De Novo Assembly of Venom Gland Transcriptome of Tropidolaemus wagleri (Temple Pit Viper, Malaysia) and Insights into the Origin of Its Major Toxin, Waglerin


Abstract
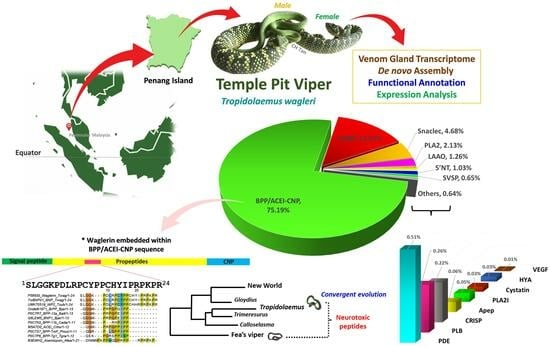
1. Introduction
2. Results and Discussion
2.1. De Novo Sequencing and Transcriptome Assembly
2.2. Toxin Gene Annotation and Expression Profile
2.3. Sequence Analysis and Characterization of Waglerin’s Precursor Gene

2.4. Divergence and Conservation of BPP/ACEI-CNP Genes among Diverse Genera
3. Conclusions
4. Materials and Methods
4.1. Preparation of T. wagleri Venom Gland Tissue
4.2. RNA Extraction and mRNA Purification
4.3. Filtration of Raw Reads
4.4. De novo Assembly of Transcriptome
4.5. Functional Annotation of Transcripts
4.6. Quantifying Transcript Abundance
4.7. Categorization of Transcripts
4.8. Sequence Alignment and Analysis
4.9. Phylogenetics
4.10. Supporting Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arbuckle, K. Evolutionary Context of Venom in Animals; Springer: Dordrecht, The Netherlands, 2017; pp. 3–31. [Google Scholar]
- Barua, A.; Mikheyev, A.S. Toxin expression in snake venom evolves rapidly with constant shifts in evolutionary rates. Proc. Biol. Sci. 2020, 287, 20200613. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N.; Nakamura, H.; Yamaguchi, K.; Hattori, S.; Matsubara, K.; Matsuda, Y.; Yamashita, A.; Isomoto, A.; et al. The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 2018, 8, 11300. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M. Accelerated evolution of toxin genes: Exonization and intronization in snake venom disintegrin/metalloprotease genes. Toxicon 2018, 148, 16–25. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wuster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 2011, 28, 2637–2649. [Google Scholar] [CrossRef]
- Ogawa, T.; Chijiwa, T.; Oda-Ueda, N.; Ohno, M. Molecular diversity and accelerated evolution of C-type lectin-like proteins from snake venom. Toxicon 2005, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deshimaru, M.; Ogawa, T.; Nakashima, K.; Nobuhisa, I.; Chijiwa, T.; Shimohigashi, Y.; Fukumaki, Y.; Niwa, M.; Yamashina, I.; Hattori, S.; et al. Accelerated evolution of crotalinae snake venom gland serine proteases. FEBS Lett. 1996, 397, 83–88. [Google Scholar] [CrossRef]
- Barua, A.; Mikheyev, A.S. Many Options, Few Solutions: Over 60 My Snakes Converged on a Few Optimal Venom Formulations. Mol. Biol. Evol. 2019, 36, 1964–1974. [Google Scholar] [CrossRef]
- Sunagar, K.; Fry, B.G.; Jackson, T.N.; Casewell, N.R.; Undheim, E.A.; Vidal, N.; Ali, S.A.; King, G.F.; Vasudevan, K.; Vasconcelos, V. Molecular evolution of vertebrate neurotrophins: Co-option of the highly conserved nerve growth factor gene into the advanced snake venom arsenalf. PLoS ONE 2013, 8, e81827. [Google Scholar] [CrossRef]
- Stroud, J.T.; Losos, J.B. Ecological Opportunity and Adaptive Radiation. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 507–532. [Google Scholar] [CrossRef]
