Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry

 ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
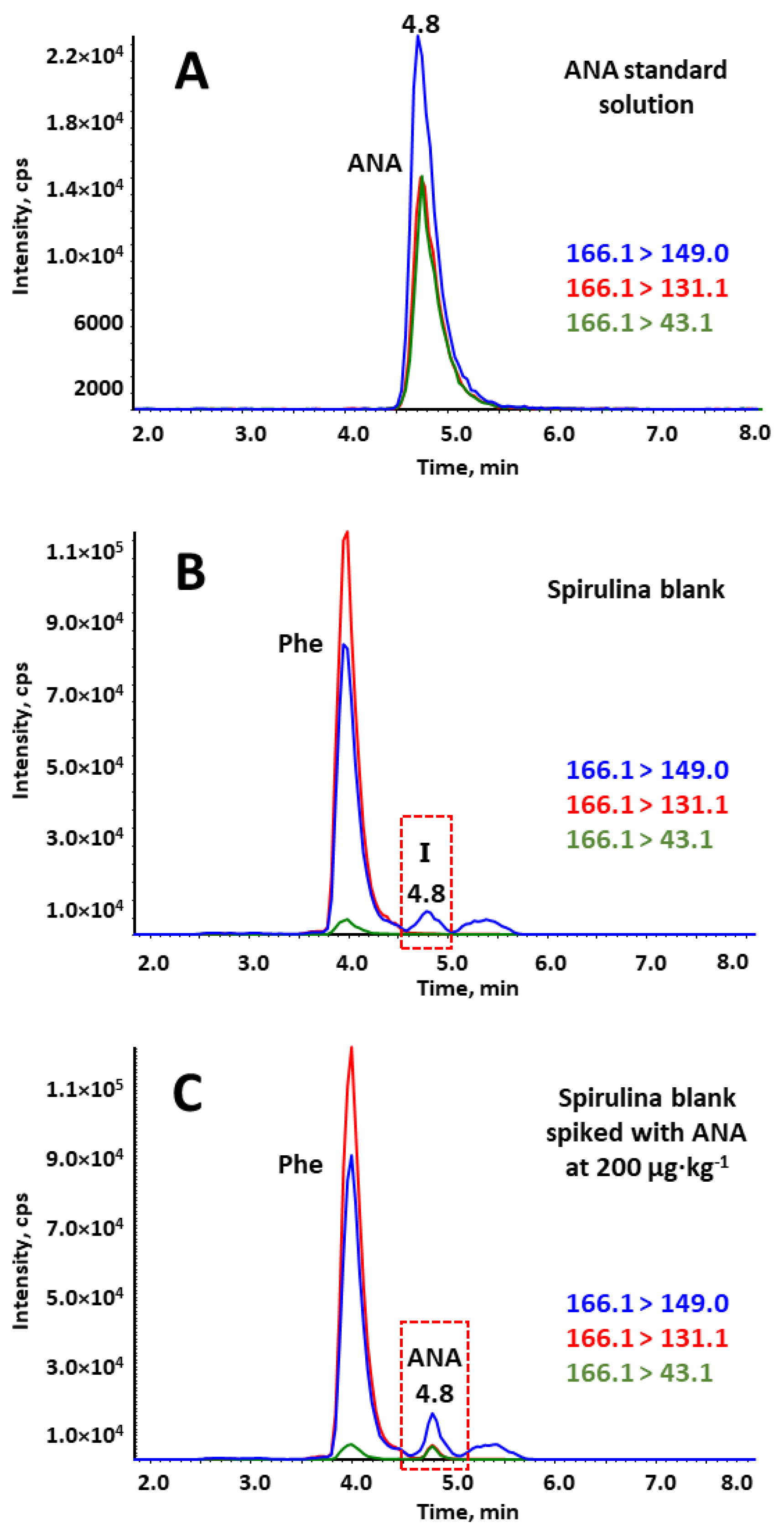
2.1. Optimization of Chromatographic and MS/MS Conditions
2.2. Multi-Toxin Extraction Procedure
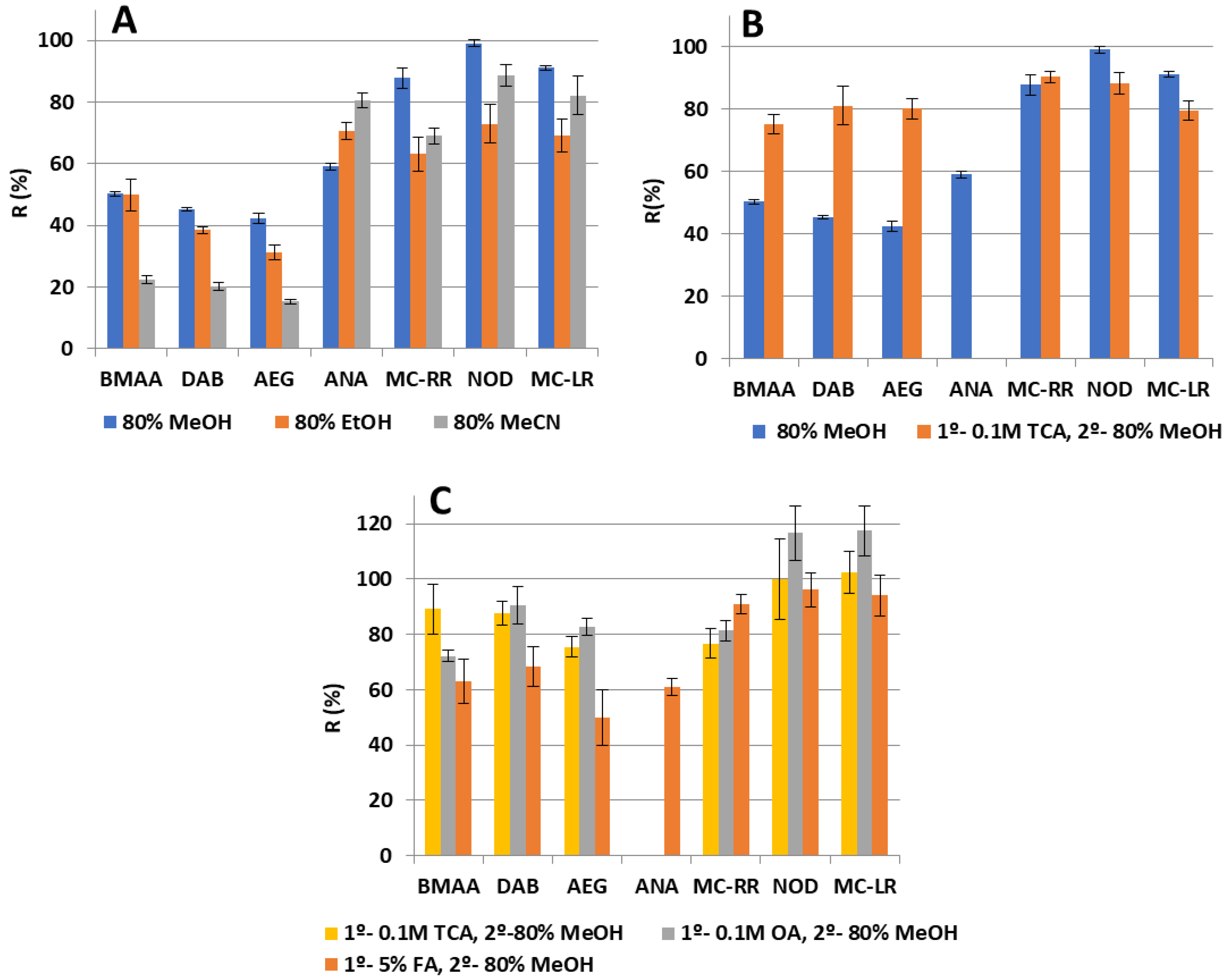
2.2.1. Study of Solvents for Solid–liquid Extraction
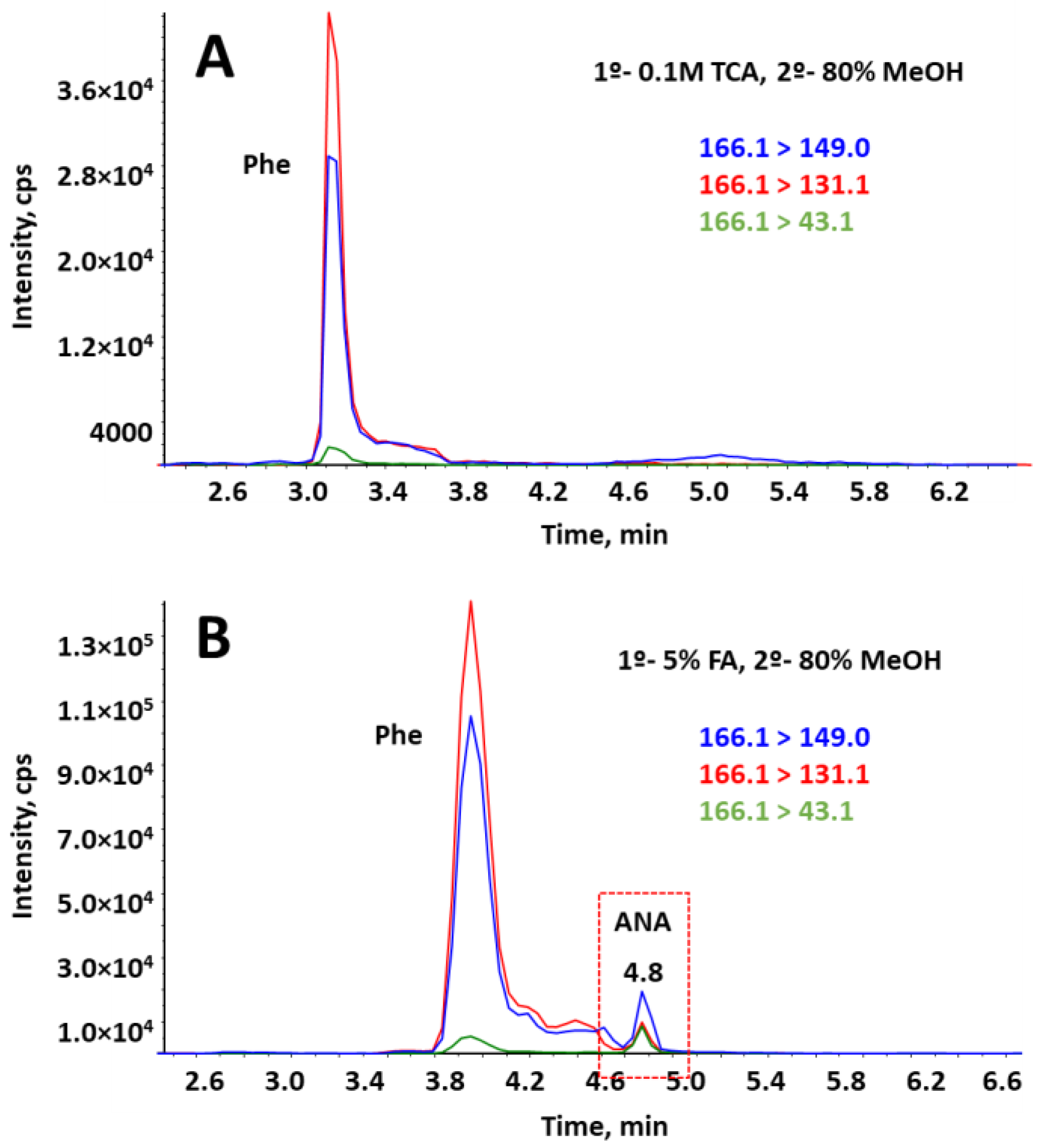
2.2.2. SPE Washing Step
2.2.3. SPE Load Volume
2.3. Validation of the Method in Spirulina Sample
2.3.1. Calibration Curves and Analytical Performance Characteristics
2.3.2. Matrix Effect
2.3.3. Precision
2.3.4. Recovery Assays
2.4. Determination of Cyanotoxins in BGA Dietary Supplements
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Instrumentation
4.3. BGA Dietary Supplements
4.4. Analyte Extraction
4.5. SPE Procedure
4.6. HILIC Separation
4.7. MS/MS Parameters
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Okamoto, K.; Fleming, L.E.; Wexler, P. Algae. In Encyclopedia of Toxicology, 2nd ed.; Oxford University Press: Oxford, UK, 2005; Volume 1, pp. 68–76. [Google Scholar]
