Cellular Uptake and Cytotoxicity of Clostridium perfringens Iota-Toxin


{kind=link}
Abstract
:1. Introduction
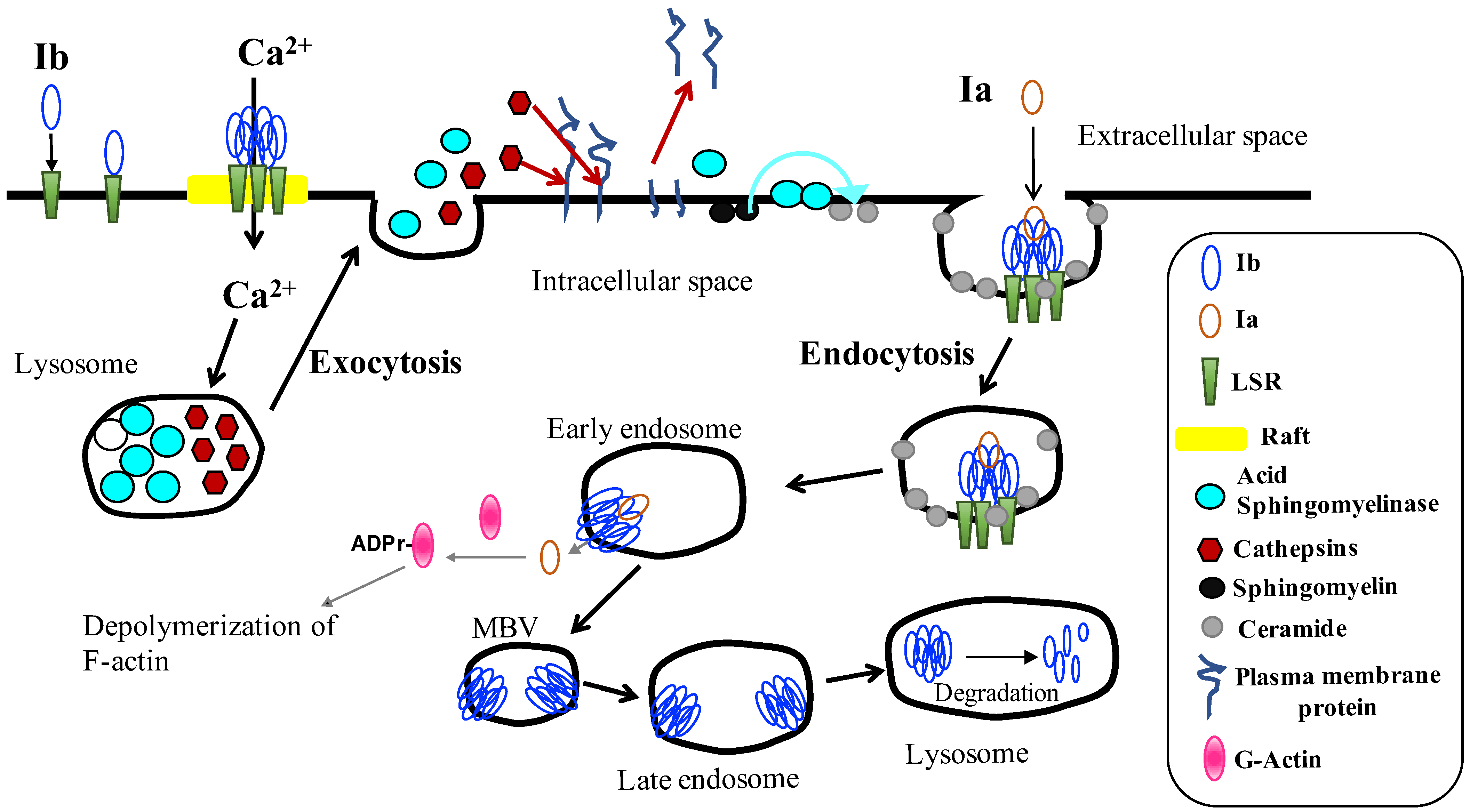
2. Binding and Internalization of Iota-Toxin
3. Cellular Trafficking of Iota-Toxin
4. Cytotoxic Effect of Ib
5. Effect of Iota-Toxin on Animals
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stiles, B.G.; Wigelsworth, D.J.; Popoff, M.R.; Barth, H. Clostridial binary toxins: Iota and C2 family portraits. Front. Cell. Infect. Microbiol. 2011, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.G.; Pradhan, K.; Fleming, J.M.; Samy, R.P.; Barth, H.; Popoff, M.R. Clostridium and bacillus binary enterotoxins: Bad for the bowels, and eukaryotic being. Toxins 2014, 6, 2626–2656. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Benz, R.; Popoff, M.R. Pore-forming activity of clostridial binary toxins. Biochim. Biophys. Acta 2016, 1858, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Aktories, K. Receptor-binding and uptake of binary actin-ADP-ribosylating toxins. Curr. Top. Microbiol. Immunol. 2017, 406, 119–133. [Google Scholar] [PubMed]
- Takehara, M.; Takagishi, T.; Seike, S.; Oda, M.; Sakaguchi, Y.; Hisatsune, J.; Ochi, S.; Kobayashi, K.; Nagahama, M. Cellular entry of Clostridium perfringens iota-toxin and Clostridium botulinum C2 toxin. Toxins 2017, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 1996, 9, 216–234. [Google Scholar] [CrossRef]
- Li, J.; Adams, V.; Bannam, T.L.; Miyamoto, K.; Garcia, J.P.; Uzal, F.A.; Rood, J.I.; McClane, B.A. Toxin plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 2013, 77, 208–333. [Google Scholar] [CrossRef] [PubMed]
- Redondo, L.M.; Farber, M.; Venzano, A.; Jost, B.H.; Parma, Y.R.; Fernandez-Miyakawa, M.E. Sudden death syndrome in adult cows associated with Clostridium perfringens type E. Anaerobe 2013, 20, 1–4. [Google Scholar] [CrossRef]
- Sakurai, J.; Nagahama, M.; Oda, M.; Tsuge, H.; Kobayashi, K. Clostridium perfringens iota-toxin: Structure and function. Toxins 2009, 1, 208–228. [Google Scholar] [CrossRef]
- Tsurumura, T.; Tsumori, Y.; Qiu, H.; Oda, M.; Sakurai, J.; Nagahama, M.; Tsuge, H. Arginine ADP-ribosylation mechanism based on structural snapshots of iota-toxin and actin complex. Proc. Natl. Acad. Sci. USA 2013, 110, 4267–4272. [Google Scholar] [CrossRef]
- Yamada, T.; Yoshida, T.; Kawamoto, A.; Mitsuoka, K.; Iwasaki, K.; Tsuge, H. Cryo-EM structures reveal translocational unfolding in the clostridial binary iota toxin complex. Nat. Struct. Mol. Biol. 2020, 27, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Marvaud, J.C.; Smith, T.; Hale, M.L.; Popoff, M.R.; Smith, L.A.; Stiles, B.G. Clostridium perfringens iota-toxin: Mapping of receptor binding and Ia docking domains on Ib. Infect. Immun. 2001, 69, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Carette, J.E.; Bell, G.W.; Schwan, C.; Guttenberg, G.; Brummelkamp, T.R.; Aktories, K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. USA 2011, 108, 16422–16427. [Google Scholar] [CrossRef]
