Myostatin Exacerbates Endothelial Dysfunction Induced by Uremic Toxin Indoxyl Sulfate and Is Associated with Hemodialysis Arteriovenous Access Complications

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
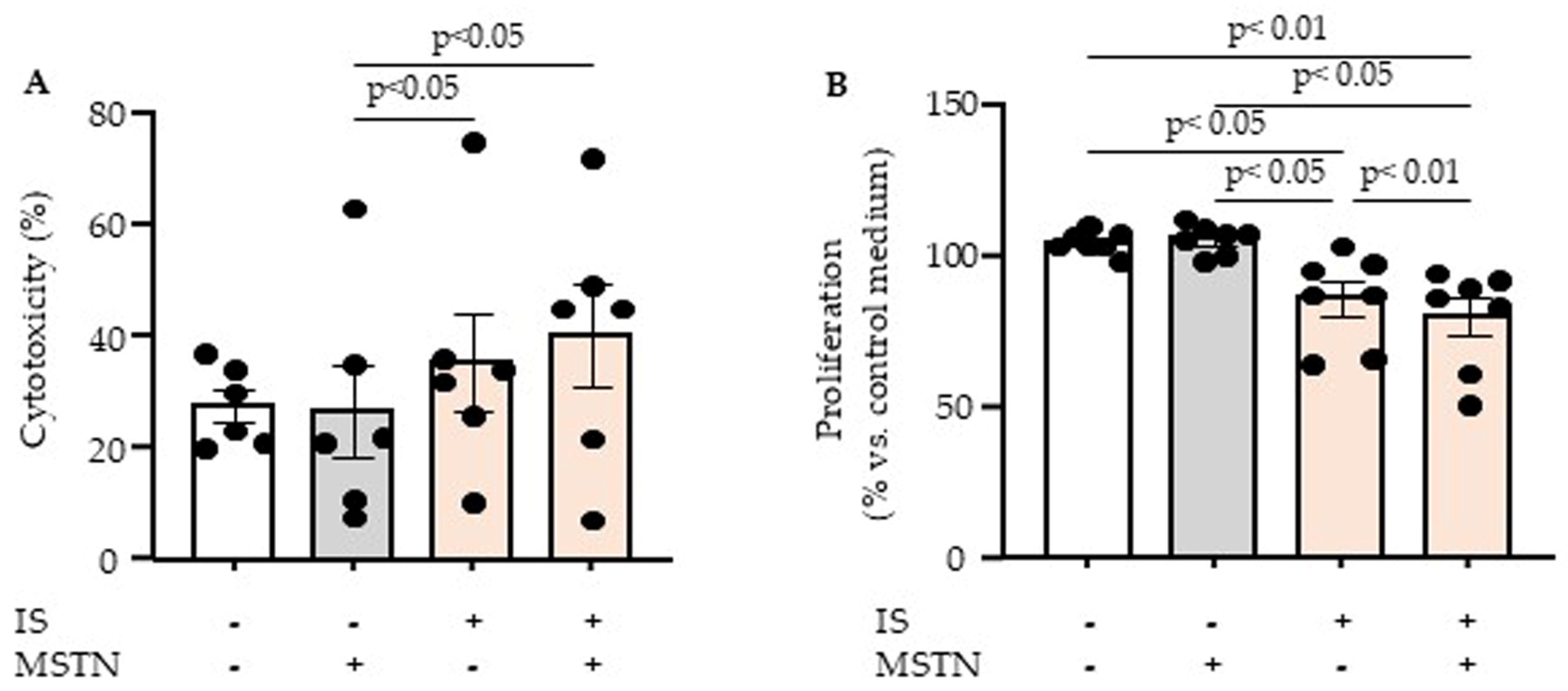
2.1. Effect of Myostatin and Indoxyl Sulfate on Endothelial Cytotoxicity and Proliferation
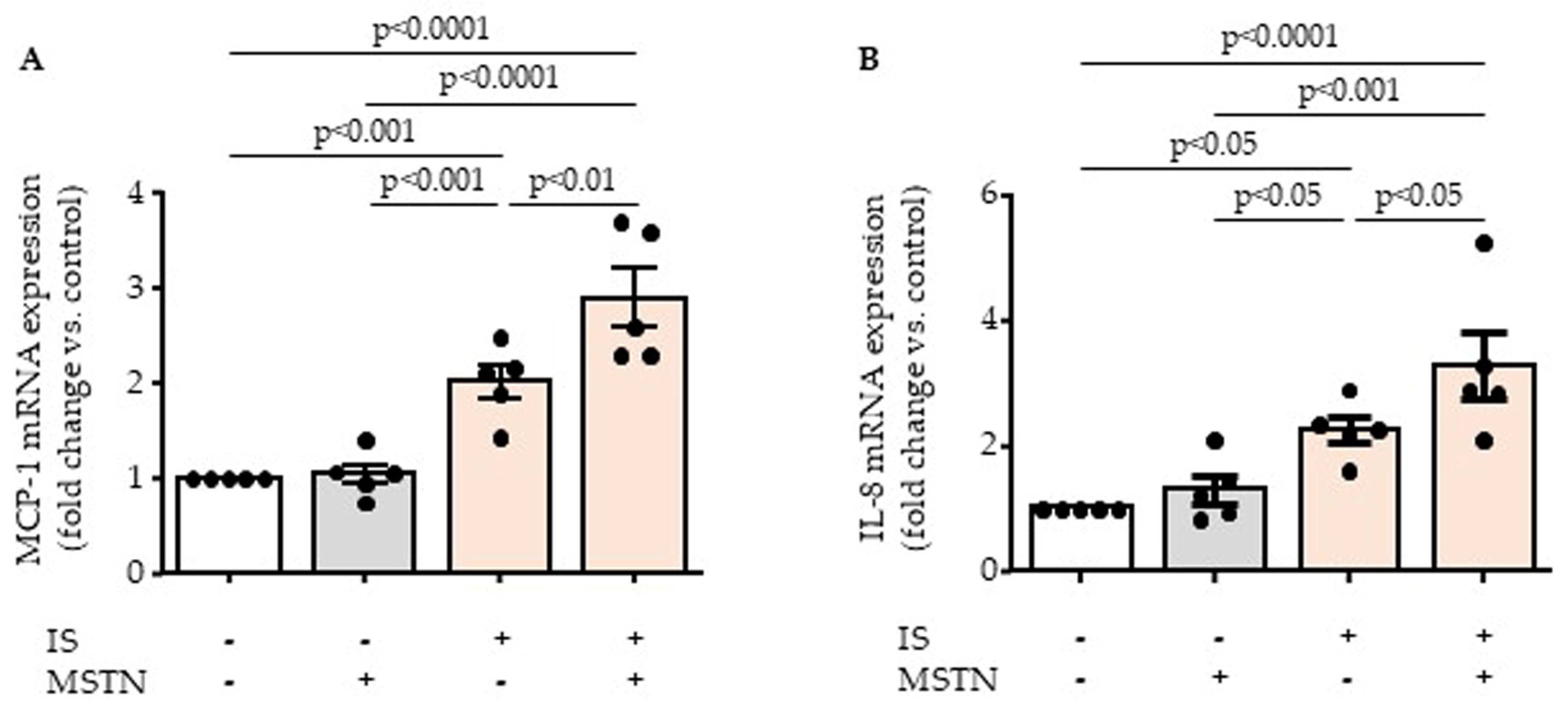
2.2. Myostatin Potentiates Inflammatory Effect of Indoxyl Sulfate by Increasing Endothelial MCP-1 and IL-8 Upregulation
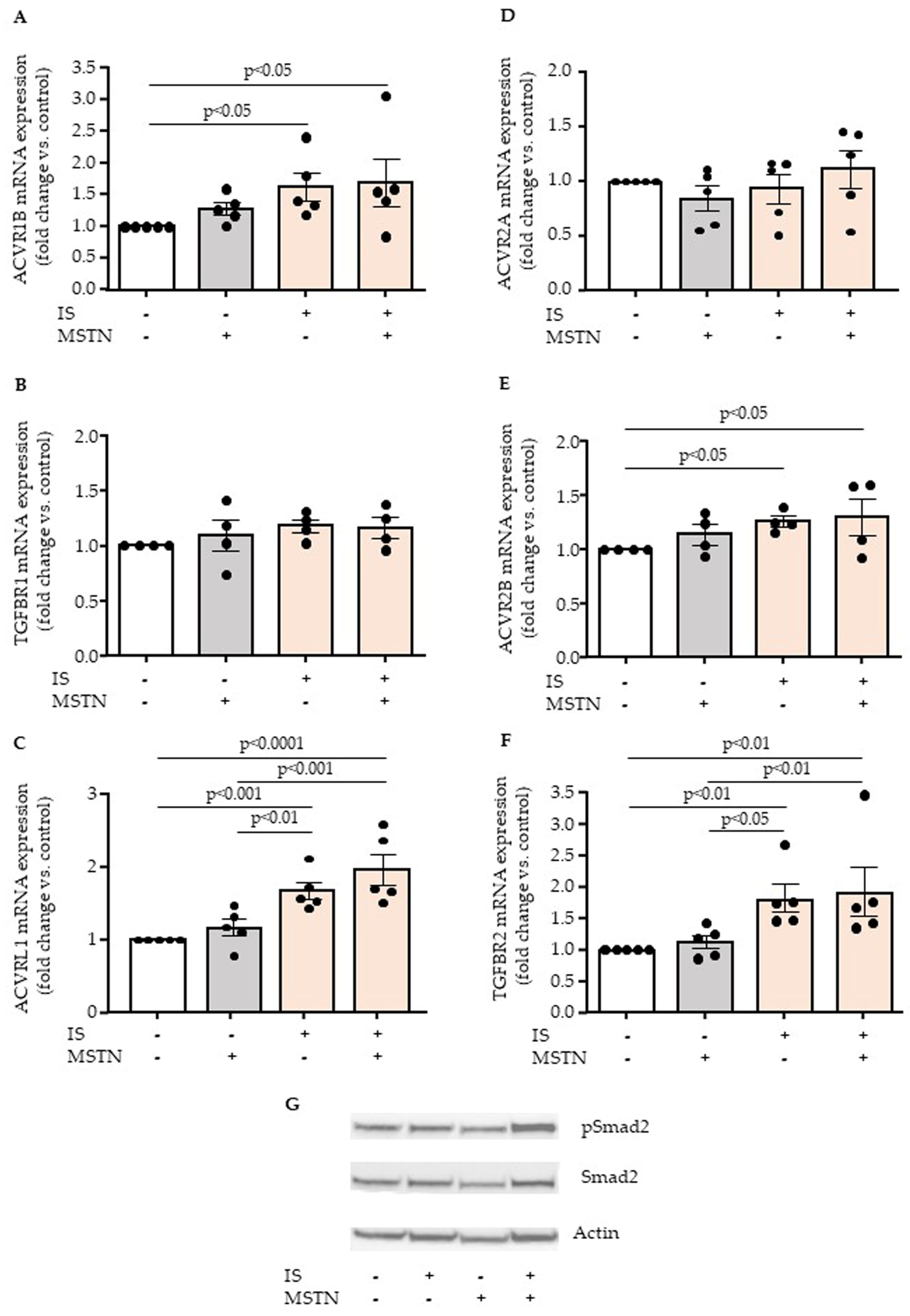
2.3. Effect of Myostatin and Indoxyl Sulfate on Endothelial Expression of Myostatin Receptors
2.4. Effects of Myostatin and Indoxyl Sulfate on Endothelial Expression and Activation of Aryl Hydrocarbon Receptor
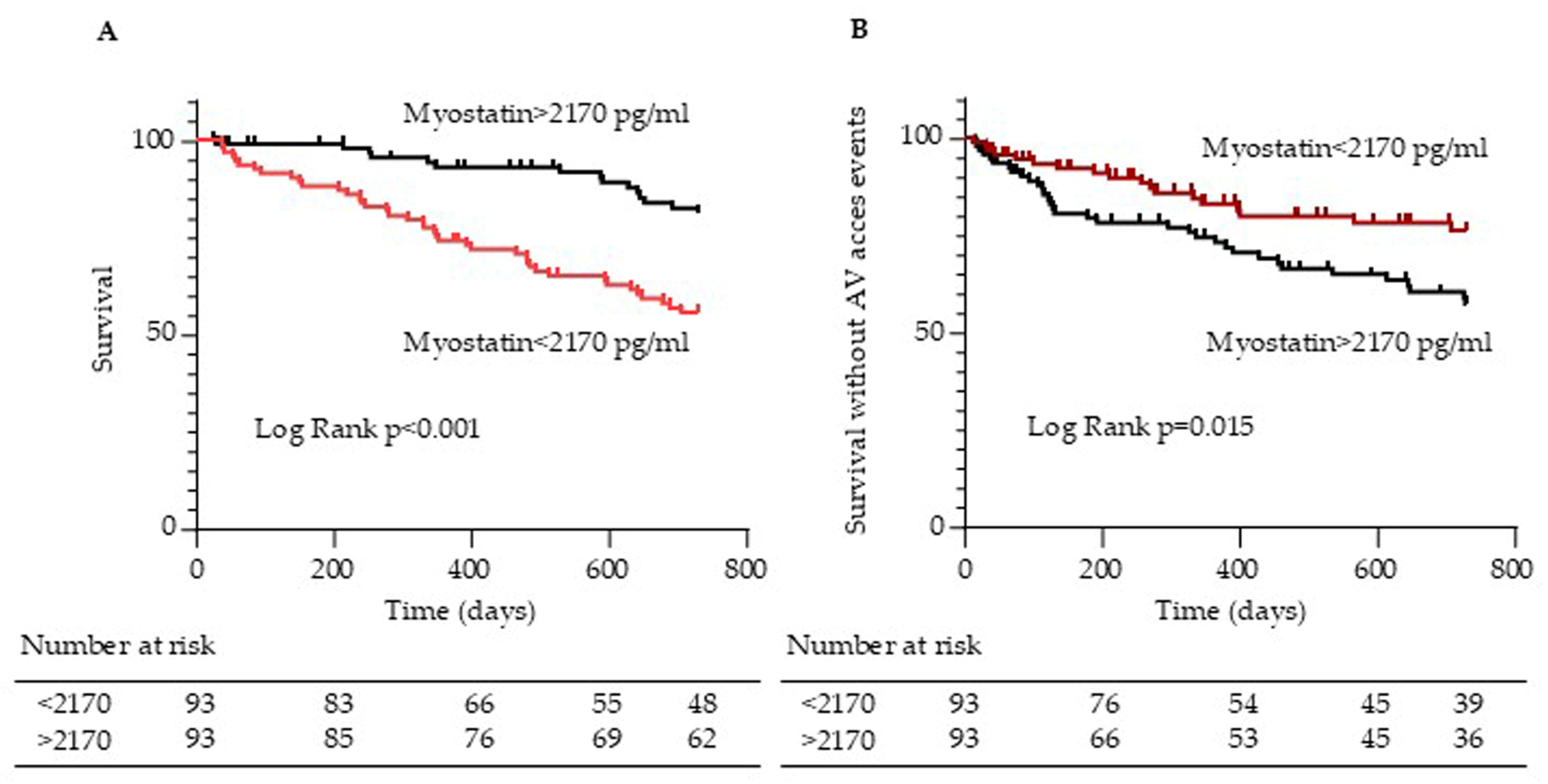
2.5. Myostatin Serum Levels Are Associated with Arteriovenous Access Events in Hemodialysis Patients
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents
5.2. Endothelial Cell Culture
5.3. Treatment of Endothelial Cells
5.4. Evaluation of Myostatin and Indoxyl Sulfate Cytotoxicity by Lactate Dehydrogenase (LDH) Assay
5.5. Endothelial Cell Proliferation Assay
5.6. Protein Extraction and Western Blot Analysis
5.7. mRNA Extraction
5.8. Quantitative RT-PCR Analysis of mRNA Expression
5.9. Patients
5.10. Laboratory Tests
5.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular Incompetence in Dialysis Patients—Protein—Bound Uremic Toxins and Endothelial Dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Chermiti, R.; Burtey, S.; Dou, L. Role of Uremic Toxins in Vascular Inflammation Associated with Chronic Kidney Disease. J. Clin. Med. 2024, 13, 7149. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Duan, S.; Liu, Y.; Wang, H. Intimal Hyperplasia of Arteriovenous Fistula. Ann. Vasc. Surg. 2022, 85, 444–453. [Google Scholar] [CrossRef]
- Bataille, S.; Chauveau, P.; Fouque, D.; Aparicio, M.; Koppe, L. Myostatin and Muscle Atrophy during Chronic Kidney Disease. Nephrol. Dial. Transpl. 2021, 36, 1986–1993. [Google Scholar] [CrossRef]
- Delanaye, P.; Bataille, S.; Quinonez, K.; Buckinx, F.; Warling, X.; Krzesinski, J.-M.; Pottel, H.; Burtey, S.; Bruyère, O.; Cavalier, E. Myostatin and Insulin-Like Growth Factor 1 Are Biomarkers of Muscle Strength, Muscle Mass, and Mortality in Patients on Hemodialysis. J. Ren. Nutr. 2019, 29, 511–520. [Google Scholar] [CrossRef]