- Corbel, S.; Mougin, C.; Bouaïcha, N. Cyanobacterial toxins: Modes of actions, fate in aquatic and soil ecosystems, phytotoxicity and bioaccumulation in agricultural crops. Chemosphere 2014, 96, 1–15. [Google Scholar] [CrossRef]
- Mutoti, M.; Gumbo, J.; Jideani, A.I.O. Occurrence of cyanobacteria in water used for food production: A review. Phys. Chem. Earth 2022, 125, 103101. [Google Scholar] [CrossRef]
- Drobac, D.; Tokodi, N.; Simeunović, J.; Baltić, V.; Stanić, D.; Svirčev, Z. Human exposure to cyanotoxins and their effects on health. Arh. Hig. Rada Toksiko. 2013, 64, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, S.A.M.; Shedid, E.S.; Saied, E.M.; Jassbi, A.R.; Jamebozorgi, F.H.; Rateb, M.E.; Du, M.; Abdel-Daim, M.M.; Kai, G.Y.; Al-Hammady, M.A.M.; et al. Cyanobacteria—From the oceans to the potential biotechnological and biomedical applications. Mar. Drugs 2021, 19, 241. [Google Scholar] [CrossRef] [PubMed]
- Chittora, D.; Meena, M.; Barupal, T.; Swapnil, P. Cyanobacteria as a source of biofertilizers for sustainable agriculture. Biochem. Biophys. Rep. 2020, 22, 100737. [Google Scholar] [CrossRef]
- Egan, B.; Hodgkins, C.; Shepherd, R.; Timotijevic, L.; Raats, M. An overview of consumer attitudes and beliefs about plant food supplements. Food Funct. 2011, 2, 747–752. [Google Scholar] [CrossRef]
- Heussner, A.H.; Mazija, L.; Fastner, J.; Dietrich, D.R. Toxin content and cytotoxicity of algal dietary supplements. Toxicol. Appl. Pharmacol. 2012, 265, 263–271. [Google Scholar] [CrossRef]
- Soni, R.A.; Sudhakar, K.; Rana, R.S. Spirulina—From growth to nutritional product: A review. Trends Food Sci. Technol. 2017, 69, 157–171. [Google Scholar] [CrossRef]
- Sierosławska, A.; Rymuszka, A.; Kalinowska, R.; Skowroński, T.; Bownik, A.; Pawlik-Skowrońska, B. Toxicity of cyanobacterial bloom in the eutrophic dam reservoir (Southeast Poland). Environ. Toxicol. Chem. 2010, 29, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Vichi, S.; Lavorini, P.; Funari, E.; Scardala, S.; Testai, E. Contamination by Microcystis and microcystins of blue-green algae food supplements (BGAS) on the Italian market and possible risk for the exposed population. Food Chem. Toxicol. 2012, 50, 4493–4499. [Google Scholar] [CrossRef]
- Miller, T.R.; Xiong, A.; Deeds, J.R.; Stutts, W.L.; Samdal, I.A.; Løvberg, K.E.; Miles, C.O. Microcystin Toxins at Potentially Hazardous Levels in Algal Dietary Supplements Revealed by a Combination of Bioassay, Immunoassay, and Mass Spectrometric Methods. J. Agric. Food Chem. 2020, 68, 8016–8025. [Google Scholar] [CrossRef]
- Chorus, I.; Bartram, J. Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring, and Management; World Health Organization: London, UK, 1999; Available online: https://cdn.who.int/media/docs/default-source/wash-documents/water-safety-and-quality/toxic-cyanobacteria---1st-ed.pdf?sfvrsn=338a8c22_1&download=true (accessed on 23 December 2022).