- Schmidt, G.; Papatheodorou, P.; Aktories, K. Novel receptors for bacterial protein toxins. Curr. Opin. Microbiol. 2015, 23, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Takehara, M.; Kobayashi, K. Interaction of Clostridium perfringens iota-toxin and lipolysis-stimulated lipoprotein receptor (LSR). Toxins 2018, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Oda, Y.; Sasaki, H.; Ikenouchi, J.; Higashi, T.; Akashi, M.; Nishi, E.; Furuse, M. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J. Cell Sci. 2011, 124, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Tokuda, S.; Kitajiri, S.; Masuda, S.; Nakamura, H.; Oda, Y.; Furuse, M. Analysis of the ‘angulin’ proteins LSR, ILDR1 and ILDR2-tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J. Cell Sci. 2013, 126, 966–977. [Google Scholar] [CrossRef]
- Furuse, M.; Izumi, Y.; Oda, Y.; Higashi, T.; Iwamoto, N. Molecular organization of tricellular tight junctions. Tissue Barriers 2014, 2, e28755. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Tachibana, K.; Krug, S.M.; Kunisawa, J.; Fromm, M.; Kondoh, M. Potential for tight junction protein-directed drug development using claudin binders and angubindin-1. Int. J. Mol. Sci. 2019, 20, 4016. [Google Scholar] [CrossRef]
- Kyuno, T.; Kyuno, D.; Kohno, T.; Konno, T.; Kikuchi, S.; Arimoto, C.; Yamaguchi, H.; Imamura, M.; Kimura, Y.; Kondoh, M.; et al. Tricellular tight junction protein LSR/angulin-1 contributes to the epithelial barrier and malignancy in human pancreatic cancer cell line. Histochem. Cell Biol. 2020, 153, 5–16. [Google Scholar] [CrossRef]
- Konno, T.; Kohno, T.; Kikuchi, S.; Shimada, H.; Satohisa, S.; Saito, T.; Kondoh, M.; Kojima, T. Epithelial barrier dysfunction and cell migration induction via JNK/cofilin/actin by angubindin-1. Tissue Barriers 2020, 8, 1695475. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Iwashita, Y.; Wakayama, E.; Nishino, I.; Nishikaji, T.; Kondoh, M. Tight junction modulating bioprobes for drug delivery system to the brain: A review. Pharmaceutics 2020, 12, 1236. [Google Scholar] [CrossRef] [PubMed]
- Wigelsworth, D.J.; Ruthel, G.; Schnell, L.; Herrlich, P.; Blonder, J.; Veenstra, T.D.; Carman, R.J.; Wilkins, T.D.; Van Nhieu, G.T.; Pauillac, S.; et al. CD44 Promotes intoxication by the clostridial iota-family toxins. PLoS ONE 2012, 7, e51356. [Google Scholar] [CrossRef] [PubMed]
- Blonder, J.; Hale, M.L.; Chan, K.C.; Yu, L.R.; Lucas, D.A.; Conrads, T.P.; Zhou, M.; Popoff, M.R.; Issaq, H.J.; Stiles, B.G.; et al. Quantitative profiling of the detergent-resistant membrane proteome of iota-b toxin induced vero cells. J. Proteome Res. 2005, 4, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Hornuss, D.; Nölke, T.; Hemmasi, S.; Castonguay, J.; Picchianti, M.; Aktories, K. Clostridium difficile binary toxin CDT induces clustering of the lipolysis-stimulated lipoprotein receptor into lipid rafts. MBio 2013, 4, e00244-13. [Google Scholar] [CrossRef]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef] [PubMed]