- Butcher, J.T.; Ali, M.I.; Ma, M.W.; McCarthy, C.G.; Islam, B.N.; Fox, L.G.; Mintz, J.D.; Larion, S.; Fulton, D.J.; Stepp, D.W. Effect of Myostatin Deletion on Cardiac and Microvascular Function. Physiol. Rep. 2017, 5, e13525. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Bhasin, S. AAV-Mediated Administration of Myostatin pro-Peptide Mutant in Adult Ldlr Null Mice Reduces Diet-Induced Hepatosteatosis and Arteriosclerosis. PLoS ONE 2013, 8, e71017. [Google Scholar] [CrossRef]
- Verzola, D.; Milanesi, S.; Bertolotto, M.; Garibaldi, S.; Villaggio, B.; Brunelli, C.; Balbi, M.; Ameri, P.; Montecucco, F.; Palombo, D.; et al. Myostatin Mediates Abdominal Aortic Atherosclerosis Progression by Inducing Vascular Smooth Muscle Cell Dysfunction and Monocyte Recruitment. Sci. Rep. 2017, 7, 46362. [Google Scholar] [CrossRef]
- Tu, P.; Bhasin, S.; Hruz, P.W.; Herbst, K.L.; Castellani, L.W.; Hua, N.; Hamilton, J.A.; Guo, W. Genetic Disruption of Myostatin Reduces the Development of Proatherogenic Dyslipidemia and Atherogenic Lesions in Ldlr Null Mice. Diabetes 2009, 58, 1739–1748. [Google Scholar] [CrossRef]
- Esposito, P.; Verzola, D.; Porta, E.L.; Milanesi, S.; Grignano, M.A.; Avella, A.; Gregorini, M.; Abelli, M.; Ticozzelli, E.; Rampino, T.; et al. Myostatin in the Arterial Wall of Patients with End-Stage Renal Disease. J. Atheroscler. Thromb. 2020, 27, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J. Myostatin: A Skeletal Muscle Chalone. Annu. Rev. Physiol. 2023, 85, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The Uremic Solute Indoxyl Sulfate Induces Oxidative Stress in Endothelial Cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The Uremic Solutes P-Cresol and Indoxyl Sulfate Inhibit Endothelial Proliferation and Wound Repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic Uremic Solutes Increase Tissue Factor Production in Endothelial Cells by the Aryl Hydrocarbon Receptor Pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells. Circ. J. 2012, 77, 224–230. [Google Scholar]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial Cells Exposed to Phosphate and Indoxyl Sulphate Promote Vascular Calcification through Interleukin-8 Secretion. Nephrol. Dial. Transpl. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Jeon, E.-Y.; Kim, S.-J. Indoxyl Sulfate-Induced Extracellular Vesicles Released from Endothelial Cells Stimulate Vascular Smooth Muscle Cell Proliferation by Inducing Transforming Growth Factor-Beta Production. J. Vasc. Res. 2019, 56, 129–138. [Google Scholar] [CrossRef]
