Advancing Enzyme-Based Detoxification Prediction with ToxZyme: An Ensemble Machine Learning Approach

 , , , ,
, , , ,  and
and
Abstract

1. Introduction
2. Results
2.1. Computing Features, Standardization, and Splitting Dataset
2.2. Compositional, Residual, and Gini Score Analysis
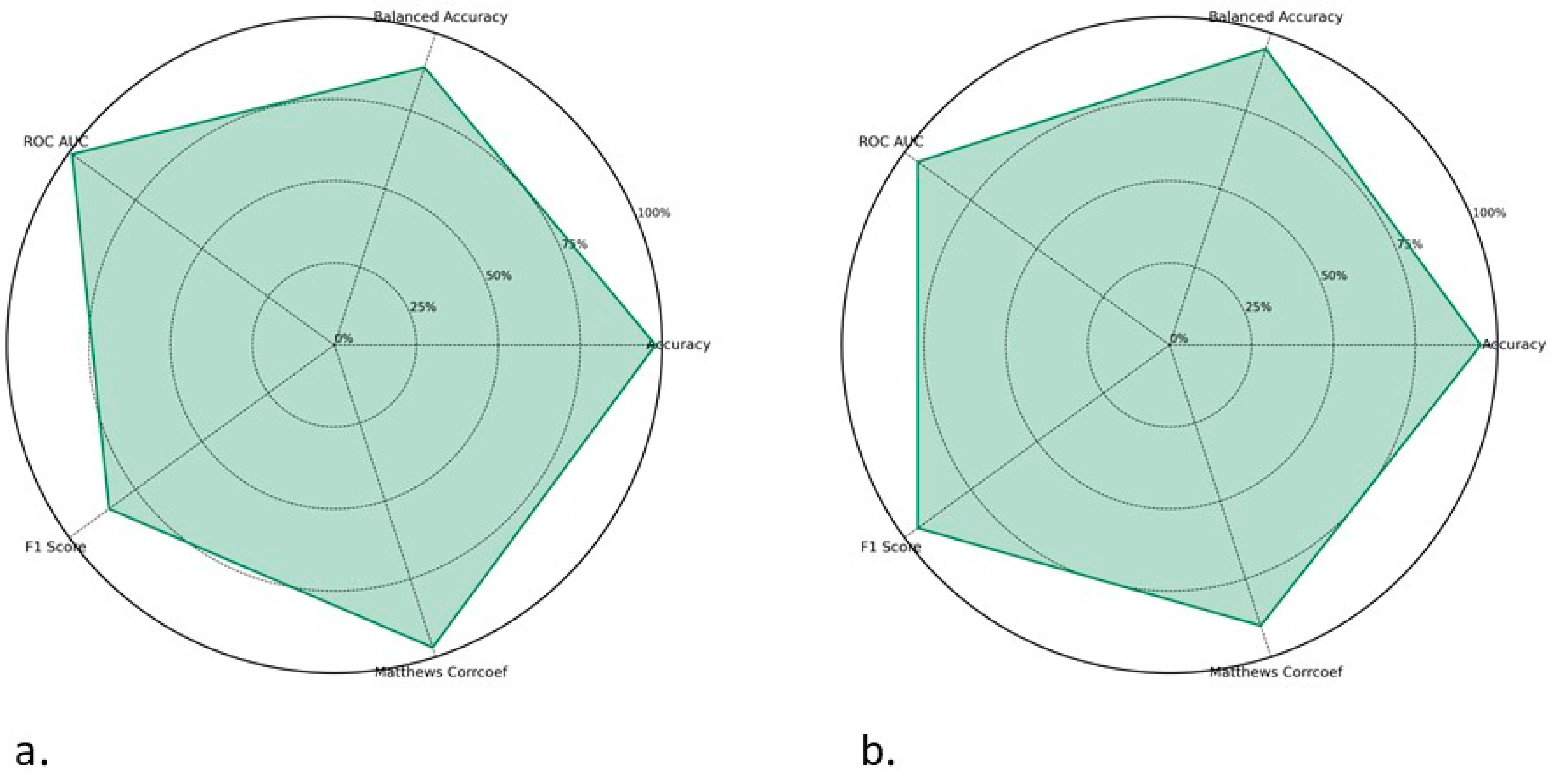
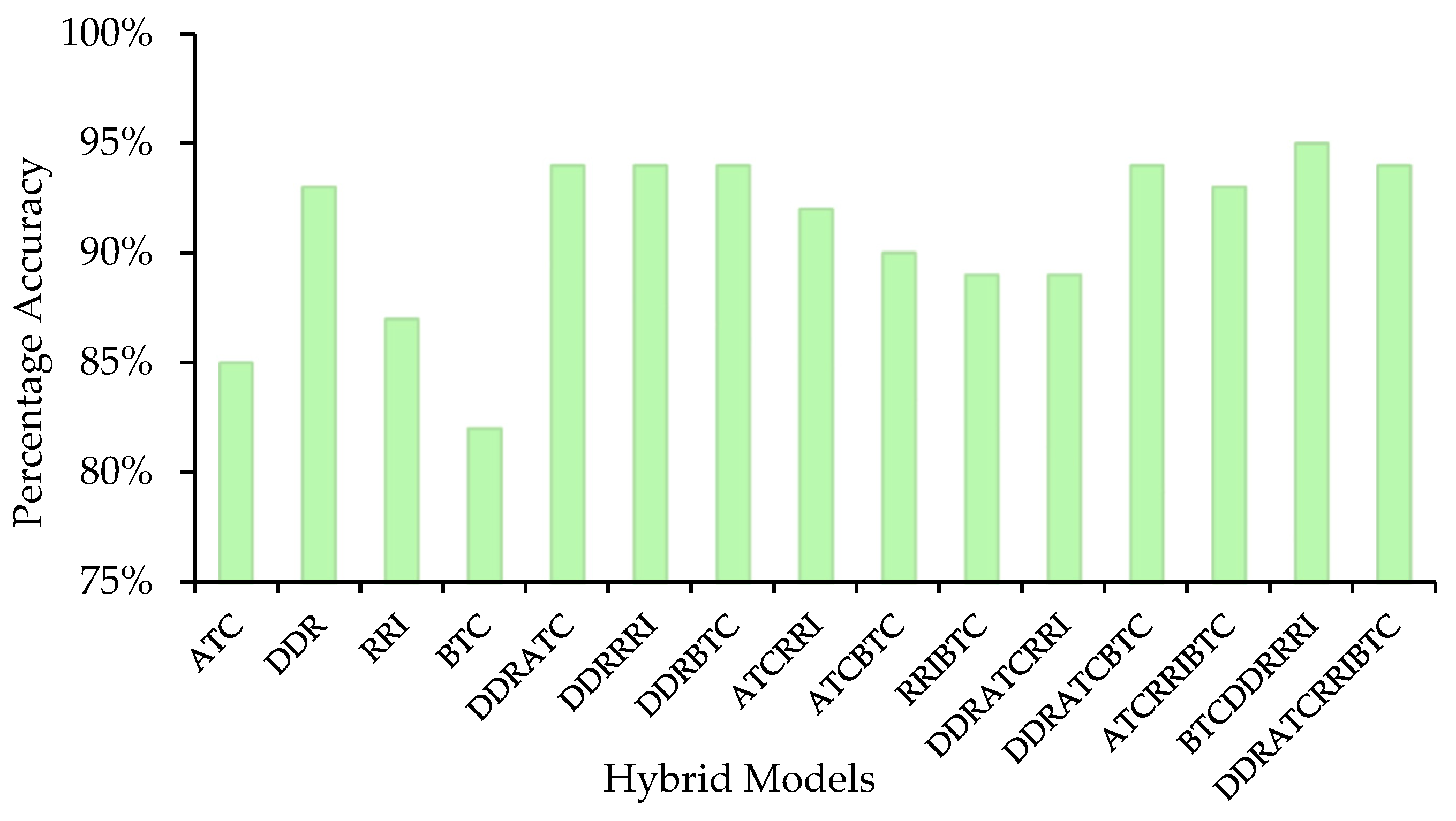
2.3. Development of Models in Main Dataset
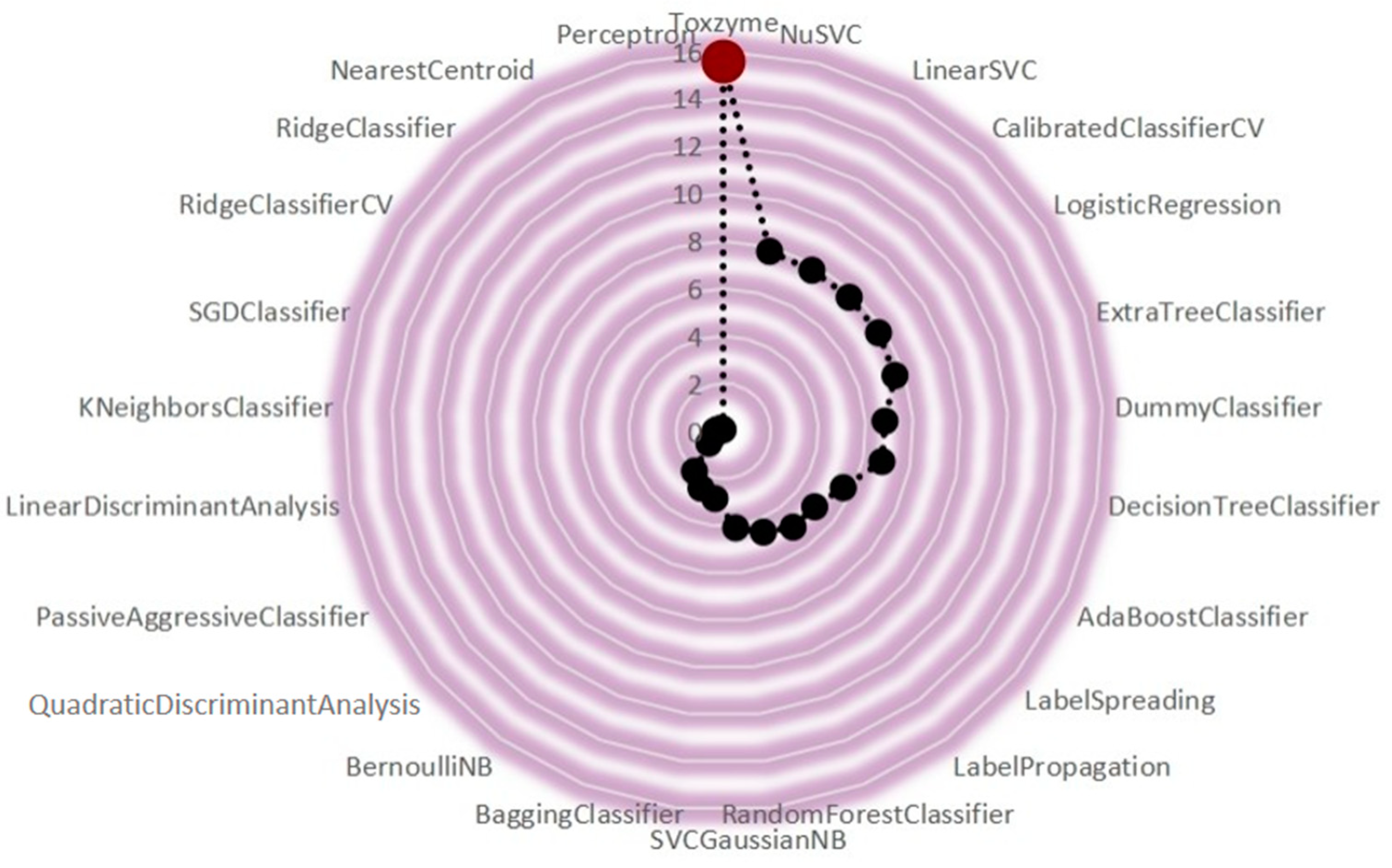
2.4. ToxZyme Workflow Performance Compared to Classical Models
2.5. ROC Curve
2.6. Graphical User Interface of ToxZyme
3. Discussion
4. Conclusions
5. Materials and Methods
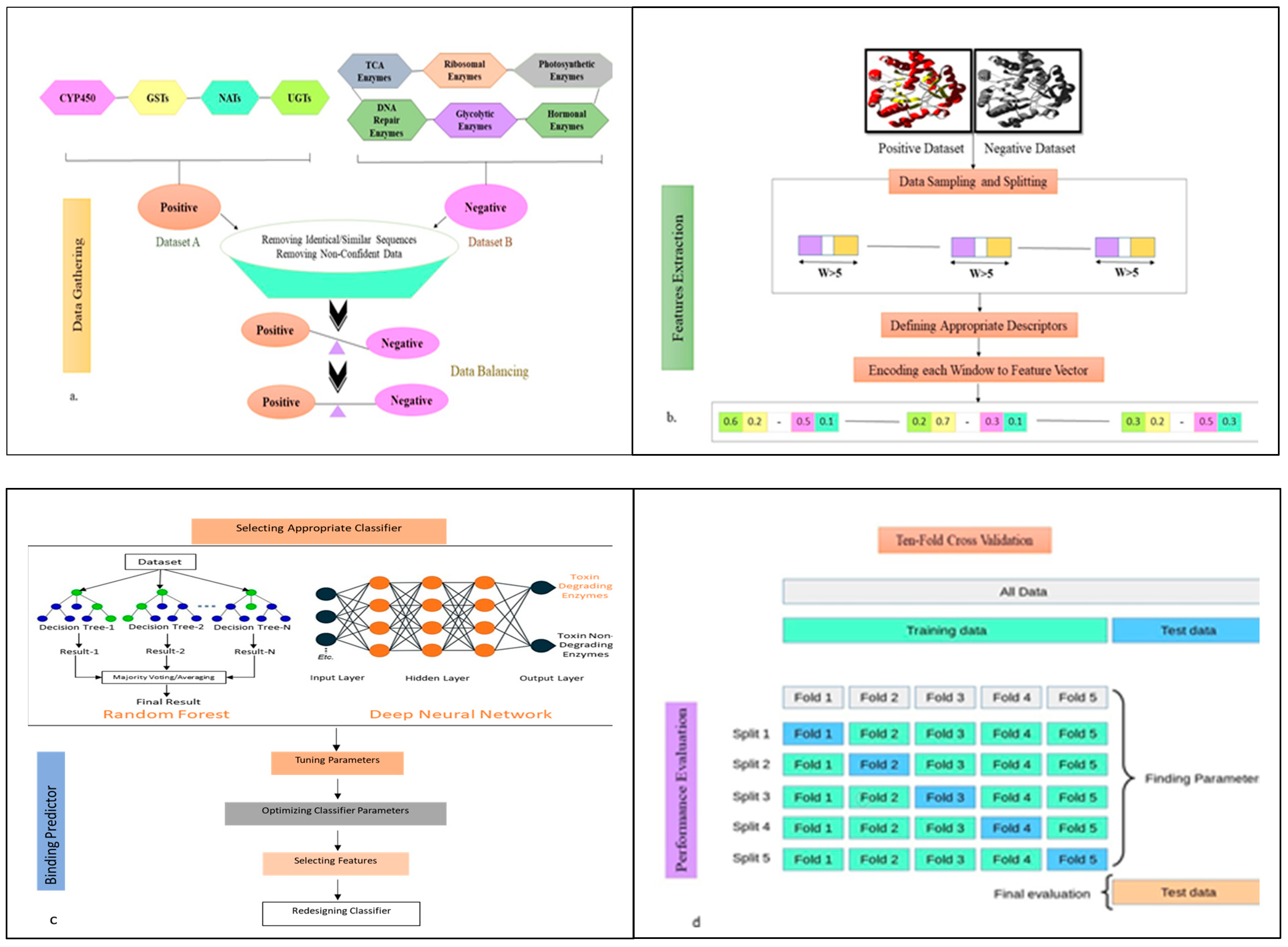
5.1. Dataset Preparation
5.2. Pipeline of ToxZyme
5.3. Features for Prediction
5.4. Class Balancing and Feature Alignment
5.5. Ensemble Model for Toxin-Degrading Enzyme Prediction and Performance Evaluation
5.6. ToxZyme Deployment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RF | Random Forest |
| DNN | Deep Neural Network |
| ML | Machine learning |
| RNN | Recurrent neural network |
| CNN | Convolutions neural network |
| BTC | Bond type composition |
| ROC AUC | Receiver operating characteristic area under the curve |
| CD-HIT | Cluster Database at High Identity with Tolerance |
| SGD | Stochastic Gradient Descent |
| SVC | Support Vector Classification |
| AAC | Amino acid composition |
| ATC | Atom type composition |
| DDR | Distance distribution of residues |
| RRI | Repeat residue information |
| GUI | Graphical user interface |
References
