Development of a New Cell-Based AP-1 Gene Reporter Potency Assay for Anti-Anthrax Toxin Therapeutics

Abstract
1. Introduction
2. Results
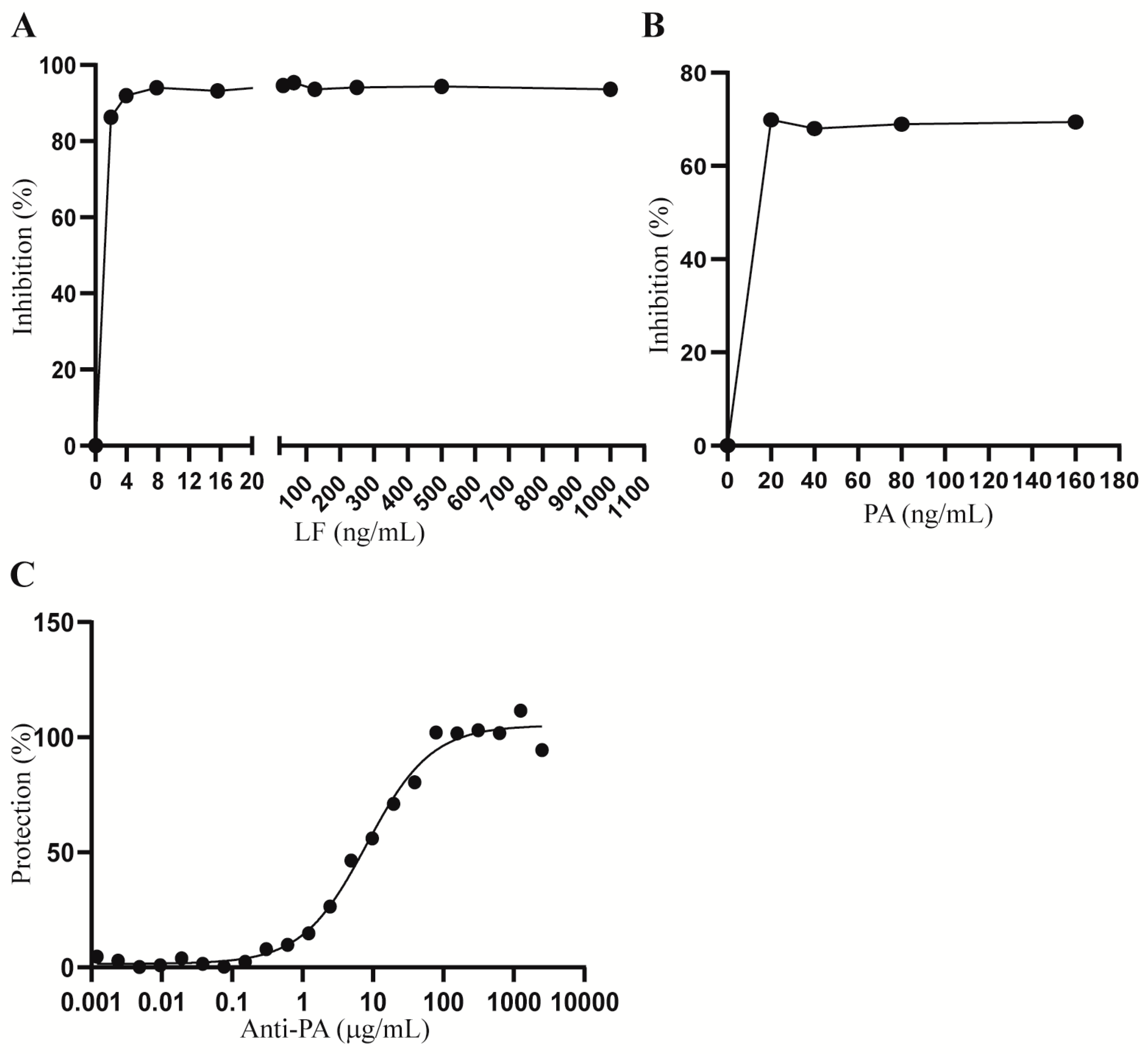
2.1. Optimization of LT and Anti-PA Antibody Concentrations for Hepa1c1c7 Cell Treatment
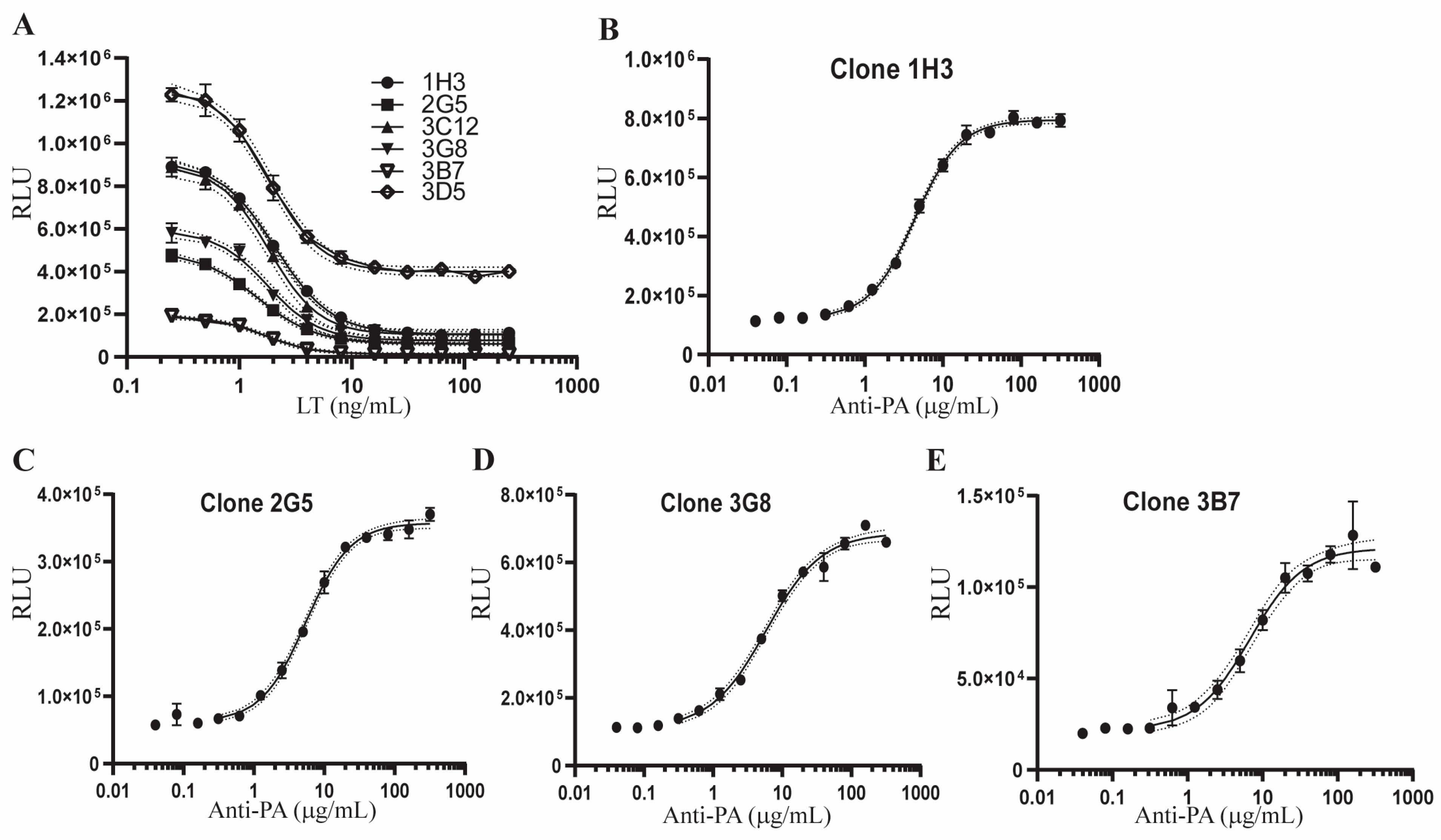
2.2. Development of Single Cell Clones of Hepa1c1c7-AP1Luc Cells for the AP-1 Reporter Assay
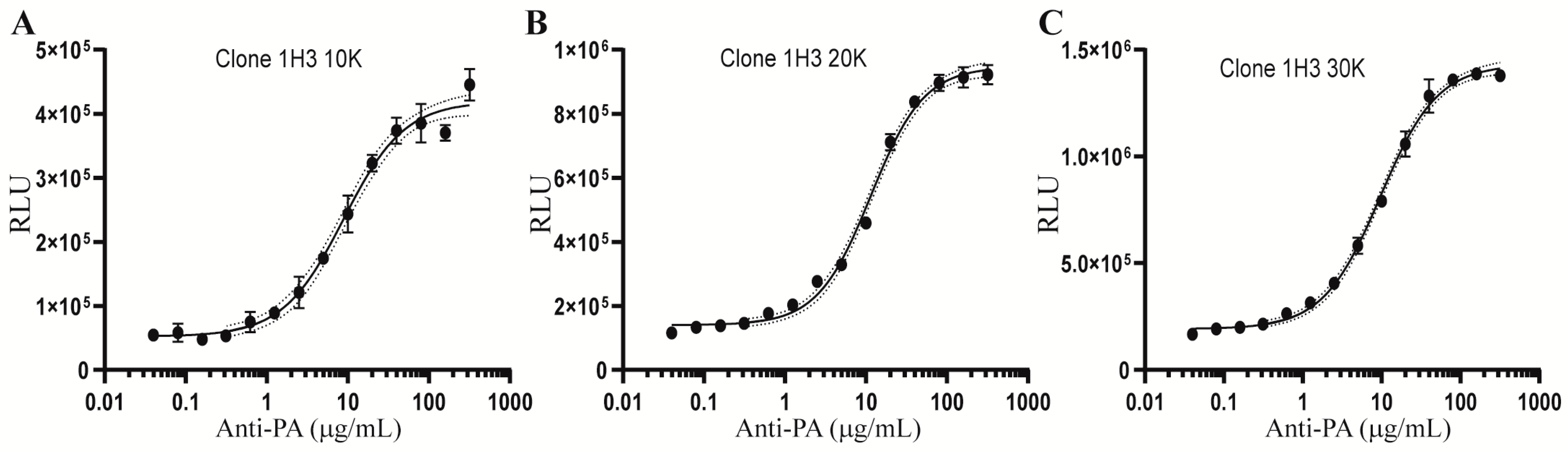
2.3. Optimization of the Cell Density for the AP-1 Reporter Assay
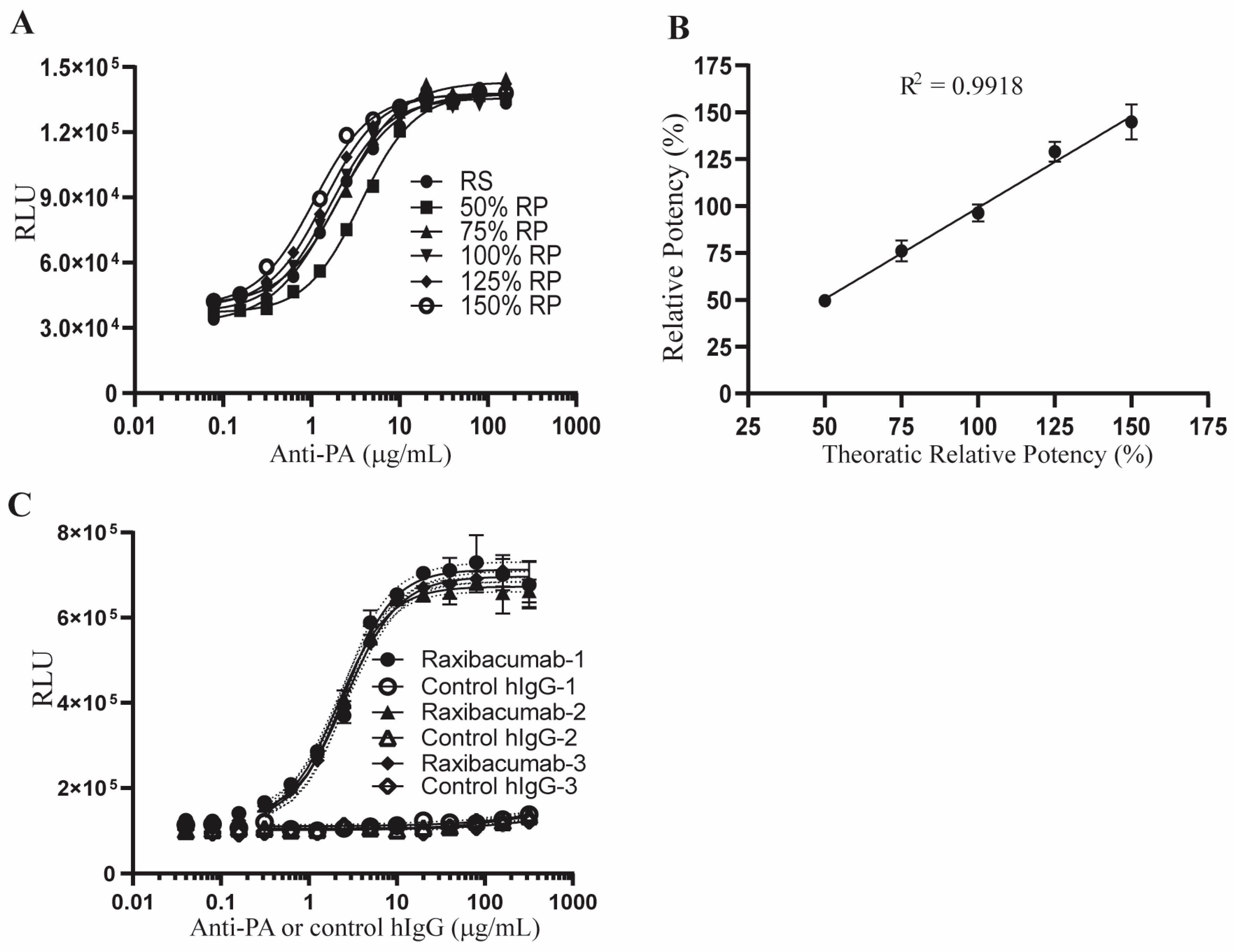
2.4. Qualification of the AP-1 Reporter Potency Assay
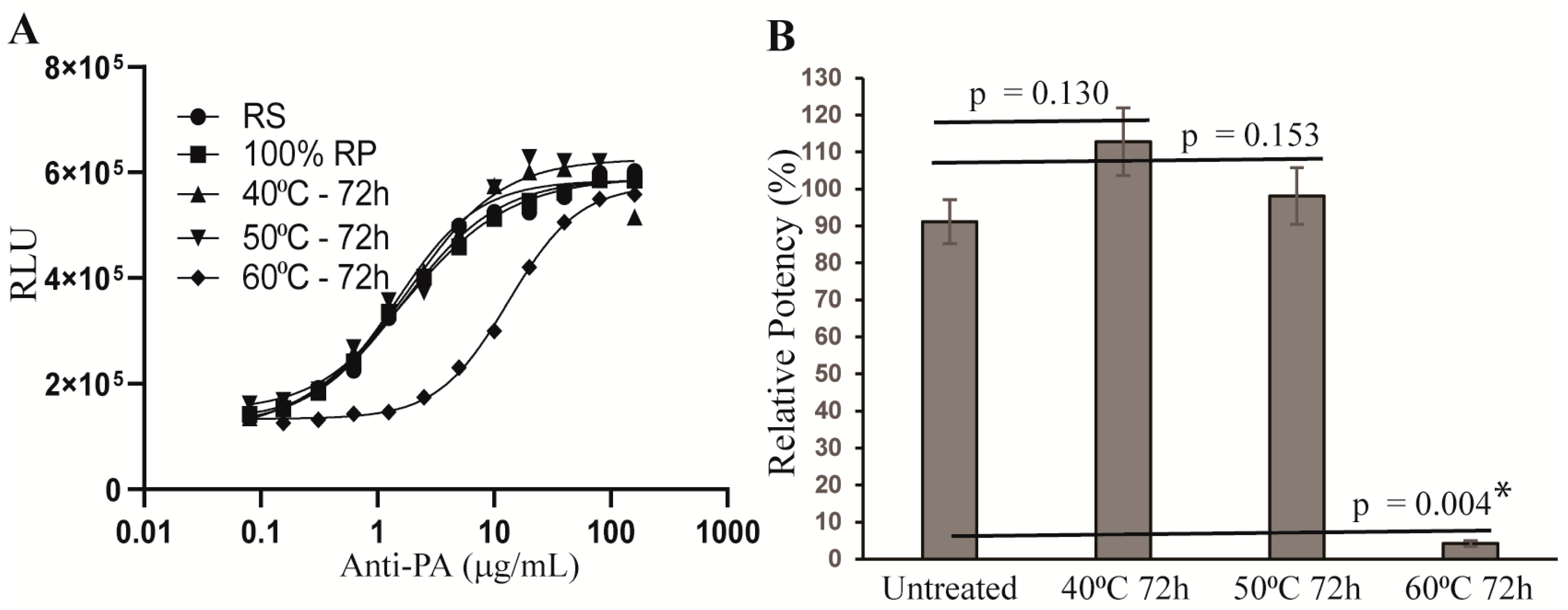