- Gilroy, D.J.; Kauffman, K.W.; Hall, R.A.; Huang, X.; Chu, F.S. Assessing potential health risks from microcystin toxins in blue-green algae dietary supplements. Environ Health Perspect. 2000, 108, 435–439. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for Drinking-Water Quality, 4th ed.; World Health Organization: Geneva, Switzerland, 2011; p. 541. [Google Scholar]
- Manolidi, K.; Triantis, T.M.; Kaloudis, T.; Hiskia, A. Neurotoxin BMAA and its isomeric amino acids in cyanobacteria and cyanobacteria-based food supplements. J. Hazard. Mater. 2019, 365, 346–365. [Google Scholar] [CrossRef]
- Bruno, M.; Fiori, M.; Mattei, D.; Melchiorre, S.; Messineo, V.; Volpi, F.; Bogialli, S.; Nazzari, M. ELISA and LC-MS/MS methods for determining cyanobacterial toxins in blue-green algae food supplements. Nat. Prod. Res. 2006, 20, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Marsan, D.W.; Conrad, S.M.; Stutts, W.L.; Parker, C.H.; Deeds, J.R. Evaluation of microcystin contamination in blue-green algal dietary supplements using a protein phosphatase inhibition-based test kit. Heliyon 2018, 4, e00573. [Google Scholar] [CrossRef]
- Turner, A.D.; Waack, J.; Lewis, A.; Edwards, C.; Lawton, L. Development and single-laboratory validation of a UHPLC-MS/MS method for quantitation of microcystins and nodularin in natural water, cyanobacteria, shellfish and algal supplement tablet powders. J. Chromatogr. B 2018, 1074–1075, 111–123. [Google Scholar] [CrossRef]
- Parker, C.H.; Stutts, W.L.; Degrasse, S.L. Development and Validation of a Liquid Chromatography-Tandem Mass Spectrometry Method for the Quantitation of Microcystins in Blue-Green Algal Dietary Supplements. J. Agric. Food Chem. 2015, 63, 10303–10312. [Google Scholar] [CrossRef] [PubMed]
- Ortelli, D.; Edder, P.; Cognard, E.; Jan, P. Fast screening and quantitation of microcystins in microalgae dietary supplement products and water by liquid chromatography coupled to time of flight mass spectrometry. Anal. Chim. Acta 2008, 617, 230–237. [Google Scholar] [CrossRef]
- Manali, K.M.; Arunraj, R.; Kumar, T.; Ramya, M. Detection of microcystin producing cyanobacteria in Spirulina dietary supplements using multiplex HRM quantitative PCR. J. Appl. Phycol. 2017, 29, 1279–1286. [Google Scholar] [CrossRef]
- Giménez-Campillo, C.; Pastor-Belda, M.; Campillo, N.; Arroyo-Manzanares, N.; Hernández-Córdoba, M.; Viñas, P. Determination of cyanotoxins and phycotoxins in seawater and algae-based food supplements using ionic liquids and liquid chromatography with time-of-flight mass spectrometry. Toxins 2019, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Scott, P.M. Determination of the cyanobacterial toxin cylindrospermopsin in algal food supplements. Food Addit. Contam. 2011, 28, 786–790. [Google Scholar] [CrossRef]
- Rellán, S.; Osswald, J.; Saker, M.; Gago-Martinez, A.; Vasconcelos, V. First detection of anatoxin-a in human and animal dietary supplements containing cyanobacteria. Food Chem. Toxicol. 2009, 47, 2189–2195. [Google Scholar] [CrossRef]