- Alencar, L.R.V.; Martins, M.; Greene, H.W. Evolutionary History of Vipers. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 1–10. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Yap, M.K.K.; Tan, N.H. Venomics of Tropidolaemus wagleri, the sexually dimorphic temple pit viper: Unveiling a deeply conserved atypical toxin arsenal. Sci. Rep. 2017, 7, 43237. [Google Scholar] [CrossRef]
- Vogel, G.; David, P.; Lutz, M.; Van Rooijen, J.; Vidal, N. Revision of the Tropidolaemus wagleri complex (Serpentes: Viperidae: Crotalinae). I. Definition of included taxa and redescription of Tropidolaemus wagleri (Boie, 1827). Zootaxa 2007, 1644, 1–40. [Google Scholar] [CrossRef]
- Tan, N.H.; Tan, K.Y.; Tan, C.H. Snakebite in Southeast Asia: Envenomation and Clinical Management. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2021; pp. 559–579. [Google Scholar]
- Wagler, J.G. Natürliches System der Amphibien: Mit. vorangehender Classification der Säugethiere und Vögel: Ein Beitrag zur vergleichenden Zoologie; J.G. Cotta Buchhandlung: München, Germany,, 1830; p. 354. [Google Scholar]
- Gray, J.E. Synopsis of the species of rattle-snakes, or family of Crotalidae. In Zoological Miscellany 2; Treuttel, Würtz & Co.: London, England, 1842; pp. 47–51. [Google Scholar]
- Brattstrom, B.H. Evolution of the pit vipers. Trans. San. Diego Soc. Nat. Hist. 1964, 13, 185–268. [Google Scholar] [CrossRef]
- Burger, W.L. Genera of Pitvipers (Serpentes: Crotalidae); University of Kansas Lawrence, University Microfilms: Ann Arbor, MI, USA, 1971. [Google Scholar]
- Vidal, N.; Lecointre, G. Weighting and congruence: A case study based on three mitochondrial genes in pitvipers. Mol. Phylogenet Evol. 1998, 9, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Thorpe, R.S. A phylogeny of the trimeresurus group of pit vipers: New evidence from a mitochondrial gene tree. Mol. Phylogenet Evol. 2000, 16, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, C.L.; Campbell, J.A.; Chippindale, P.T. Multigene phylogenetic analysis of pitvipers, with comments on their biogeography. In Biology of the Vipers; Schuett, G.W., Höggren, M., Douglas, M.E., Greene, H.W., Eds.; Eagle Mountain Publishing: Eagle Mountain, UT, USA, 2002; pp. 93–110. [Google Scholar]
- Malhotra, A.; Thorpe, R.S. A phylogeny of four mitochondrial gene regions suggests a revised taxonomy for Asian pitvipers (Trimeresurus and Ovophis). Mol. Phylogenet Evol. 2004, 32, 83–100. [Google Scholar] [CrossRef]
- Creer, S.; Pook, C.E.; Malhotra, A.; Thorpe, R.S. Optimal intron analyses in the Trimeresurus radiation of Asian pitvipers. Syst. Biol. 2006, 55, 57–72. [Google Scholar] [CrossRef]
- Kuch, U.; Gumprecht, A.; Melaun, C. A new species of Temple Pitviper (Tropidolaemus Wagler, 1830) from Sulawesi, Indonesia (Squamata: Viperidae: Crotalinae). Zootaxa 2007, 1446, 1–20. [Google Scholar] [CrossRef]
- Uetz, P.; Freed, P.; Aguilar, R.; Reyes, F.; Hošek, J. The Reptile Database. Available online: https://reptile-database.reptarium.cz/species?genus=Tropidolaemus&species=wagleri (accessed on 24 April 2023).