- Singh, B.K.; Walker, A. Microbial Degradation of Organophosphorus Compounds. FEMS Microbiol. Rev. 2006, 30, 428–471. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.M.; Hashemi, S.A.; Iman Moezzi, S.M.; Ravan, N.; Gholami, A.; Lai, C.W.; Chiang, W.H.; Omidifar, N.; Yousefi, K.; Behbudi, G. Recent Advances in Enzymes for the Bioremediation of Pollutants. Biochem. Res. Int. 2021, 2021, 5599204. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, X.; Luan, H.; Zhang, Y.; Xu, W.; Feng, W.; Song, P. Bioenzymatic Detoxification of Mycotoxins. Front. Microbiol. 2024, 15, 1434987. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.H. Occurrence, Toxicity, and Analysis of Major Mycotoxins in Food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, Toxicology, and Exposure Assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Čolović, R.; Puvača, N.; Cheli, F.; Avantaggiato, G.; Greco, D.; Đuragić, O.; Pinotti, L. Decontamination of Mycotoxin-Contaminated Feedstuffs and Compound Feed. Toxins 2019, 11, 617. [Google Scholar] [CrossRef]
- Kaul, P.; Asano, Y. Strategies for Discovery and Improvement of Enzyme Function: State of the Art and Opportunities. Microb. Biotechnol. 2012, 5, 18–33. [Google Scholar] [CrossRef]
- Xu, H.; Wang, L.; Sun, J.; Wang, L.; Guo, H.; Ye, Y.; Sun, X. Microbial Detoxification of Mycotoxins in Food and Feed. Crit. Rev. Food Sci. Nutr. 2022, 62, 4951–4969. [Google Scholar] [CrossRef]
- Gratz, S.W. Do Plant-Bound Masked Mycotoxins Contribute to Toxicity? Toxins 2017, 9, 85. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, D.; Wei, N. Enzyme Discovery and Engineering for Sustainable Plastic Recycling. Trends Biotechnol. 2022, 40, 22–37. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2019, 10, 1210–1223. [Google Scholar] [CrossRef]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Samui, P.; Kumar, D.R.; Asteris, P.G. State-of-the-Art XGBoost, RF, and DNN-Based Soft-Computing Models for PGPN Piles. Geomech. Geoeng. 2024, 19, 975–990. [Google Scholar] [CrossRef]
- Sahibzada, K.I.; Shahid, S.; Akhter, M.; Abid, R.; Azhar, M.; Hu, Y.; Wei, D.Q. HIV OctaScanner: A Machine Learning Approach to Unveil Proteolytic Cleavage Dynamics in HIV-1 Protease Substrates. J. Chem. Inf. Model. 2025, 65, 640–648. [Google Scholar] [CrossRef]
- Noviandy, T.R.; Maulana, A.; Irvanizam, I.; Idroes, G.M.; Maulydia, N.B.; Tallei, T.E.; Subianto, M.; Idroes, R. Interpretable Machine Learning Approach to Predict Hepatitis C Virus NS5B Inhibitor Activity Using Voting-Based LightGBM and SHAP. Intell. Syst. Appl. 2025, 25, 200481. [Google Scholar] [CrossRef]
- Tijare, P.; Kumar, N.; Raghava, G.P. Designing of Thermostable Proteins with a Desired Melting Temperature. bioRxiv 2024. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification Using Support Vector Machines. Mach. Learn. 2003, 46, 389–422. [Google Scholar] [CrossRef]
- Aldayel, M.F.; Alrajeh, H.S.; Sallam, N.M.A.; Imran, M. Bacillus amyloliquefaciens IKMM and zinc nanoparticles as biocontrol candidate induce the systemic resistance by producing antioxidants in tomato plants challenged with early blight pathogen. J. Crop Health 2024, 76, 87–103. [Google Scholar] [CrossRef]
- Ndiaye, S.; Zhang, M.; Fall, M.; Ayessou, N.M.; Zhang, Q.; Li, P. Current review of mycotoxin biodegradation and bioadsorption: Microorganisms, mechanisms, and main important applications. Toxins 2022, 14, 729. [Google Scholar] [CrossRef]