- Rawn, D.F.K.; Lau, B.P.Y.; Niedzwiadek, B.; Lawrence, J.F. Improved method for the determination of anatoxin-a and two of its metabolites in blue-green algae using liquid chromatography with fluorescence detection. J. AOAC Int. 2005, 88, 1741–1747. [Google Scholar] [CrossRef]
- Rawn, D.F.K.; Niedzwiadek, B.; Lau, B.P.Y.; Saker, M. Anatoxin-a and its metabolites in blue-green algae food supplements from Canada and Portugal. J. Food Prot. 2007, 70, 776–779. [Google Scholar] [CrossRef]
- Draisci, R.; Ferretti, E.; Palleschi, L.; Marchiafava, C. Identification of anatoxins in blue-green algae food supplements using liquid chromatography-tandem mass spectrometry. Food Addit. Contam. 2001, 18, 525–531. [Google Scholar] [CrossRef]
- Roy-Lachapelle, A.; Solliec, M.; Bouchard, M.F.; Sauvé, S. Detection of Cyanotoxins in Algae Dietary Supplements. Toxins 2017, 9, 76. [Google Scholar] [CrossRef]
- Rzymski, P.; Niedzielski, P.; Kaczmarek, N.; Jurczak, T.; Klimaszyk, P. The multidisciplinary approach to safety and toxicity assessment of microalgae-based food supplements following clinical cases of poisoning. Harmful Algae 2015, 46, 34–42. [Google Scholar] [CrossRef]
- Klijnstra, M.D.; Faassen, E.J.; Gerssen, A. A generic LC-HRMS screening method for marine and freshwater phycotoxins in fish, shellfish, water, and supplements. Toxins 2021, 13, 823. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Parra, E.; Boutarfa, S.; Aboal, M. Are Cyanotoxins the Only Toxic Compound Potentially Present in Microalgae Supplements? Results from a Study of Ecological and Non-Ecological Products. Toxins 2020, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- van Pamel, E.; Henrottin, J.; van Poucke, C.; Gillard, N.; Daeseleire, E. Multi-Class UHPLC-MS/MS Method for Plant Toxins and Cyanotoxins in Food Supplements and Application for Belgian Market Samples. Planta Med. 2021, 87, 1069–1079. [Google Scholar] [CrossRef]
- Aparicio-Muriana, M.M.; Carmona-Molero, R.; Lara, F.J.; García-Campaña, A.M.; del Olmo-Iruela, M. Multiclass cyanotoxin analysis in reservoir waters: Tandem solid-phase extraction followed by zwitterionic hydrophilic interaction liquid chromatography-mass spectrometry. Talanta 2022, 237, 122929. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.L. Cyanobacterial peptides beyond microcystins—A review on co-occurrence, toxicity, and challenges for risk assessment. Water Res. 2019, 151, 488–499. [Google Scholar] [CrossRef]
- Buratti, F.M.; Manganelli, M.; Vichi, S.; Stefanelli, M.; Scardala, S.; Testai, E.; Funari, E. Cyanotoxins: Producing organisms, occurrence, toxicity, mechanism of action and human health toxicological risk evaluation. Arch. Toxicol. 2017, 91, 1049–1130. [Google Scholar] [CrossRef]
- Schneider, T.; Simpson, C.; Desai, P.; Tucker, M.; Lobner, D. Neurotoxicity of isomers of the environmental toxin L-BMAA. Toxicon 2020, 184, 175–179. [Google Scholar] [CrossRef]
