corporation (Lakewood, NJ, USA). N-Tosyl-L-phenylalanine chloromethyl ketone [Synonym: TPCK] chymotrypsin inhibitor was purchased from Sigma (St. Louis, MO, USA), as was N-a-tosyl-L-lysine chloromethyl ketone [Synonym: TLCK] trypsin inhibitor. Protease Inhibitor Cocktail III Mammalian [AEBSF HCl: 100 mM, Aprotinin: 80 uM, Bestatin: 5 mM, E-64: 1.5 mM, Leupeptin: 2 mM, Pepstatin A: 1 mM] was purchased from RPI (Mt Prospect, IL, USA) [Research Products International].
4.2. Clostridium Perfringens Enterotoxin [CPE]
As described previously [
39], CPE was purified from type F strain NCTC8238 [ATCC 12916] using sequential ammonium sulfate precipitations and gel filtration chromatography. This purified CPE was homogeneous as assessed by SDS-PAGE.
4.3. Treatment of CPE with Purified Trypsin In Vitro
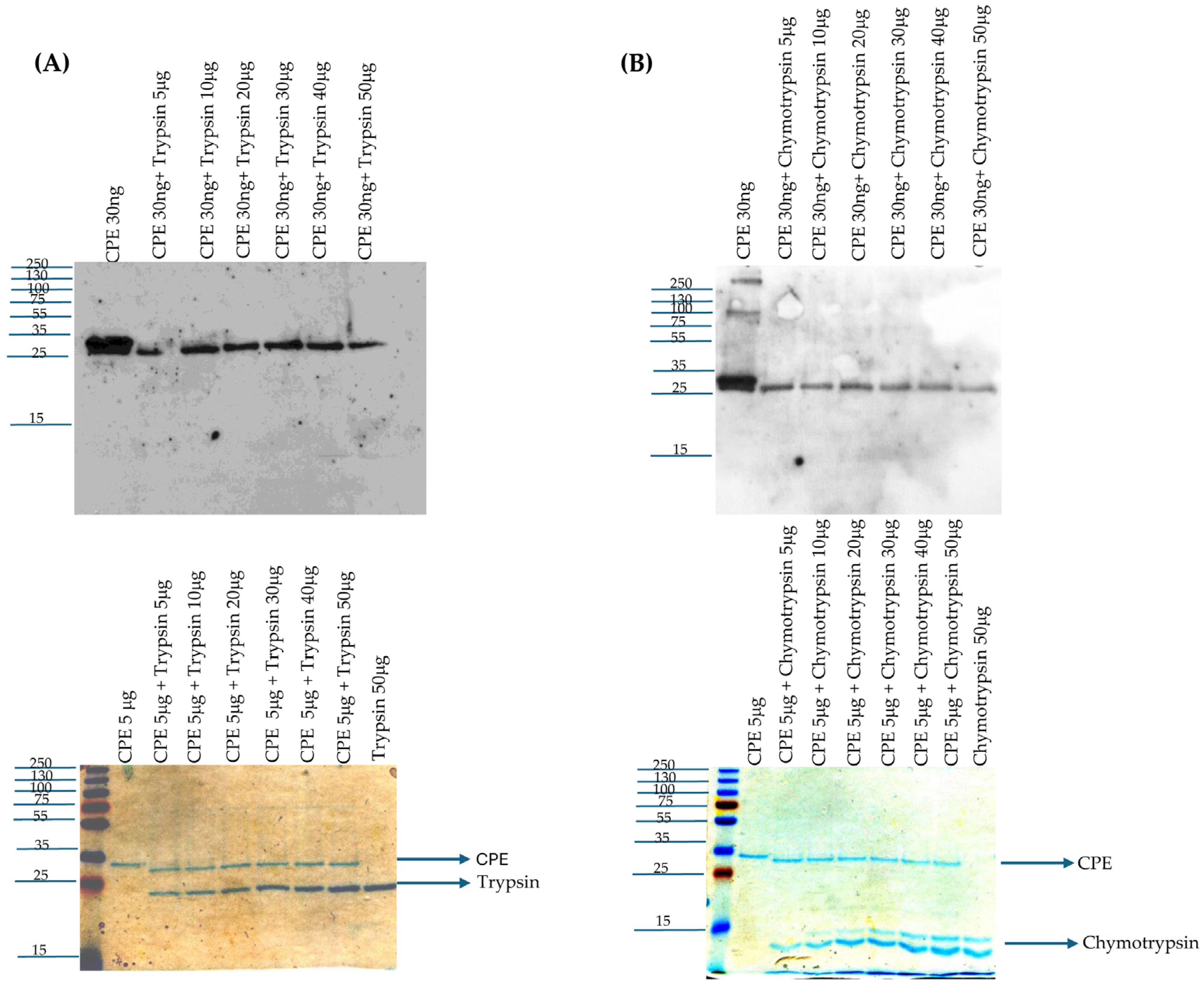
Purified CPE [30 ng] was treated with 5, 10, 20, 30, 40, or 50 μg of trypsin in PBS [total volume 25 μL] for 10 min at 37 °C. After this incubation, 5 μL of 5x loading buffer (which contains both SDS and β-mercaptoethanol) was added to each sample and the samples were then loaded onto a 12% acrylamide gel containing SDS. After electrophoresis, the separated proteins on these gels were electrotransferred onto nitrocellulose membranes [0.45 μm pore size, Biorad (Hercules, CA, USA)], which were then blocked with 5% milk in Tris-buffer saline with 0.1% Tween-20 [Fisher Scientific, Fair Lawn, NJ, USA]. The blocked membranes were incubated overnight at 4 °C with a 1:1000 dilution of polyclonal CPE antibody [
40]. On the next day, the blot was incubated with a 1:10,000 dilution of goat anti-rabbit IgG antibody conjugated with horseradish peroxidase [Invitrogen (Eugene, OR, USA)] at RT for 1 h with gentle shaking. The blot was developed using SuperSignal West Pico PLUS Chemiluminescent Substrate [Thermo Scientific (Rockford, IL, USA)].
The above experiment was also performed using a reaction containing 5 μg of CPE in PBS [total volume 25 μL] with 5–50 μg of trypsin in PBS that was incubated for 10 min at 37 °C, followed by electrophoresis, as above, and staining the gels with Coomassie blue [G-250, Biorad (Hercules, CA, USA)].