- Ryu, J.H.; Park, H.; Kim, S.J. The Effects of Indoxyl Sulfate-Induced Endothelial Microparticles on Neointimal Hyperplasia Formation in an Ex Vivo Model. Ann. Surg. Treat. Res. 2017, 93, 11–17. [Google Scholar] [CrossRef]
- Bataille, S.; Chermiti, R.; Giaime, P.; Pedinielli, N.; Kay, N.M.; Lano, G.; Tawfik, A.; Mancini, J.; Burtey, S.; Dou, L. Levels of Interleukin 8 and Monocyte Chemoattractant Protein-1 Are Associated with Arteriovenous Fistula Events and Upregulated in the Endothelium by Indoxyl Sulfate and TGF-β Pathway: FR-PO469. J. Am. Soc. Nephrol. 2023, 34, 532. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Q.; Fei, T.; Han, J.-D.J.; Chen, Y.-G. MCP-1 Mediates TGF-Beta-Induced Angiogenesis by Stimulating Vascular Smooth Muscle Cell Migration. Blood 2007, 109, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Spinetti, G.; Wang, M.; Monticone, R.; Zhang, J.; Zhao, D.; Lakatta, E.G. Rat Aortic MCP-1 and Its Receptor CCR2 Increase with Age and Alter Vascular Smooth Muscle Cell Function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1397–1402. [Google Scholar] [CrossRef]
- Grudzinska, M.K.; Kurzejamska, E.; Hagemann, N.; Bojakowski, K.; Soin, J.; Lehmann, M.H.; Reinecke, H.; Murry, C.E.; Soderberg—Naucler, C.; Religa, P. Monocyte Chemoattractant Protein 1-Mediated Migration of Mesenchymal Stem Cells Is a Source of Intimal Hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Juncos, J.P.; Grande, J.P.; Kang, L.; Ackerman, A.W.; Croatt, A.J.; Katusic, Z.S.; Nath, K.A. MCP-1 Contributes to Arteriovenous Fistula Failure. J. Am. Soc. Nephrol. 2011, 22, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Chen, T.-Y.; Hsieh, M.-Y.; Lin, L.; Yang, C.-W.; Chuang, S.-Y.; Tarng, D.-C. Monocyte Chemoattractant Protein-1 Levels and Postangioplasty Restenosis of Arteriovenous Fistulas. Clin. J. Am. Soc. Nephrol. 2017, 12, 113–121. [Google Scholar] [CrossRef]
- Lux, A.; Salway, F.; Dressman, H.K.; Kröner-Lux, G.; Hafner, M.; Day, P.J.R.; Marchuk, D.A.; Garland, J. ALK1 Signalling Analysis Identifies Angiogenesis Related Genes and Reveals Disparity between TGF-Beta and Constitutively Active Receptor Induced Gene Expression. BMC Cardiovasc. Disord. 2006, 6, 13. [Google Scholar] [CrossRef]