2.5. Stability-Indicating Capacity of the AP-1 Reporter Potency Assay
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents for Development of the AP-1 Reporter Assay
4.2. Transduction and Establishment of the Hepa1c1c7-AP1Luc Cell Line for the AP-1 Reporter Assay
4.3. Development of Single Cell Clones and a Cell Bank for the AP1 Reporter Assay
4.4. Cell Treatment and Measurement of Luciferase Activity in Treated Cells
4.5. Measurement of Anti-PA Potency
4.6. Qualification of the AP-1 Reporter Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Disclaimer
References
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Hicks, C.W.; Cui, X.; Li, Y.; Eichacker, P.Q. Anthrax infection. Am. J. Respir. Crit. Care Med. 2011, 184, 1333–1341. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Bower, W.A.; Hendricks, K.A.; Vieira, A.R.; Traxler, R.M.; Weiner, Z.; Lynfield, R.; Hoffmaster, A. What Is Anthrax? Pathogens 2022, 11, 690. [Google Scholar] [CrossRef]
- Doganay, M.; Dinc, G.; Kutmanova, A.; Baillie, L. Human Anthrax: Update of the Diagnosis and Treatment. Diagnostics 2023, 13, 1056. [Google Scholar] [CrossRef]
- Person, M.K.; Cook, R.; Bradley, J.S.; Hupert, N.; Bower, W.A.; Hendricks, K. Systematic Review of Hospital