4.4. Treatment of CPE with Purified Chymotrypsin In Vitro
Purified CPE [30 ng] was treated with 5, 10, 20, 30, 40, or 50 μg of chymotrypsin in PBS [total volume 25 μL] for 10 min at 37 °C. After this treatment, CPE Western blotting was performed as described above for trypsin treatment. Similarly, the above experiment was repeated using a reaction containing 5 μg of CPE in 25 μL of PBS with 5–50 μg of purified chymotrypsin that was incubated for 10 min at 37 °C, followed by electrophoresis [as described above] and staining the gels with Coomassie blue stain.
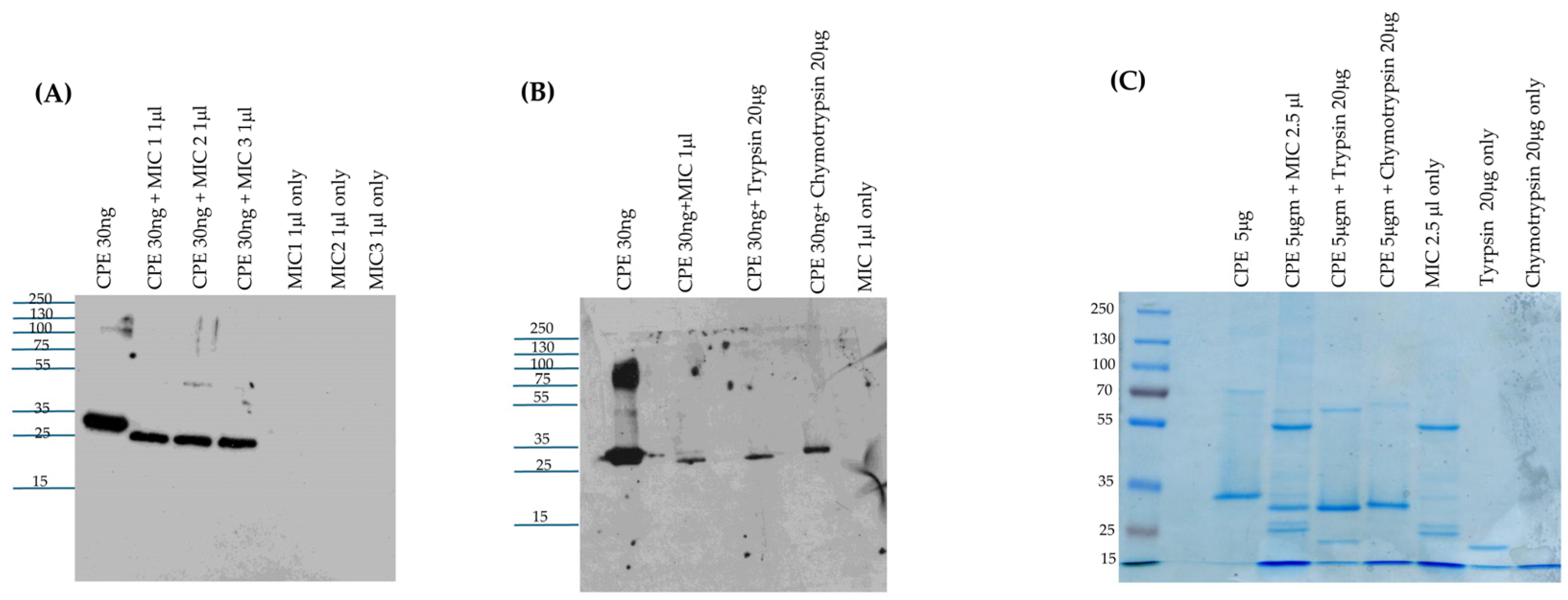
4.5. Preparation of MICs
Small intestinal lumen contents were obtained from healthy ~4-month-old BALB/c mice after they had been euthanized by means of CO2 asphyxiation for colony management. Equal numbers of male and female mice were used. After euthanasia, small intestinal lumen contents were immediately harvested from excised small intestine by gentle squeezing and stored at −80 °C. For use in experiments, each intestinal content sample was thawed, weighed, and mixed with PBS buffer at a ratio of 1:1. The mixture was then centrifuged for 5 min and supernatants [now designated as MIC] were collected, aliquoted, and stored at −80 °C until use in the current study. Typically, one MIC preparation was used for each experimental repetition.
4.6. Comparison of CPE Processing by MICs from Different Mice
To evaluate the ex vivo effects of MICs from different mice on CPE, 30 ng of native CPE was incubated in 25 μL of PBS with 1 μL of MICs from three different mice [2 female and 1 male] for 10 min at 37 °C. After incubation, 5 μL of 5x loading buffer was added to each sample and the samples were then boiled for 5 min before loading onto a 12% acrylamide gel containing SDS. After electrophoresis, separated proteins were electrotransferred onto a 0.45 μm pore size nitrocellulose membrane and processed for CPE Western blot analysis as described earlier.
4.7. Comparison of CPE Processing by MICs, Purified Trypsin, or Purified Chymotrypsin