- Wu, X.; Ma, J.; Han, J.-D.; Wang, N.; Chen, Y.-G. Distinct Regulation of Gene Expression in Human Endothelial Cells by TGF-Beta and Its Receptors. Microvasc. Res. 2006, 71, 12–19. [Google Scholar] [CrossRef]
- Qin, Y.; Fan, F.; Zhao, Y.; Cui, Y.; Wei, X.; Kohama, K.; Gordon, J.R.; Li, F.; Gao, Y. Recombinant Human CXCL8(3-72)K11R/G31P Regulates Smooth Muscle Cell Proliferation and Migration through Blockage of Interleukin-8 Receptor. IUBMB Life 2013, 65, 67–75. [Google Scholar] [CrossRef]
- Zeller, I.; Knoflach, M.; Seubert, A.; Kreutmayer, S.B.; Stelzmüller, M.E.; Wallnoefer, E.; Blunder, S.; Frotschnig, S.; Messner, B.; Willeit, J.; et al. Lead Contributes to Arterial Intimal Hyperplasia through Nuclear Factor Erythroid 2-Related Factor-Mediated Endothelial Interleukin 8 Synthesis and Subsequent Invasion of Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1733–1740. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Sabatino, A.; Cuppari, L.; Stenvinkel, P.; Lindholm, B.; Avesani, C.M. Sarcopenia in Chronic Kidney Disease: What Have We Learned so Far? J. Nephrol. 2021, 34, 1347–1372. [Google Scholar] [CrossRef]
- Lok, C.E.; Huber, T.S.; Orchanian-Cheff, A.; Rajan, D.K. Arteriovenous Access for Hemodialysis: A Review. JAMA 2024, 331, 1307. [Google Scholar] [CrossRef] [PubMed]
- Roy-Chaudhury, P.; Sukhatme, V.P.; Cheung, A.K. Hemodialysis Vascular Access Dysfunction: A Cellular and Molecular Viewpoint. J. Am. Soc. Nephrol. 2006, 17, 1112–1127. [Google Scholar] [CrossRef] [PubMed]
- Rothuizen, T.C.; Wong, C.; Quax, P.H.A.; van Zonneveld, A.J.; Rabelink, T.J.; Rotmans, J.I. Arteriovenous Access Failure: More than Just Intimal Hyperplasia? Nephrol. Dial. Transpl. 2013, 28, 1085–1092. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, T.H.; Cheng, O.H.; Peden, E.; Li, Q.; Wang, J.; Huang, F.; Melancon, M.P.; Sheikh-Hamad, D.; Wang, T.; et al. Interplay between Skeletal Muscle Catabolism and Remodeling of Arteriovenous Fistula via YAP1 Signaling. J. Am. Soc. Nephrol. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Taniguchi, R.; Ohashi, Y.; Lee, J.S.; Hu, H.; Gonzalez, L.; Zhang, W.; Langford, J.; Matsubara, Y.; Yatsula, B.; Tellides, G.; et al. Endothelial Cell TGF-β (Transforming Growth Factor-Beta) Signaling Regulates Venous Adaptive Remodeling to Improve Arteriovenous Fistula Patency. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 868–883. [Google Scholar] [CrossRef]