Treatment Outcomes for Naturally Acquired and Bioterrorism-Related Anthrax, 1880–2018. Clin. Infect. Dis. 2022, 75 (Suppl. S3), S392–S401. [Google Scholar] [CrossRef]
- Williams, M.; Armstrong, L.; Sizemore, D.C. Biologic, Chemical, and Radiation Terrorism Review. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rathjen, N.A.; Shahbodaghi, S.D. Bioterrorism. Am. Fam. Phys. 2021, 104, 376–385. [Google Scholar]
- Inglesby, T.V.; O’Toole, T.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Friedlander, A.M.; Gerberding, J.; Hauer, J.; Hughes, J.; et al. Anthrax as a biological weapon, 2002: Updated recommendations for management. JAMA 2002, 287, 2236–2252. [Google Scholar] [CrossRef]
- Peters, C.J.; Hartley, D.M. Anthrax inhalation and lethal human infection. Lancet 2002, 359, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the United States. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar] [CrossRef]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef]
- Liu, W.; Nestorovich, E.M. Anthrax toxin channel: What we know based on over 30 years of research. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183715. [Google Scholar] [CrossRef]
- Obiltoxaximab (Anthim) for inhalation anthrax. Med. Lett. Drugs Ther. 2018, 60, 150–151.