For CPE Western blot analyses, 30 ng of native CPE was added to PBS containing 1 μL of MICs, 20 μg of purified trypsin, or 20 μg of purified chymotrypsin [reaction volumes 25 μL] for 10 min at 37 °C. Identical MIC, trypsin, or chymotrypsin samples were similarly incubated without CPE. After this incubation, all samples were treated with 5 μL of 5x loading buffer and this sample was then boiled for 5 min; the trypsin- or chymotrypsin-treated samples were not boiled to reduce CPE aggregation. All samples were then electrophoresed on 12% acrylamide gels containing SDS and processed for CPE Western blotting, as described earlier. For Coomassie blue staining analyses, 5 μg of native CPE was incubated in PBS [reaction volume of 25 μL] containing 2.5 μL of MICs, 20 μg of purified trypsin, or 20 μg of purified chymotrypsin. The samples were then processed and electrophoresed on a 12% acrylamide gel containing SDS as described above for CPE Western blots. After electrophoresis, gels were stained with Coomassie blue.
4.8. Effects of Protease Inhibitors on MIC Processing of CPE Ex Vivo
Purified trypsin [10 μg] was preincubated with 200 μM TLCK or 200 μM TPCK in PBS [total reaction volume was 20 μL], while 10 μg of purified chymotrypsin was preincubated with 200 μM TPCK or 200 μM TLCK in PBS [total reaction volume was 20 μL]. The control sample contained only 20 μL of PBS. All samples were preincubated at 37 °C for 30 min. After this preincubation, 5 μg of CPE in 5 μL of PBS was added to each sample before incubation for 10 min at 37 °C. The incubated samples were then treated with 5 μL of 5x loading buffer and electrophoresed on 12% acrylamide gels containing SDS, followed by staining these gels with Coomassie blue. For MIC treatments, MICs [1 μL] in PBS [total reaction volume of 20 μL] were preincubated with 200 μM TLCK, 200 μM TPCK, 200 μM each of TCPK and TLCK, or 5 μL of Protease Inhibitor Cocktail III. As a control, 1 μL of MICs was preincubated in 20 μL of PBS. These preincubations were performed at 37 °C for 30 min. After this preincubation, 5 μL of PBS containing 5 μg of CPE was added to each sample and the samples were incubated for 10 min at 37 °C. These samples were then treated with 5 μL of 5x loading buffer, boiled for 5 min, and then electrophoresed on 12% acrylamide gels containing SDS, followed by staining the gels with Coomassie blue staining.