- Hu, H.; Lee, S.-R.; Bai, H.; Guo, J.; Hashimoto, T.; Isaji, T.; Guo, X.; Wang, T.; Wolf, K.; Liu, S.; et al. TGFβ (Transforming Growth Factor—Beta)—Activated Kinase 1 Regulates Arteriovenous Fistula Maturation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e203–e213. [Google Scholar] [CrossRef]
- Zhang, W.; Gonzalez, L.; Li, X.; Bai, H.; Li, Z.; Taniguchi, R.; Langford, J.; Ohashi, Y.; Thaxton, C.; Aoyagi, Y.; et al. Endothelial TGF-β Signaling Regulates Endothelial-Mesenchymal Transition During Arteriovenous Fistula Remodeling in Mice with Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 2509–2526. [Google Scholar] [CrossRef]
- Mauro, C.R.; Ding, K.; Xue, H.; Tao, M.; Longchamp, A.; Belkin, M.; Kristal, B.S.; Ozaki, C.K. Adipose Phenotype Predicts Early Human Autogenous Arteriovenous Hemodialysis Remodeling. J. Vasc. Surg. 2016, 63, 171–176.e1. [Google Scholar] [CrossRef]
- Franzoni, M.; Cattaneo, I.; Longaretti, L.; Figliuzzi, M.; Ene-Iordache, B.; Remuzzi, A. Endothelial Cell Activation by Hemodynamic Shear Stress Derived from Arteriovenous Fistula for Hemodialysis Access. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H49–H59. [Google Scholar] [CrossRef]
- Kaizu, Y.; Ohkawa, S.; Odamaki, M.; Ikegaya, N.; Hibi, I.; Miyaji, K.; Kumagai, H. Association between Inflammatory Mediators and Muscle Mass in Long-Term Hemodialysis Patients. Am. J. Kidney Dis. 2003, 42, 295–302. [Google Scholar] [CrossRef]
- Bataille, S.; Landrier, J.-F.; Astier, J.; Giaime, P.; Sampol, J.; Sichez, H.; Ollier, J.; Gugliotta, J.; Serveaux, M.; Cohen, J.; et al. The “Dose-Effect” Relationship Between 25-Hydroxyvitamin D and Muscle Strength in Hemodialysis Patients Favors a Normal Threshold of 30 ng/mL for Plasma 25-Hydroxyvitamin D. J. Ren. Nutr. 2016, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Sallée, M.; Pelletier, M.; Bataille, S.; Fraisse, M.; Berda-Haddad, Y.; Brunet, P.; Burtey, S. Mean Platelet Volume Predicts Vascular Access Events in Hemodialysis Patients. J. Clin. Med. 2019, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Calaf, R.; Cerini, C.; Genovesio, C.; Verhaeghe, P.; Jourde-Chiche, N.; Berge-Lefranc, D.; Gondouin, B.; Dou, L.; Morange, S.; Argiles, A.; et al. Determination of Uremic Solutes in Biological Fluids of Chronic Kidney Disease Patients by HPLC Assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2281–2286. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients (n = 186) | Myostatin < 2170 pg/mL (n = 93) | Myostatin > 2170 pg/mL (n = 93) | p | |