- Díez-Quijada, L.; Puerto, M.; Gutiérrez-Praena, D.; Llana-Ruiz-Cabello, M.; Jos, A.; Cameán, A.M. Microcystin-RR: Occurrence, content in water and food and toxicological studies. A review. Environ. Res. 2019, 168, 467–489. [Google Scholar] [CrossRef]
- Dimitrakopoulos, I.K.; Kaloudis, T.S.; Hiskia, A.E.; Thomaidis, N.S.; Koupparis, M.A. Development of a fast and selective method for the sensitive determination of anatoxin-a in lake waters using liquid chromatography-tandem mass spectrometry and phenylalanine-d5 as internal standard. Anal. Bioanal. Chem. 2010, 397, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Liestianty, D.; Rodianawati, I.; Arfah, R.A.; Assa, A.; Patimah; Sundari; Muliadi. Nutritional analysis of spirulina sp to promote as superfood candidate. IOP Conf. Ser. Mater. Sci. Eng. 2019, 509, 012031. [Google Scholar] [CrossRef]
- Gurbuz, F.; Uzunmehmetoğlu, O.Y.; Diler, Ö.; Metcalf, J.S.; Codd, G.A. Occurrence of microcystins in water, bloom, sediment and fish from a public water supply. Sci. Total Environ. 2016, 562, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.R.; Fischer, A.; Michel, C.; Hoeger, S. Chapter 39: Toxin mixture in cyanobacterial blooms—A critical comparison of reality with current procedures employed in human health risk assessment. In Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs. Advances in Experimental Medicine and Biology; Hudnell, H.K., Ed.; Springer: New York, NY, USA, 2008; Volume 619, pp. 885–912. [Google Scholar] [CrossRef]
- Saker, M.L.; Jungblut, A.D.; Neilan, B.A.; Rawn, D.F.K.; Vasconcelos, V.M. Detection of microcystin synthetase genes in health food supplements containing the fresh water cyanobacterium Aphanizomenon flos-aquae. Toxicon 2005, 46, 555–562. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Linear Range (μg·kg−1) | LOD (μg·kg−1) | LOQ (μg·kg−1) | R2 | Intra-Day Precision (RSD %) n = 9 | Inter-Day Precision (RSD %) n = 15 | Matrix Effect (%) n = 9 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L1 | L2 | L3 | L1 | L2 | L3 | |||||
| MC-LR | 60–500 | 18 | 60 | 0.9906 | 8.3 | 11.5 | 11.4 | 14.9 | 14.7 | 14.8 | 467.8 | 401.2 | 356.4 |
| NOD | 50–500 | 15 | 50 | 0.9872 | 11.9 | 13.1 | 16.3 | 15.2 | 15.3 | 13.9 | 138.2 | 118.7 | 123.3 |
| MC-RR | 50–500 | 15 | 50 | 0.9910 | 8.8 | 7.0 | 3.7 | 8.2 | 10.1 | 5.6 | 41.0 | 43.7 | 38.7 |
| ANA | 150–750 | 45 | 150 | 0.9900 | 9.1 | 8.0 | 7.1 | 8.5 | 8.6 | 8.2 | 33.0 | 27.6 | 26.0 |
| BMAA | 300–2500 | 90 | 300 | 0.9817 | 15.2 | 19.5 | 19.2 | 25.1 | 19.6 | 18.8 | 23.0 | 18.1 | 9.1 |
| DAB | 300–2500 | 90 | 300 | 0.9938 | 9.8 | 15.4 | 9.2 | 17.3 | 16.3 | 11.9 | 24.4 | 29.7 | 21.5 |
| AEG | 150–2500 | 45 | 150 | 0.9940 | 18.3 | 9.5 | 15.2 | 16.3 | 16.1 | 19.3 | 88.0 | 86.0 | 63.8 |
| Recoveries (%) (n = 6) | ||||||
|---|---|---|---|---|---|---|
| L1 | L2 | L3 | ||||
| Analyte | R % | RSD % | R % | RSD % | R % | RSD % |
| MC-LR | 74.0 | 15.1 | 76.6 | 9.9 | 81.6 | 9.2 |