4.9. Caco-2 Cell Culture
Authenticated Caco-2 cells were purchased from ATCC. Genetic information about these cells (lot number 61777387) is available at
https://www.atcc.org/products/htb-37., as accessed on 15 March 2025. These Caco-2 cells were grown in Eagle’s essential medium [Lonza (Walkersville, MD, USA)] supplemented with 10% fetal bovine serum [Gibco (Grand Island, NY, USA)], 1% MEM nonessential amino acids [Cytiva (Logan, UT, USA)], 100 μg/mL of penicillin-streptomycin solution [Corning (Manassas, VA, USA)], and 1% glutamine [Corning]. These cultures were maintained at 37 °C in a 5% CO
2 atmosphere.
4.10. MICs’ Effects on Large CPE Complex Formation by Caco-2 Cells
Caco-2 cells were grown in 6-well cell culture dishes for 5–6 days. Cultures were then treated for 15 min at 37 °C with CPE [0.5 μg/mL] in the presence of 1 μL of MICs in 1 mL of HBSS; the MICs were first added into the culture, immediately followed by the addition of CPE. Other Caco-2 cell wells were treated with 1 mL of HBSS alone or with 1 mL of HBSS containing 1 μL of MICs [no CPE]. After a 15 or 30 min of incubation, each sample was gently collected using a cell scrapper and lysed with RIPA buffer [Boston BioProducts, Inc (Millford, MA, USA).] in the presence of Benzonase Nuclease [EMD Millipore (Burlington, MA, USA)] and Protease Inhibitor Cocktail III at RT for 30 min with gentle absorption. The samples were centrifuged and the lysates were collected in sterile tubes. A 1 μL aliquot of lysate from each treatment was then mixed with 24 μL of HBSS buffer plus 5 μL of 5x loading buffer and the total 30 μL final volume was loaded onto a 6% SDS PAGE gel and processed for CPE Western blotting as already described.
4.11. Effects of MIC Pretreatment on CPE Cytotoxicity and Large CPE Complex Formation for Caco-2 Cells
HBSS containing native CPE [3 μg] in the presence or absence of 2.5 μL of MICs, HBSS containing 2.5 μL of MICs, or HBSS alone [total sample volumes of 25 μL] was preincubated at 37 °C for 10 min. After that preincubation, samples were adjusted with HBSS to a 1 μg/mL CPE concentration, with a similar dilution of the MIC alone [no CPE] sample. Each sample [1 mL] was then added onto confluent Caco-2 cell cultures for 1 h at 37 °C. After this treatment, culture supernatants were collected, centrifuged and the lysates were processed for LDH-release, as an indicator of cytotoxicity, using the CyQUANT LDH Cytotoxicity Assay Kit [Invitrogen].
Adherent cells in these cultures were gently collected using a cell scrapper and lysed with RIPA buffer with Benzonase Nuclease and Protease Inhibitor Cocktail III at RT for 30 min. The samples were centrifuged and a 1 μL aliquot of lysate from each treatment was then mixed with 24 μL of HBSS buffer plus 5 μL of 5x loading buffer; the total final volume was then loaded onto a 6% acrylamide gel with SDS and processed for CPE Western blotting as already described.