|---|---|---|---|---|
| Age (years) | 71.4 [18; 94] | 76 [25; 94] | 62 [18; 94] | <0.0001 |
| Gender ratio (F/M) | 68/118 | 37/56 | 31/62 | 0.44 |
| Body Mass Index (kg/m2) | 24.6 [12.9; 41.5] | 23.9 [12.9; 34.6] | 26.3 [16.4; 41.5] | 0.015 |
| Weight | 71.5 [30; 112] | 66 [30; 92.5] | 74 [37; 112] | 0.001 |
| Arteriovenous graft | 32 (17%) | 20 (21%) | 12 (13%) | 0.17 |
| SBP before dialysis (mmHg) | 140 [83; 210] | 135 [92; 210] | 146 [83; 195] | 0.032 |
| DBP before dialysis (mmHg) | 70 [40; 119] | 68 [41; 119] | 74 [40; 115] | <0.0001 |
| Dialysis vintage (month) | 44 [3; 432] | 45 [3; 432] | 42 [3; 421] | 0.933 |
| History of hypertension | 157 (84%) | 79 (85%) | 78 (84%) | >0.999 |
| History of diabetes | 71 (38%) | 36 (39%) | 35 (38%) | >0.999 |
| History of CAD | 64 (34%) | 38 (41%) | 26 (28%) | 0.089 |
| History of heart failure | 40 (22%) | 21 (23%) | 19 (20%) | 0.86 |
| History of atrial fibrillation | 56 (30%) | 40 (43%) | 16 (17%) | 0.0002 |
| History of PAD | 46 (25%) | 27 (29%) | 19 (20%) | 0.23 |
| History of stroke/TIA | 28 (15%) | 19 (20%) | 9 (10%) | 0.064 |
| History of DVT/PE | 22 (12%) | 13 (14%) | 9 (10%) | 0.49 |
| History of renal transplantation | 18 (10%) | 5 (5%) | 13 (14%) | 0.080 |
| History of dyslipidemia | 59 (32%) | 26 (28%) | 33 (35%) | 0.34 |
| Antihypertensive drugs | 129 (69%) | 58 (62%) | 71 (76%) | 0.056 |
| Antiplatelet drugs | 95 (51%) | 51 (55%) | 44 (47%) | 0.38 |
| Anticoagulant drugs | 35 (19%) | 22 (24%) | 13 (14%) | 0.132 |
| Antidiabetic treatments | 71 (38%) | 36 (39%) | 35 (38%) | >0.999 |
| Hypolipidemic drugs | 55 (29%) | 26 (28%) | 29 (31%) | 0.75 |
| Erythropoiesis stimulating agents | 139 (74%) | 66 (71%) | 73 (78%) | 0.31 |
| Hemoglobin (g/dL) | 10.7 [6.1; 13.4] | 10.7 [8; 13.4] | 10.7 [6.1; 13.3] | 0.716 |
| Serum CRP (mg/L) | 6.6 [0.2; 418.9] | 11.6 [0.6; 418.9] | 3.6 [0.2; 247.1] | <0.0001 |
| Serum IL-6 (pg/mL) | 4 [0; 232] | 6.5 [0; 232.2] | 2.8 [0; 107.7] | 0.001 |
| Serum ferritin (ng/mL) | 394 [26; 5000] | 447 [26; 5000] | 352 [26; 1216] | 0.037 |
| Serum albumin (g/L) | 38.7 [21.6; 47.2] | 37.8 [21.6; 47.2] | 39.5 [27.3; 46.6] | 0.001 |