- Mazumdar, S. Raxibacumab. MAbs 2009, 1, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt 3, 739–745. [Google Scholar] [CrossRef]
- Ouyang, W.; Guo, P.; Fang, H.; Frucht, D.M. Anthrax lethal toxin rapidly reduces c-Jun levels by inhibiting c-Jun gene transcription and promoting c-Jun protein degradation. J. Biol. Chem. 2017, 292, 17919–17927. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, M.A.; Liu, S.; Portley, M.K.; O’Mard, D.; Fattah, R.; Szabo, R.; Bugge, T.H.; Khillan, J.S.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleaves regulatory subunits of phosphoinositide-3 kinase to contribute to toxin lethality. Nat. Microbiol. 2020, 5, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638. [Google Scholar] [CrossRef]
- Chavarria-Smith, J.; Vance, R.E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 2013, 9, e1003452. [Google Scholar] [CrossRef]
- Newman, Z.L.; Printz, M.P.; Liu, S.; Crown, D.; Breen, L.; Miller-Randolph, S.; Flodman, P.; Leppla, S.H.; Moayeri, M. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 2010, 6, e1000906. [Google Scholar] [CrossRef]
- Newman, Z.L.; Crown, D.; Leppla, S.H.; Moayeri, M. Anthrax lethal toxin activates the inflammasome in sensitive rat macrophages. Biochem. Biophys. Res. Commun. 2010, 398, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, R.; Batra, S. Anthrax toxin. Crit. Rev. Microbiol. 2001, 27, 167–200. [Google Scholar] [CrossRef]
- Brachman, P.S. Inhalation anthrax. Ann. N. Y. Acad. Sci. 1980, 353, 83–93. [Google Scholar] [CrossRef]
- Hesse, E.M.; Godfred-Cato, S.; Bower, W.A. Antitoxin Use in the Prevention and Treatment of Anthrax Disease: A Systematic Review. Clin. Infect. Dis. 2022, 75 (Suppl. S3), S432–S440. [Google Scholar] [CrossRef]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal antibody therapies against anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Raxibacumab for anthrax. Med. Lett. Drugs Ther. 2013, 55, 27–28.
- Raxibacumab. In Drugs and Lactation Database; LactMed(R): Bethesda, MD, USA, 2006.
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Raxibacumab for inhalational anthrax: An effective specific therapeutic approach? Expert. Opin. Investig. Drugs 2010, 19, 909–911. [Google Scholar] [CrossRef]
- Kummerfeldt, C.E. Raxibacumab: Potential role in the treatment of inhalational anthrax. Infect. Drug Resist. 2014, 7, 101–109. [Google Scholar] [CrossRef]
- Greig, S.L. Obiltoxaximab: First Global Approval. Drugs 2016, 76, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.F.; Leach, T.S.; King, A.; Guttendorf, R. Safety, Pharmacokinetics, and Immunogenicity of Obiltoxaximab After Intramuscular Administration to Healthy Humans. Clin. Pharmacol. Drug Dev. 2018, 7, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.W.; Morrill, A.M. Obiltoxaximab: Adding to the Treatment Arsenal for Bacillus anthracis Infection. Ann. Pharmacother. 2017, 51, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Ngundi, M.M.; Meade, B.D.; Lin, T.L.; Tang, W.J.; Burns, D.L. Comparison of three anthrax toxin neutralization assays. Clin. Vaccine Immunol. 2010, 17, 895–903. [Google Scholar] [CrossRef]