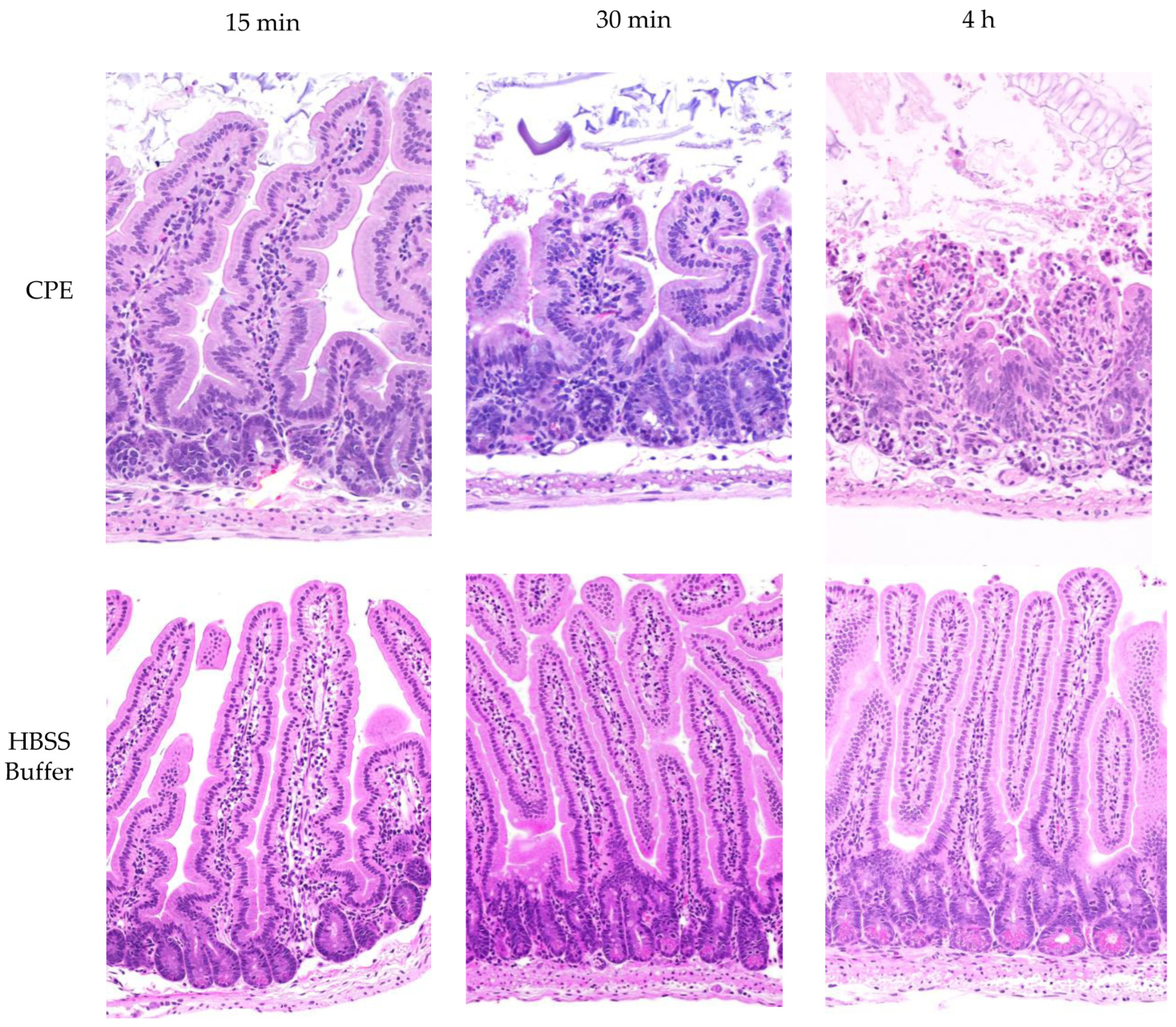
4.12. CPE Treatment of Mouse Small Intestinal Loops
To assess the effects of intestinal proteases on in vivo CPE processing, large CPE complex formation and enterotoxicity, the mouse small intestinal loop model was used. Balb/C mouse small intestine segments [~10 cm] were surgically ligated under general anesthesia, as previously described [
18]. These intestinal loop experiments were conducted using 6 groups of mice [
n = 6 per group, with equal numbers of male and female mice]. Each loop was challenged with 1 mL of HBSS that did or did not contain 100 μg of purified CPE. After 15 min, 30 min, or 4 h of incubation, the mice were euthanized. Samples from these treated intestinal loops were collected, fixed in 10% buffered formalin at pH 7.2 for 72 h and then processed into 4 μm sections before staining with hematoxylin and eosin [H&E]. These samples were examined by a pathologist in a blinded fashion. Contents of the intestinal loops were collected by gentle squeezing and stored at −80 °C until their use in the experiments.
4.13. Analysis of Large CPE Complex Formation and Stability In Vivo
Intestinal contents from control or CPE-treated mice, as described above, were centrifuged and half of the supernatant volume was boiled for 5 min while the other half was not. These samples were then electrophoresed on 12% acrylamide gels with SDS or on 6% acrylamide gel with SDS and processed for CPE Western blotting as described above for Caco-2 cells. Samples run on 12% acrylamide gels with SDS were boiled for 5 min before electrophoresis and Western blotting.
4.14. Statistical Analyses
The statistical significance of CPE cytotoxicity results was assessed by means of one-way ANOVA with Dunnett’s multiple-comparison test.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}