| Parathyroid hormone (ng/L) | 25 [1; 2217] | 24 [2; 2217] | 26 [1; 901] | 0.62 |
| Serum calcium (mmol/L) | 2.34 [1.84; 3.09] | 2.36 [1.85; 3.09] | 2.33 [1.84; 2.76] | 0.24 |
| Serum phosphate (mmol/L) | 1.50 [0.38; 4.03] | 1.38 [0.64; 3.15] | 1.62 [0.38; 4.03] | 0.001 |
| Serum potassium (mmol/L) | 5.01 [2.91; 6.82] | 4.83 [2.91; 6.82] | 5.07 [3.40; 6.82] | 0.025 |
| Serum bicarbonate (mmol/L) | 21.8 [15.4; 44.2] | 22.2 [15.4; 44.2] | 21.5 [16.6; 32.0] | 0.27 |
| Serum indoxyl sulfate (µM) | 87.7 [0; 301] | 81 [0; 219] | 100 [25; 301] | 0.002 |
| Serum indole-3 acetic acid (µM) | 3.1 [0.2; 33.5] | 2.9 [0.2; 33.5] | 3.5 [1; 30.7] | 0.017 |
| Serum p-cresyl sulfate (µM) | 149 [0; 1227] | 149 [0; 1227] | 151 [0; 559] | 0.79 |
| Serum myostatin (pg/mL) | 2170 [92; 14,085] | 1409 [92; 2157] | 3233 [2178; 14,085] | <0.0001 |
| Mortality Risk | Crude HR | 95% CI | p-Value |
|---|---|---|---|
| Myostatin > 2170 pg/mL | 0.34 | 0.19 to 0.61 | <0.0001 |
| Indoxyl sulfate > 87.7 µM | 0.84 | 0.49 to 1.43 | 0.518 |
| Age > 65 years | 2.46 | 1.27 to 4.76 | 0.008 |
| BMI = 18.5 to 24.9 kg/m2 (ref.) | 0.651 | ||
| BMI < 18.5 kg/m2 | 2.05 | 0.49 to 8.63 | 0.329 |
| BMI 25.0 to 29.9 kg/m2 | 1.54 | 0.34 to 6.89 | 0.571 |
| BMI ≥ 30 kg/m2 | 2.15 | 0.47 to 9.81 | 0.323 |
| SBP before dialysis > 140 mmHg | 1.10 | 0.64 to 1.87 | 0.729 |
| Serum albumin > 35 g/L | 0.42 | 0.24 to 0.73 | 0.002 |
| Serum CRP > 6.6 mg/L | 2.96 | 1.63 to 5.36 | <0.0001 |
| History of diabetes | 1.60 | 0.94 to 2.71 | 0.083 |
| Mortality Risk | Adjusted HR | 95% CI | p-Value |
|---|---|---|---|
| Myostatin > 2170 pg/mL | 0.48 | 0.26 to 0.90 | 0.022 |
| Age > 65 years | 1.59 | 0.80 to 3.17 | 0.184 |
| Serum albumin (g/L) | 0.62 | 0.35 to 1.09 | 0.094 |
| Serum CRP (mg/L) | 2.22 | 1.19 to 4.13 | 0.012 |
| History of diabetes | 1.53 | 0.90 to 2.61 | 0.119 |
| Risk of AV Access Event | Crude HR | 95% CI | p-Value |
|---|---|---|---|
| Myostatin > 2170 pg/mL | 2.00 | 1.13 to 3.54 | 0.018 |
| Indoxyl sulfate > 87.7 µM | 1.06 | 0.62 to 1.83 | 0.827 |
| Age > 65 years | 0.88 | 0.50 to 1.53 | 0.645 |
| BMI = 18.5 to 24.9 kg/m2 (ref.) | 0.018 | ||
| BMI < 18.5 kg/m2 | 0.35 | 0.05 to 2.60 | 0.302 |
| BMI 25.0 to 29.9 kg/m2 | 1.66 | 0.85 to 3.25 | 0.139 |
| BMI ≥ 30 kg/m2 | 2.74 | 1.35 to 5.55 | 0.005 |
| SBP before dialysis > 140 mmHg | 1.56 | 0.89 to 2.74 | 0.124 |