- Orlov, N.; Ananjeva, N.; Khalikov, R. Natural history of pitvipers in Eastern and Southeastern Asia. In Biology of the Vipers; Schuett, G.W., Hoggren, M., Douglas, M.E., Greene, H.W., Eds.; Eagle Mountain Publishing: Eagle Mountain, UT, USA, 2002; pp. 345–360. [Google Scholar]
- World Health Organization. Guidelines for the Management of Snakebites, 2nd ed.; WHO Regional Office for South-East: New Delhi, India, 2016. [Google Scholar]
- Ismail, A.K.; Abd Hamid, M.N.H.; Ariff, N.A.; Frederic Ng, V.E.R.; Goh, W.C.; Abdul Samat, N.S.; Osman, A.M.Z.; Safferi, R.S.; Mohamed Ismail, Z. Frequency, clinical characteristics and outcomes of Tropidolaemus species bite envenomations in Malaysia. PLoS Negl. Trop. Dis. 2023, 17, e0010983. [Google Scholar] [CrossRef]
- Tan, N.H.; Tan, C.S. The enzymatic activities and lethal toxins of Trimeresurus wagleri (speckled pit viper) venom. Toxicon 1989, 27, 349–357. [Google Scholar] [CrossRef]
- Minton, S.A., Jr. Preliminary observations on the venom of Wagler’s pit viper (Trimeresurus wagleri). Toxicon 1968, 6, 93–97. [Google Scholar] [CrossRef]
- Tsai, M.C.; Hsieh, W.H.; Smith, L.A.; Lee, C.Y. Effects of waglerin-I on neuromuscular transmission of mouse nerve-muscle preparations. Toxicon 1995, 33, 363–371. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Weinstein, S.A. Structure-function studies of waglerin I, a lethal peptide from the venom of Wagler’s pit viper, Trimeresurus wagleri. Toxicon 1995, 33, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Smith, L.A.; Lee, C.Y. A study on the cause of death due to waglerin-I, a toxin from Trimeresurus wagleri. Toxicon 1995, 33, 111–114. [Google Scholar] [CrossRef]
- Tasoulis, T.; Isbister, G.K. A review and database of snake venom proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.K.; Tan, C.H.; Sim, S.M.; Fung, S.Y.; Sumana, K.; Sitprija, V.; Tan, N.H. Cross neutralization of common Southeast Asian viperid venoms by a Thai polyvalent snake antivenom (Hemato Polyvalent Snake Antivenom). Acta Trop. 2014, 132, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, N.H.; Sim, S.M.; Fung, S.Y.; Gnanathasan, C.A. Immunological properties of Hypnale hypnale (hump-nosed pit viper) venom: Antibody production with diagnostic and therapeutic potentials. Acta Trop. 2012, 122, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.P.; Tan, K.Y.; Tan, N.H.; Tan, C.H. Exploring the diversity and novelty of toxin genes in Naja sumatrana, the Equatorial spitting cobra from Malaysia through de novo venom-gland transcriptomics. Toxins 2019, 11, 104. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Tan, K.Y.; Tan, C.H. De novo venom gland transcriptomics of Calliophis bivirgata flaviceps: Uncovering the complexity of toxins from the Malayan blue coral snake. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20210024. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y. De Novo venom-gland transcriptomics of spine-bellied sea snake (Hydrophis curtus) from Penang, Malaysia—Next-generation sequencing, functional annotation and toxinological correlation. Toxins 2021, 13, 127. [Google Scholar] [CrossRef]
- Hofmann, E.P.; Rautsaw, R.M.; Strickland, J.L.; Holding, M.L.; Hogan, M.P.; Mason, A.J.; Rokyta, D.R.; Parkinson, C.L. Comparative venom-gland transcriptomics and venom proteomics of four Sidewinder Rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Sci. Rep. 2018, 8, 15534. [Google Scholar] [CrossRef]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Dashevsky, D.; Rokyta, D.; Frank, N.; Nouwens, A.; Fry, B.G. Electric Blue: Molecular Evolution of Three-Finger Toxins in the Long-Glanded Coral Snake Species Calliophis bivirgatus. Toxins 2021, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.T.; Macrander, J.; Vasieva, O.; Rojnuckarin, P. Exploring Toxin Genes of Myanmar Russell’s Viper, Daboia siamensis, through De Novo Venom Gland Transcriptomics. Toxins 2023, 15, 309. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; McGivern, J.J.; Wray, K.P.; Seavy, M.; Calvin, K.; Rokyta, D.R. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J. Proteom. 2014, 96, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wagstaff, S.C.; Wüster, W.; Cook, D.A.N.; Bolton, F.M.S.; King, S.I.; Pla, D.; Sanz, L.; Calvete, J.J.; Harrison, R.A. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 9205–9210. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genom. 2015, 16, 687. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, K.Y.; Ng, T.S.; Tan, N.H.; Chong, H.P. De Novo Venom Gland Transcriptome Assembly and Characterization for Calloselasma rhodostoma (Kuhl, 1824), the Malayan Pit Viper from Malaysia: Unravelling Toxin Gene Diversity in a Medically Important Basal Crotaline. Toxins 2023, 15, 315. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: Elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef]
- Xie, B.; Dashevsky, D.; Rokyta, D.; Ghezellou, P.; Fathinia, B.; Shi, Q.; Richardson, M.K.; Fry, B.G. Dynamic genetic differentiation drives the widespread structural and functional convergent evolution of snake venom proteinaceous toxins. BMC Biol. 2022, 20, 4. [Google Scholar] [CrossRef]
- Weinstein, S.A.; Schmidt, J.J.; Bernheimer, A.W.; Smith, L.A. Characterization and amino acid sequences of two lethal peptides isolated from venom of Wagler’s pit viper, Trimeresurus wagleri. Toxicon 1991, 29, 227–236. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Weinstein, S.A.; Smith, L.A. Molecular properties and structure-function relationships of lethal peptides from venom of Wagler’s pit viper, Trimeresurus wagleri. Toxicon 1992, 30, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.M.; Chuang, C.C.; Chuang, L.C.; Yu, H.M.; Wang, K.T.; Chiou, S.H.; Wu, S.H. Protein engineering of venom toxins by synthetic approach and NMR dynamic simulation: Status of basic amino acid residues in waglerin I. Biochem. Biophys. Res. Commun. 1996, 227, 59–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ondetti, M.A.; Williams, N.J.; Sabo, E.F.; Pluscec, J.; Weaver, E.R.; Kocy, O. Angiotensin-converting enzyme inhibitors from the venom of Bothrops jararaca. Isolation, elucidation of structure, and synthesis. Biochemistry 1971, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.S.; Cushman, D.W. Inhibition of homogeneous angiotensin-converting enzyme of rabbit lung by synthetic venom peptides of Bothrops jararaca. Biochim. Biophys. Acta 1973, 293, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, D.C.; Spencer, P.J. Bradykinin-potentiating and related peptides from reptile venoms. In Handbook of Venoms and Toxins of Reptiles; CRC Press: Boca Raton, FL, USA, 2021; pp. 241–250. [Google Scholar]
- Graham, R.L.; Graham, C.; McClean, S.; Chen, T.; O’Rourke, M.; Hirst, D.; Theakston, D.; Shaw, C. Identification and functional analysis of a novel bradykinin inhibitory peptide in the venoms of New World Crotalinae pit vipers. Biochem. Biophys. Res. Commun. 2005, 338, 1587–1592. [Google Scholar] [CrossRef]
- Soares, M.R.; Oliveira-Carvalho, A.L.; Wermelinger, L.S.; Zingali, R.B.; Ho, P.L.; Junqueira-de-Azevedo, I.L.; Diniz, M.R. Identification of novel bradykinin-potentiating peptides and C-type natriuretic peptide from Lachesis muta venom. Toxicon 2005, 46, 31–38. [Google Scholar] [CrossRef]