- Etxaniz, A.; González-Bullón, D.; Martín, C.; Ostolaza, H. Membrane repair mechanisms against permeabilization by pore-forming toxins. Toxins 2018, 10, 234. [Google Scholar] [CrossRef]
- Brito, C.; Cabanes, D.; Sarmento Mesquita, F.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef]
- Espiritu, R.A. Repairing plasma membrane damage in regulated necrotic cell death. Mol. Biol. Rep. 2021, 48, 2751–2759. [Google Scholar] [CrossRef]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gomes, T.; Corrotte, M.; Tam, C.; Andrews, N.W. Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS ONE 2016, 11, e0152583. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Takehara, M.; Miyamoto, K.; Ishidoh, K.; Kobayashi, K. Acid sphingomyelinase promotes cellular internalization of Clostridium perfringens iota-toxin. Toxins 2018, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Kobayashi, K.; Takehara, M. Cathepsin release from lysosomes promotes endocytosis of Clostridium perfringens iota-toxin. Toxins 2021, 13, 721. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Babiychuk, E.B. Ceramide in plasma membrane repair. Handb. Exp. Pharmacol. 2013, 216, 341–353. [Google Scholar]
- Nagahama, M.; Takehara, M.; Takagishi, T.; Seike, S.; Miyamoto, K.; Kobayashi, K. Cellular uptake of Clostridium botulinum C2 toxin requires acid sphingomyelinase activity. Infect. Immun. 2017, 85, e00966-16. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Kobayashi, K.; Ochi, S.; Takehara, M. Internalization of Clostridium botulinum C2 toxin is regulated by cathepsin B released from lysosomes. Toxins 2021, 13, 272. [Google Scholar] [CrossRef]
- Hale, M.L.; Marvaud, J.C.; Popoff, M.R.; Stiles, B.G. Detergent-resistant membrane microdomains facilitate Ib oligomer formation and biological activity of Clostridium perfringens iota-toxin. Infect. Immun. 2004, 72, 2186–2193. [Google Scholar] [CrossRef]
- Gibert, M.; Marvaud, J.C.; Pereira, Y.; Hale, M.L.; Stiles, B.G.; Boquet, P.; Lamaze, C.; Popoff, M.R. Differential requirement for the translocation of clostridial binary toxins: Iota toxin requires a membrane potential gradient. FEBS Lett. 2007, 581, 1287–1296. [Google Scholar] [CrossRef]
- Gibert, M.; Monier, M.N.; Ruez, R.; Hale, M.L.; Stiles, B.G.; Benmerah, A.; Johannes, L.; Lamaze, C.; Popoff, M.R. Endocytosis and toxicity of clostridial binary toxins depend on a clathrin-independent pathway regulated by Rho-GDI. Cell. Microbiol. 2011, 13, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Kroll, C.; Ernst, K.; Schwan, C.; Popoff, M.; Fischer, G.; Buchner, J.; Aktories, K.; Barth, H. Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect. Immun. 2011, 79, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Liebscher, M.; Mathea, S.; Granzhan, A.; Schmid, J.; Popoff, M.R.; Ihmels, H.; Barth, H.; Schiene-Fischer, C. A novel Hsp70 inhibitor prevents cell intoxication with the actin ADP-ribosylating Clostridium perfringens iota toxin. Sci. Rep. 2016, 6, 20301. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Schmid, J.; Beck, M.; Hägele, M.; Hohwieler, M.; Hauff, P.; Ückert, A.K.; Anastasia, A.; Fauler, M.; Jank, T.; et al. Hsp70 facilitates trans-membrane transport of bacterial ADP-ribosylating toxins into the cytosol of mammalian cells. Sci. Rep. 2017, 7, 2724. [Google Scholar] [CrossRef] [PubMed]
- Ernst, K.; Sailer, J.; Braune, M.; Barth, H. Intoxication of mammalian cells with binary clostridial enterotoxins is inhibited by the combination of pharmacological chaperone inhibitors. Naunyn-Schmiedebergs Arch. Pharmacol. 2021, 394, 941–954. [Google Scholar] [CrossRef]
- Ernst, K. Requirement of Peptidyl-Prolyl Cis/Trans isomerases and chaperones for cellular uptake of bacterial AB-type toxins. Front. Cell. Infect. Microbiol. 2022, 12, 938015. [Google Scholar] [CrossRef]
- Wegner, A.; Aktories, K. ADP-ribosylated actin caps the barbed ends of actin filaments. J. Biol. Chem. 1988, 263, 13739–13742. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Papatheodorou, P.; Lang, A.E. Bidirectional attack on the actin cytoskeleton. Bacterial protein toxins causing polymerization or depolymerization of actin. Toxicon 2012, 60, 572–581. [Google Scholar] [CrossRef]
- Eckhardt, M.; Barth, H.; Blöcker, D.; Aktories, K. Binding of Clostridium botulinum C2 toxin to asparagine-linked complex and hybrid carbohydrates. J. Biol. Chem. 2000, 275, 2328–2334. [Google Scholar] [CrossRef]
- Barth, H.; Blocker, D.; Behlke, J.; Bergsma-Schutter, W.; Brisson, A.; Benz, R.; Aktories, K. Cellular uptake of Clostridium botulinum C2 toxin requires oligomerization and acidification. J. Biol. Chem. 2000, 275, 18704–18711. [Google Scholar] [CrossRef] [PubMed]
- Pust, S.; Barth, H.; Sandvig, K. Clostridium botulinum C2 toxin is internalized by clathrin- and Rho-dependent mechanisms. Cell. Microbiol. 2010, 12, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Barth, H. Exploring the role of host cell chaperones/PPIases during cellular up-take of bacterial ADP-ribosylating toxins as basis for novel pharmacological strategies to protect mammalian cells against these virulence factors. Naunyn-Schmiedebergs Arch. Pharmacol. 2011, 383, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Böhm, N.; Ernst, K.; Langer, S.; Schwan, C.; Aktories, K.; Popoff, M.; Fischer, G.; Barth, H. FK506-binding protein 51 interacts with Clostridium botulinum C2 toxin and FK506 inhibits membrane translocation of the toxin in mammalian cells. Cell. Microbiol. 2012, 14, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Bouvet, P. Clostridial toxins. Future Microbiol. 2009, 4, 1021–1064. [Google Scholar] [CrossRef]