- Liu, L.; Xie, M.; Wei, D. Biological detoxification of mycotoxins: Current status and future advances. Int. J. Mol. Sci. 2022, 23, 1064. [Google Scholar] [CrossRef]
- Lin, L.; Wang, L.; Zhao, X.; Li, J.; Wong, K.F. Indivec: An Exploration of Leveraging Large Language Models for Media Bias Detection with Fine-Grained Bias Indicators. arXiv 2024, arXiv:2402.00345. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Oversampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Rana, M.S.; Hossain, M.M.; Li, F. Comparative Analysis of Machine Learning Models for Predicting the Compressive Strength of Ultra-High-Performance Steel Fiber Reinforced Concrete. J. Eng. Res. 2025, 16, 1–6. [Google Scholar] [CrossRef]
- Escuder-Rodríguez, J.J.; DeCastro, M.E.; Becerra, M.; Rodríguez-Belmonte, E.; González-Siso, M.I. Advances of Functional Metagenomics in Harnessing Thermozymes. In Metagenomics; Academic Press: Cambridge, MA, USA, 2025; pp. 493–516. [Google Scholar] [CrossRef]
- Gaur, V.K.; Sharma, P.; Gaur, P.; Varjani, S.; Ngo, H.H.; Guo, W.; Chaturvedi, P.; Singhania, R.R. Sustainable mitigation of heavy metals from effluents: Toxicity and fate with recent technological advancements. Bioengineered 2021, 12, 7297–7313. [Google Scholar] [CrossRef]
- Chica, R.A.; Doucet, N.; Pelletier, J.N. Semi-Rational Approaches to Engineering Enzyme Activity. Biotechnol. Adv. 2005, 23, 545–555. [Google Scholar]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef]
- Ji, C.; Fan, Y.; Zhao, L. Review on biological degradation of mycotoxins. Anim. Nutr. 2016, 2, 127–133. [Google Scholar] [CrossRef]