- Zmuda, J.F.; Zhang, L.; Richards, T.; Pham, Q.; Zukauskas, D.; Pierre, J.L.; Laird, M.W.; Askins, J.; Choi, G.H. Development of an edema factor-mediated cAMP-induction bioassay for detecting antibody-mediated neutralization of anthrax protective antigen. J. Immunol. Methods 2005, 298, 47–60. [Google Scholar] [CrossRef]
- Hering, D.; Thompson, W.; Hewetson, J.; Little, S.; Norris, S.; Pace-Templeton, J. Validation of the anthrax lethal toxin neutralization assay. Biologicals 2004, 32, 17–27. [Google Scholar] [CrossRef]
- Li, H.; Soroka, S.D.; Taylor, T.H., Jr.; Stamey, K.L.; Stinson, K.W.; Freeman, A.E.; Abramson, D.R.; Desai, R.; Cronin, L.X.; Oxford, J.W.; et al. Standardized, mathematical model-based and validated in vitro analysis of anthrax lethal toxin neutralization. J. Immunol. Methods 2008, 333, 89–106. [Google Scholar] [CrossRef]
- Mabry, R.; Brasky, K.; Geiger, R.; Carrion, R., Jr.; Hubbard, G.B.; Leppla, S.; Patterson, J.L.; Georgiou, G.; Iverson, B.L. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin. Vaccine Immunol. 2006, 13, 671–677. [Google Scholar] [CrossRef]
- Molin, F.D.; Fasanella, A.; Simonato, M.; Garofolo, G.; Montecucco, C.; Tonello, F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon 2008, 52, 824–828. [Google Scholar] [CrossRef]
- Xie, T.; Rotstein, D.; Sun, C.; Fang, H.; Frucht, D.M. Gastric pH and Toxin Factors Modulate Infectivity and Disease Progression After Gastrointestinal Exposure to Bacillus anthracis. J. Infect. Dis. 2017, 216, 1471–1475. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Kappelmann, M.; Bosserhoff, A.; Kuphal, S. AP-1/c-Jun transcription factors: Regulation and function in malignant melanoma. Eur. J. Cell Biol. 2014, 93, 76–81. [Google Scholar] [CrossRef]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef]
- Ouyang, W.; Frucht, D.M. Erk1/2 Inactivation-Induced c-Jun Degradation Is Regulated by Protein Phosphatases, UBE2d3, and the C-Terminus of c-Jun. Int. J. Mol. Sci. 2021, 22, 3889. [Google Scholar] [CrossRef]
- Ouyang, W.; Guo, P.; Takeda, K.; Fu, Q.; Fang, H.; Frucht, D.M. Erk1/2 inactivation promotes a rapid redistribution of COP1 and degradation of COP1 substrates. Proc. Natl. Acad. Sci. USA 2020, 117, 4078–4087. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A.; Morachis, J.M. High efficiency sorting and outgrowth for single-cell cloning of mammalian cell lines. Biotechnol. Lett. 2022, 44, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Sun, C.; Uslu, K.; Auth, R.D.; Fang, H.; Ouyang, W.; Frucht, D.M. A New Murine Model for Gastrointestinal Anthrax Infection. PLoS ONE 2013, 8, e66943. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Mytle, N.; Hopkins, R.J.; Malkevich, N.V.; Basu, S.; Meister, G.T.; Sanford, D.C.; Comer, J.E.; Van Zandt, K.E.; Al-Ibrahim, M.; Kramer, W.G.; et al. Evaluation of intravenous anthrax immune globulin for treatment of inhalation anthrax. Antimicrob. Agents Chemother. 2013, 57, 5684–5692. [Google Scholar] [CrossRef]
- Clark, A.; Wolfe, D.N. Current State of Anthrax Vaccines and Key R&D Gaps Moving Forward. Microorganisms 2020, 8, 651. [Google Scholar] [CrossRef]