| Serum albumin > 35 g/L | 1.11 | 0.56 to 2.22 | 0.760 |
| Serum CRP > 6.6 mg/L | 0.86 | 0.50 to 1.48 | 0.590 |
| History of diabetes | 1.64 | 0.95 to 2.83 | 0.074 |
| Risk of AV Access Event | Adjusted HR | 95% CI | p-Value |
|---|---|---|---|
| Myostatin > 2170 pg/mL | 1.90 | 1.02 to 3.55 | 0.044 |
| BMI = 18.5 to 24.9 kg/m2 (ref.) | 0.141 | ||
| BMI < 18.5 kg/m2 | 0.36 | 0.05 to 2.71 | 0.319 |
| BMI 25.0 to 29.9 kg/m2 | 1.59 | 0.81 to 3.11 | 0.179 |
| BMI ≥ 30 kg/m2 | 2.01 | 0.95 to 4.26 | 0.066 |
| SBP before dialysis > 140 mmHg | 1.18 | 0.64 to 2.17 | 0.602 |
| History of diabetes | 1.39 | 0.76 to 2.53 | 0.286 |
| Cox Model | Fine and Gray Model * | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | |
| Survival | 0.34 | 0.19 to 0.61 | 0.000 | - | - | - |
| AV access event | 2.00 | 1.13 to 3.54 | 0.018 | 2.32 | 1.31 to 4.11 | 0.004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solignac, J.; Dou, L.; Chermiti, R.; McKay, N.; Giaime, P.; Pedinielli, N.; Benjelloun, H.; Lano, G.; Mancini, J.; Burtey, S.; et al. Myostatin Exacerbates Endothelial Dysfunction Induced by Uremic Toxin Indoxyl Sulfate and Is Associated with Hemodialysis Arteriovenous Access Complications. Toxins 2025, 17, 159. https://doi.org/10.3390/toxins17040159
Solignac J, Dou L, Chermiti R, McKay N, Giaime P, Pedinielli N, Benjelloun H, Lano G, Mancini J, Burtey S, et al. Myostatin Exacerbates Endothelial Dysfunction Induced by Uremic Toxin Indoxyl Sulfate and Is Associated with Hemodialysis Arteriovenous Access Complications. Toxins. 2025; 17(4):159. https://doi.org/10.3390/toxins17040159
Chicago/Turabian StyleSolignac, Justine, Laetitia Dou, Rania Chermiti, Nathalie McKay, Philippe Giaime, Nathalie Pedinielli, Hamza Benjelloun, Guillaume Lano, Julien Mancini, Stéphane Burtey, and et al. 2025. "Myostatin Exacerbates Endothelial Dysfunction Induced by Uremic Toxin Indoxyl Sulfate and Is Associated with Hemodialysis Arteriovenous Access Complications" Toxins 17, no. 4: 159. https://doi.org/10.3390/toxins17040159
APA StyleSolignac, J., Dou, L., Chermiti, R., McKay, N., Giaime, P., Pedinielli, N., Benjelloun, H., Lano, G., Mancini, J., Burtey, S., & Bataille, S. (2025). Myostatin Exacerbates Endothelial Dysfunction Induced by Uremic Toxin Indoxyl Sulfate and Is Associated with Hemodialysis Arteriovenous Access Complications. Toxins, 17(4), 159. https://doi.org/10.3390/toxins17040159