- Alberts, J.F.; Engelbrecht, Y.; Steyn, P.S.; Holzapfel, W.H.; van Zyl, W.H. Biological degradation of aflatoxin B1 by Rhodococcus erythropolis. Appl. Environ. Microbiol. 2006, 72, 309–318. [Google Scholar] [CrossRef]
- Wu, Q.; Dohnal, V.; Huang, L.; Kuca, K.; Wang, X.; Chen, G.; Yuan, Z. Metabolic pathways of ochratoxin A. Curr. Drug Metab. 2011, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Das, M. A review on biological control and metabolism of aflatoxin. Crit. Rev. Food Sci. Nutr. 2003, 43, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Chabikwa, T.G.; Barbier, F.F.; Tanurdzic, M.; Beveridge, C.A. De Novo Transcriptome Assembly and Annotation for Gene Discovery in Avocado, Macadamia, and Mango. Sci. Data 2020, 7, 9. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ML Models Classifiers | Accuracy | Balanced Accuracy | ROC AUC | F1 Score | Matthews Corr. Coef. | Time Taken (s) |
|---|---|---|---|---|---|---|
| ToxZyme | 0.9533 | 0.9533 | 0.9533 | 0.9533 | 0.9070 | 11.5715 |
| LGBM | 0.9496 | 0.9496 | 0.9496 | 0.9496 | 0.8995 | 0.9148 |
| Extra Trees | 0.9475 | 0.9475 | 0.9475 | 0.9475 | 0.8961 | 1.9171 |
| Random Forest | 0.9467 | 0.9467 | 0.9467 | 0.9466 | 0.8953 | 6.1708 |
| SVC | 0.9383 | 0.9383 | 0.9283 | 0.9383 | 0.8768 | 2.7257 |
| Label Spreading | 0.9283 | 0.9283 | 0.9283 | 0.9283 | 0.8569 | 7.8394 |
| Label Propagation | 0.9283 | 0.9283 | 0.9283 | 0.9283 | 0.8569 | 4.9729 |
| K Neighbors | 0.9208 | 0.9208 | 0.9208 | 0.9208 | 0.8432 | 0.3244 |
| Bagging | 0.9133 | 0.9133 | 0.9133 | 0.9132 | 0.8289 | 4.2002 |
| Ada Boost | 0.9088 | 0.9088 | 0.9088 | 0.9087 | 0.8175 | 2.8612 |