- Fang, H.; Cordoba-Rodriguez, R.; Lankford, C.S.; Frucht, D.M. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J. Immunol. 2005, 174, 4966–4971. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Xie, T.; Rotstein, D.; Fang, H.; Frucht, D.M. Passive Immunotherapy Protects against Enteric Invasion and Lethal Sepsis in a Murine Model of Gastrointestinal Anthrax. Toxins 2015, 7, 3960–3976. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeatability | Test Results | ||||||||||
| Theoretical RP | Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 | Exp. 6 | Exp. 7 | Average | SD | RB% | CV |
| 50 | 46.68 | 51.42 | 45.33 | 49.27 | 49.83 | 49.94 | 54.27 | 49.53 | 2.95 | −0.93 | 5.95 |
| 75 | 71.39 | 86.74 | 74.05 | 79.53 | 73.44 | 70.47 | 76.26 | 75.98 | 5.63 | 1.31 | 7.41 |
| 100 | 95.49 | 93.57 | 101.21 | 102.26 | 97.41 | 95.58 | 88.98 | 96.36 | 4.53 | −3.64 | 4.70 |
| 125 | 123.09 | 125.32 | 129.06 | 126.69 | 126.74 | 138.00 | 134.28 | 129.03 | 5.29 | 3.22 | 4.10 |
| 150 | 152.88 | 149.39 | 155.5 | 130.85 | 136.83 | 149.77 | 138.91 | 144.88 | 9.29 | −3.42 | 6.42 |
| Intermediate Precision | Test Results | ||||||||||
| Theoretical RP | Analyst | Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 | Exp. 6 | Exp. 7 | Average | SD | CV |
| 100 | 1 | 95.49 | 93.57 | 101.21 | 102.26 | 97.41 | 95.58 | 88.98 | |||
| 100 | 2 | 99.83 | 103.02 | 116.27 | 108.98 | 94.97 | 101.30 | 101.83 | 100.05 | 6.80 | 6.79 |
| Attribute | Experiment | Acceptance Criteria | Results | Pass/Fail |
|---|---|---|---|---|
| Accuracy | Reference standard (RS) assessed at five relative potency (RP) levels (50%, 75%, 100%, 125% and 150% by 7 independent experiments | ±25% relative bias (RB) | −3.64 ≤ RB ≤ 3.22 | Pass |
| Repeatability | CV ≤ 25% | CV ≤ 7.41% | Pass | |
| Linearity | R² ≥ 0.97 | R² = 0.9918 | Pass | |
| Intermediate precision | RS assessed at the 100% RP level by 2 analyst and 7 independent experiments for each analyst | CV ≤ 30% | CV = 6.79 | Pass |
| Specificity | Anti-PA antibody and normal human IgG were tested at same concentrations by 3 independent experiments | 4-parameter logistic (4PL) convergence for Anti-PA antibody but not control human IgG | 4PL convergence for anti-PA antibody, but not for control human IgG | Pass |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, W.; Xie, T.; Fang, H.; Frucht, D.M. Development of a New Cell-Based AP-1 Gene Reporter Potency Assay for Anti-Anthrax Toxin Therapeutics. Toxins 2023, 15, 528. https://doi.org/10.3390/toxins15090528
Ouyang W, Xie T, Fang H, Frucht DM. Development of a New Cell-Based AP-1 Gene Reporter Potency Assay for Anti-Anthrax Toxin Therapeutics. Toxins. 2023; 15(9):528. https://doi.org/10.3390/toxins15090528
Chicago/Turabian StyleOuyang, Weiming, Tao Xie, Hui Fang, and David M. Frucht. 2023. "Development of a New Cell-Based AP-1 Gene Reporter Potency Assay for Anti-Anthrax Toxin Therapeutics" Toxins 15, no. 9: 528. https://doi.org/10.3390/toxins15090528
APA StyleOuyang, W., Xie, T., Fang, H., & Frucht, D. M. (2023). Development of a New Cell-Based AP-1 Gene Reporter Potency Assay for Anti-Anthrax Toxin Therapeutics. Toxins, 15(9), 528. https://doi.org/10.3390/toxins15090528