- Molles, B.E.; Rezai, P.; Kline, E.F.; McArdle, J.J.; Sine, S.M.; Taylor, P. Identification of residues at the alpha and epsilon subunit interfaces mediating species selectivity of Waglerin-1 for nicotinic acetylcholine receptors. J. Biol. Chem. 2002, 277, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J.; Zdenek, C.N.; Debono, J.; Harrich, D.; Fry, B.G. Evolutionary Interpretations of nicotinic acetylcholine receptor targeting venom effects by a clade of Asian Viperidae snakes. Neurotox. Res. 2020, 38, 312–318. [Google Scholar] [CrossRef]
- Utkin, Y.N.; Weise, C.; Kasheverov, I.E.; Andreeva, T.V.; Kryukova, E.V.; Zhmak, M.N.; Starkov, V.G.; Hoang, N.A.; Bertrand, D.; Ramerstorfer, J.; et al. Azemiopsin from Azemiops feae viper venom, a novel polypeptide ligand of nicotinic acetylcholine receptor. J. Biol. Chem. 2012, 287, 27079–27086. [Google Scholar] [CrossRef]
- Debono, J.; Xie, B.; Violette, A.; Fourmy, R.; Jaeger, M.; Fry, B.G. Viper Venom Botox: The molecular origin and evolution of the waglerin peptides used in anti-wrinkle skin cream. J. Mol. Evol. 2017, 84, 8–11. [Google Scholar] [CrossRef]
- Mackessy, P.S. Reptile Venoms and Toxins: Unlimited Opportunities for Basic and Applied Research. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; Mackessy, P.S., Ed.; CRC Press: Boca Raton, FL, United States, 2021; pp. 3–18. [Google Scholar]
- Rotenberg, D.; Bamberger, E.; Kochva, E. Studies on ribonucleic acid synthesis in the venom glands of Vipera palaestinae (Ophidia, Reptilia). Biochem. J. 1971, 121, 609–612. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wery, M.; Descrimes, M.; Thermes, C.; Gautheret, D.; Morillon, A. Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-Seq. Methods 2013, 63, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Total raw reads | 56,736,590 |
| Total clean reads | 52,761,436 |
| Total clean nucleotides (nt) | 4,748,529,240 |
| Q20 percentage | 97.58% |
| N percentage | 0.00% |
| GC percentage | 49.68% |
| Contigs created | 114,027 |
| Total length (nt) | 39,412,125 |
| Mean length (nt) | 346 |
| N50 | 546 |
| Transcripts assembled | 58,914 |
| Total length (nt) | 39,179,883 |
| Mean length (nt) | 665 |
| N50 | 1032 |
| Unigene/transcripts assembled (FPKM > 1) | 54,781 |
| Unidentified | 30,661 |
| Non-toxin | 24,079 |
| Toxin | 41 |
| Transcript Code | Annotated Protein Subtype | Matched Accession Code | Matched Species | Relative Abundance (%) |
|---|---|---|---|---|
| Bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide (BPP/ACEI-CNP) | 75.19% | |||
| TwBNP01 | Waglerin peptide 2 | UMK70519 | Tropidolaemus subannulatus | 75.19% |
| Snake venom metalloproteinase (SVMP): PIII class | 13.91% | |||
| Tw_SMP01 | Zinc metalloproteinase-disintegrin-like VAP1 | XP_039210133 | Crotalus tigris | 7.44% |
| Tw_SMP02 | Zinc metalloproteinase-disintegrin HV1 | Q90ZI3 | Protobothrops flavoviridis | 4.17% |
| Tw_SMP03 | Zinc metalloproteinase-disintegrin-like 2d | J3SDW6 | Crotalus adamanteus | 2.30% |
| Snaclec (SNL) | 4.68% | |||
| Tw_SCL01 | Snaclec agglucetin subunit alpha-2 | Q8AYA5 | Deinagkistrodon acutus | 2.97% |
| Tw_SCL02 | Snaclec alboaggregin-D subunit beta | P0DM39 | Trimeresurus albolabris | 1.64% |
| Tw_SCL03 | C-type lectin TsL | Q9YGP1 | Trimeresurus stejnegeri | 0.06% |
| Phospholipase A2 (PLA2) | 2.13% | |||
| Tw_PLA01 | Basic phospholipase A2 homolog acutohaemolysin | O57385 | Deinagkistrodon acutus | 1.47% |
| Tw_PLA02 | Acidic phospholipase A2 Tgc-E6 | A8E2V8 | Trimeresurus gracilis | 0.60% |
| Tw_PLA03 | Basic phospholipase A2 Sms-N6 | Q6EER6 | Sistrurus miliarius | 0.06% |
| Tw_PLA04 | Basic phospholipase A2 Tbo-G6D49 | Q2YHJ2 | Trmeresurus borneensis | 0.0005% |
| L-amino acid oxidase (LAAO) | 1.26% | |||
| Tw_LAO01 | L-amino acid oxidase Lm29 | J7H670 | Lachesis muta | 1.26% |
| 5′nucleotidase (5′NT) | 1.03% | |||
| Tw_5NT01 | Snake venom 5′-nucleotidase | F8S0Z7 | Crotalus adamanteus | 0.56% |