| NuSVC | 0.8946 | 0.8946 | 0.8946 | 0.8943 | 0.7932 | 6.9112 |
| Linear SVC | 0.8917 | 0.8917 | 0.8917 | 0.8917 | 0.7835 | 1.4990 |
| Logistic Regression | 0.8913 | 0.8913 | 0.8913 | 0.8912 | 0.7826 | 0.1096 |
| Calibrated CV | 0.8913 | 0.8913 | 0.8913 | 0.8912 | 0.7827 | 6.1418 |
| SGD | 0.8804 | 0.8804 | 0.8804 | 0.8804 | 0.7612 | 0.1666 |
| Ridge | 0.8717 | 0.8717 | 0.8717 | 0.8717 | 0.7434 | 0.0427 |
| Decision Tree | 0.8713 | 0.8713 | 0.8713 | 0.8712 | 0.7425 | 0.9473 |
| Linear Discriminant Analysis | 0.8713 | 0.8713 | 0.8713 | 0.8712 | 0.7427 | 0.9291 |
| Ridge CV | 0.8692 | 0.8692 | 0.8692 | 0.8692 | 0.7385 | 0.1671 |
| Extra Tree | 0.8550 | 0.8550 | 0.8550 | 0.8550 | 0.7100 | 0.0583 |
| Passive Aggressive | 0.8475 | 0.8475 | 0.8475 | 0.8473 | 0.6971 | 0.0681 |
| Nearest Centroid | 0.8475 | 0.8475 | 0.8475 | 0.8475 | 0.6952 | 0.0433 |
| Bernoulli NB | 0.8442 | 0.8442 | 0.8442 | 0.8440 | 0.6895 | 0.0501 |
| Perceptron | 0.8438 | 0.8438 | 0.8438 | 0.8437 | 0.6876 | 0.0559 |
| Quadratic Discriminant Analysis | 0.8075 | 0.8075 | 0.8075 | 0.8048 | 0.6327 | 0.0708 |
| Gaussian NB | 0.8021 | 0.8021 | 0.8021 | 0.8001 | 0.6165 | 0.0412 |
| Dummy | 0.5000 | 0.5000 | 0.5000 | 0.3333 | 0.0000 | 0.0385 |
| Model | Accuracy (%) | Precision (%) | Recall (%) | ROC-AUC (%) |
|---|---|---|---|---|
| ToxZyme | 92.7 | 92.0 | 91.5 | 91.0 |
| DeepEC | 85.4 | 84.1 | 83.8 | 82.5 |
| PRIAM | 83.1 | 81.7 | 81.2 | 80.8 |
| EFICAz | 80.6 | 79.5 | 78.9 | 77.3 |
| Model Approach | Featured Models | Features |
|---|---|---|
| Monohybrids | Monohybrid I | ATC |
| Monohybrid II | DDR | |
| Monohybrid III | RRI | |