| NOD | 102.9 | 16.1 | 94.1 | 19.2 | 77.7 | 15.4 |
| MC-RR | 91.2 | 11.6 | 89.9 | 5.1 | 88.0 | 4.4 |
| ANA | 87.8 | 11.8 | 80.1 | 10.2 | 78.7 | 9.0 |
| BMAA | 70.2 | 17.5 | 64.2 | 16.8 | 80.7 | 16.5 |
| DAB | 91.0 | 11.8 | 76.6 | 12.5 | 72.9 | 11.7 |
| AEG | 82.7 | 11.1 | 67.9 | 18.2 | 81.6 | 14.0 |
| Sample | MC-LR | NOD | MC-RR | ANA | BMAA | DAB | AEG |
|---|---|---|---|---|---|---|---|
| 1 | n.d. | n.d. | n.d. | n.d. | n.d. | 432 | n.d. |
| 2 | n.d. | n.d. | n.d. | n.d. | n.d. | 1065 | n.d. |
| 3 | n.d. | n.d. | n.d. | n.d. | n.d. | 2408 | 194 |
| 4 | n.d. | n.d. | n.d. | n.d. | n.d. | 900 | n.d. |
| 5 | n.d. | n.d. | n.d. | n.d. | n.d. | 605 | n.d. |
| 6 | n.d. | n.d. | n.d. | n.d. | n.d. | 2038 | 71 |
| 7 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 8 | n.d. | n.d. | n.d. | n.d. | n.d. | 331 | n.d. |
| 9 | 6583 | n.d. | 5508 | n.d. | n.d. | n.d. | n.d. |
| Analyte | Retention Time (min) | Molecular Ion | Precursor Ion (m/z) | DP (V) | EP (V) | CEP (V) | Product Ions (m/z) | CE (V) | CXP (V) |
|---|---|---|---|---|---|---|---|---|---|
| MC-LR | 2.8 | [M + H]+ | 995.6 | 136.0 | 10.5 | 32.0 | Qion 135.2 | 93.0 | 4.0 |
| 136.0 | 10.5 | 32.0 | Iion 105.0 | 129.0 | 4.0 | ||||
| NOD | 3.0 | [M + H]+ | 825.4 | 96.0 | 6.5 | 24.0 | Qion 135.1 | 89.0 | 2.0 |
| 96.0 | 6.5 | 100.0 | Iion 103.2 | 129.0 | 4.0 | ||||
| MC-RR | 4.0 | [M + 2H]2+ | 519.8 | 41.0 | 6.0 | 18.0 | Qion 135.2 | 39.0 | 4.0 |
| 41.0 | 6.0 | 18.0 | Iion 103.2 | 91.0 | 6.0 | ||||
| ANA | 4.8 | [M + H]+ | 166.2 | 36.0 | 4.0 | 10.0 | Qion 131.1 | 21.0 | 4.0 |
| 36.0 | 4.0 | 10.0 | Iion 43.1 | 39.0 | 4.0 | ||||
| BMAA | 8.9 | [M + H]+ | 119.2 | 26.0 | 4.0 | 6.0 | Qion 44.1 | 27.0 | 2.0 |
| 26.0 | 4.0 | 6.0 | Iion 102.1 | 15.0 | 4.0 | ||||
| DAB | 9.7 | [M + H]+ | 119.1 | 21.0 | 3.0 | 14.0 | Qion 101.0 | 11.0 | 4.0 |
| 21.0 | 3.0 | 14.0 | Iion 56.0 | 31.0 | 2.0 | ||||
| AEG | 11.3 | [M + H]+ | 119.1 | 26.0 | 3.5 | 10.0 | Qion 102.0 | 11.0 | 4.0 |
| 26.0 | 3.5 | 10.0 | Iion 56.0 | 25.0 | 4.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aparicio-Muriana, M.d.M.; Lara, F.J.; Olmo-Iruela, M.D.; García-Campaña, A.M. Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry. Toxins 2023, 15, 127. https://doi.org/10.3390/toxins15020127
Aparicio-Muriana MdM, Lara FJ, Olmo-Iruela MD, García-Campaña AM. Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry. Toxins. 2023; 15(2):127. https://doi.org/10.3390/toxins15020127
Chicago/Turabian StyleAparicio-Muriana, María del Mar, Francisco J. Lara, Monsalud Del Olmo-Iruela, and Ana M. García-Campaña. 2023. "Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry" Toxins 15, no. 2: 127. https://doi.org/10.3390/toxins15020127
APA StyleAparicio-Muriana, M. d. M., Lara, F. J., Olmo-Iruela, M. D., & García-Campaña, A. M. (2023). Determination of Multiclass Cyanotoxins in Blue-Green Algae (BGA) Dietary Supplements Using Hydrophilic Interaction Liquid Chromatography-Tandem Mass Spectrometry. Toxins, 15(2), 127. https://doi.org/10.3390/toxins15020127