| Tw_5NT02 | Snake venom 5′-nucleotidase | F8S0Z7 | Crotalus adamanteus | 0.47% |
| Snake venom serine proteinase (SVSP) | 0.65% | |||
| Tw_SSP01 | Venom plasminogen activator | E5L0E5 | Agkistrodon piscivorus leucostoma | 0.40% |
| Tw_SSP02 | Thrombin-like enzyme gyroxin analog | P33589 | Lachesis muta muta | 0.14% |
| Tw_SSP03 | Beta-fibrinogenase mucrofibrase-3 | Q91509 | Protobothrops mucrosquamatus | 0.11% |
| Tw_SSP04 | Beta-fibrinogenase mucrofibrase-3 | Q91509 | Protobothrops mucrosquamatus | 0.09% |
| Tw_SSP05 | Bradykinin-releasing enzyme KR-E-1 | Q7SZE2 | Gloydius ussuriensis | 0.09% |
| Tw_SSP06 | Thrombin-like enzyme stejnobin | Q8AY81 | Trimeresurus stejnegeri | 0.07% |
| Tw_SSP07 | Thrombin-like enzyme acutobin | Q9I8X2 | Deinagkistrodon acutus | 0.07% |
| Tw_SSP08 | Serine protease VLSP-3 | E0Y420 | Macrovipera lebetina | 0.05% |
| Tw_SSP09 | Thrombin-like enzyme KN-BJ 2 | O13069 | Bothrops jararaca | 0.02% |
| Phosphodiesterase (PDE) | 0.51% | |||
| Tw_PDE01 | Venom phosphodiesterase 1 | J3SEZ3 | Crotalus adamanteus | 0.33% |
| Tw_PDE02 | Venom phosphodiesterase 1 | J3SEZ3 | Crotalus adamanteus | 0.18% |
| Phospholipase B (PLB) | 0.26% | |||
| Tw_PLB01 | Phospholipase B | F8S101 | Crotalus adamanteus | 0.13% |
| Tw_PLB02 | Phospholipase B | F8S101 | Crotalus adamanteus | 0.12% |
| Cysteine-rich secretory protein (CRISP) | 0.22% | |||
| Tw_CRP01 | cysteine-rich secretory protein Og-CRPa | Q7ZTA0 | Agkistrodon piscivorus piscivorus | 0.22% |
| Phospholipase A2 inhibitor (PLA2 inhibitor) | 0.05% | |||
| Tw_PLI01 | Phospholipase A2 inhibitor | O93233 | Gloydius brevicaudus siniticus | 0.05% |
| Aminopeptidase | 0.06% | |||
| Tw_AP01 | Aminopeptidase A | A5HUI5 | Gloydius brevicaudus | 0.06% |
| Cystatin | 0.03% | |||
| Tw_CYS01 | Cystatin-C | J3SE80 | Crotalus adamanteus | 0.02% |
| Tw_CYS02 | Cystatin-1 | J3RYX9 | Crotalus adamanteus | 0.01% |
| Hyaluronidase (HYA) | 0.03% | |||
| Tw_HYA01 | Hyaluronidase | J3S820 | Crotalus adamanteus | 0.03% |
| Vascular endothelial growth factor (VEGF) | 0.005 | |||
| Tw_VGF01 | Snake venom vascular endothelial growth factor toxin | Q330K6 | Protobothrops mucrosquamatus | <0.005 |
| Tw_VGF02 | Snake venom vascular endothelial growth factor toxin | Q90X24 | Bothrops insularis | <0.005 |
| Tw_VGF03 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF04 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF05 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF06 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF07 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF08 | Vascular endothelial growth factor A | C0K3N4 | Agkistrodon piscivorus piscivorus | <0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.H.; Tan, K.Y.; Tan, N.H. De Novo Assembly of Venom Gland Transcriptome of Tropidolaemus wagleri (Temple Pit Viper, Malaysia) and Insights into the Origin of Its Major Toxin, Waglerin. Toxins 2023, 15, 585. https://doi.org/10.3390/toxins15090585
Tan CH, Tan KY, Tan NH. De Novo Assembly of Venom Gland Transcriptome of Tropidolaemus wagleri (Temple Pit Viper, Malaysia) and Insights into the Origin of Its Major Toxin, Waglerin. Toxins. 2023; 15(9):585. https://doi.org/10.3390/toxins15090585
Chicago/Turabian StyleTan, Choo Hock, Kae Yi Tan, and Nget Hong Tan. 2023. "De Novo Assembly of Venom Gland Transcriptome of Tropidolaemus wagleri (Temple Pit Viper, Malaysia) and Insights into the Origin of Its Major Toxin, Waglerin" Toxins 15, no. 9: 585. https://doi.org/10.3390/toxins15090585
APA StyleTan, C. H., Tan, K. Y., & Tan, N. H. (2023). De Novo Assembly of Venom Gland Transcriptome of Tropidolaemus wagleri (Temple Pit Viper, Malaysia) and Insights into the Origin of Its Major Toxin, Waglerin. Toxins, 15(9), 585. https://doi.org/10.3390/toxins15090585