| Monohybrid IV | BTC | |
| Dihybrids | Dihybrid I | ATC + DDR |
| Dihybrid II | DDR + RRI | |
| Dihybrid III | DDR + BTC | |
| Dihybrid IV | ATC + RRI | |
| Dihybrid V | ATC + BTC | |
| Dihybrid VI | RRI + BTC | |
| Trihybrids | Trihybrid I | ATC + DDR + RRI |
| Trihybrid II | ATC + DDR + BTC | |
| Trihybrid III | ATC + RRI + BTC | |
| Trihybrid III | BTC + DDR + RRI | |
| Tetrahybrid | Tetrahybrid | ATC + DDR + RRI + BTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahibzada, K.I.; Shahid, S.; Akhter, M.; Faisal, M.; Abd El Rahman, R.A.; Imran, M.; Lv, Y.; Wei, D.; Hu, Y. Advancing Enzyme-Based Detoxification Prediction with ToxZyme: An Ensemble Machine Learning Approach. Toxins 2025, 17, 171. https://doi.org/10.3390/toxins17040171
Sahibzada KI, Shahid S, Akhter M, Faisal M, Abd El Rahman RA, Imran M, Lv Y, Wei D, Hu Y. Advancing Enzyme-Based Detoxification Prediction with ToxZyme: An Ensemble Machine Learning Approach. Toxins. 2025; 17(4):171. https://doi.org/10.3390/toxins17040171
Chicago/Turabian StyleSahibzada, Kashif Iqbal, Shumaila Shahid, Mohsina Akhter, Muhammad Faisal, Reham A. Abd El Rahman, Muhammad Imran, Yangyong Lv, Dongqing Wei, and Yuansen Hu. 2025. "Advancing Enzyme-Based Detoxification Prediction with ToxZyme: An Ensemble Machine Learning Approach" Toxins 17, no. 4: 171. https://doi.org/10.3390/toxins17040171
APA StyleSahibzada, K. I., Shahid, S., Akhter, M., Faisal, M., Abd El Rahman, R. A., Imran, M., Lv, Y., Wei, D., & Hu, Y. (2025). Advancing Enzyme-Based Detoxification Prediction with ToxZyme: An Ensemble Machine Learning Approach. Toxins, 17(4), 171. https://doi.org/10.3390/toxins17040171