- Richard, J.F.; Mainguy, G.; Gibert, M.; Marvaud, J.C.; Stiles, B.G.; Popoff, M.R. Transcytosis of iota-toxin across polarized Caco-2 cells. Mol. Microbiol. 2002, 43, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Umezaki, M.; Oda, M.; Kobayashi, K.; Tone, S.; Suda, T.; Ishidoh, K.; Sakurai, J. Clostridium perfringens iota-toxin b induces rapid cell necrosis. Infect. Immun. 2011, 79, 4353–4360. [Google Scholar] [CrossRef]
- Martínez-Meléndez, A.; Cruz-López, F.; Morfin-Otero, R.; Maldonado-Garza, H.J.; Garza-González, E. An update on Clostridioides difficile binary toxin. Toxins 2022, 14, 305. [Google Scholar] [CrossRef]
- Fischer, S.; Ückert, A.K.; Landenberger, M.; Papatheodorou, P.; Hoffmann-Richter, C.; Mittler, A.K.; Ziener, U.; Hägele, M.; Schwan, C.; Müller, M.; et al. Human peptide α-defensin-1 interferes with Clostridioides difficile toxins TcdA, TcdB, and CDT. FASEB J. 2020, 34, 6244–6261. [Google Scholar] [CrossRef]
- Ernst, K.; Landenberger, M.; Nieland, J.; Nørgaard, K.; Frick, M.; Fois, G.; Benz, R.; Barth, H. Characterization and pharmacological inhibition of the pore-forming Clostridioides difficile CDTb toxin. Toxins 2021, 13, 390. [Google Scholar] [CrossRef] [PubMed]
- Landenberger, M.; Nieland, J.; Roeder, M.; Norgaard, K.; Papatheodorou, P.; Ernst, K.; Barth, H. The cytotoxic effect of Clostridioides difficile pore-forming toxin CDTb. Biochim. Biophys. Acta 2021, 1863, 183603. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Benz, R.; Just, I.; Aktories, K. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J. Biol. Chem. 1994, 269, 16706–16711. [Google Scholar] [CrossRef] [PubMed]
- Bachmeyer, C.; Benz, R.; Barth, H.; Aktories, K.; Gilbert, M.; Popoff, M.R. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: Inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J. 2001, 15, 1658–1660. [Google Scholar] [CrossRef] [PubMed]
- Blöcker, D.; Bachmeyer, C.; Benz, R.; Aktories, K.; Barth, H. Channel formation by the binding component of Clostridium botulinum C2 toxin: Glutamate 307 of C2II affects channel properties in vitro and pH-dependent C2I translocation in vivo. Biochemistry 2003, 42, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Eisele, J.; Schreiner, S.; Borho, J.; Fischer, S.; Heber, S.; Endres, S.; Fellermann, M.; Wohlgemuth, L.; Huber-Lang, M.; Fois, G.; et al. The pore-forming subunit C2IIa of the binary Clostridium botulinum C2 Toxin reduces the chemotactic translocation of human polymorphonuclear leukocytes. Front. Pharmacol. 2022, 13, 810611. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Kobayashi, K. Lethal and dermonecrotic activities of Clostridium perfringens iota toxin: Biological activities induced by cooperation of two nonlinked components. Microbiol. Immunol. 1995, 39, 249–253. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagahama, M.; Takehara, M.; Seike, S.; Sakaguchi, Y. Cellular Uptake and Cytotoxicity of Clostridium perfringens Iota-Toxin. Toxins 2023, 15, 695. https://doi.org/10.3390/toxins15120695
Nagahama M, Takehara M, Seike S, Sakaguchi Y. Cellular Uptake and Cytotoxicity of Clostridium perfringens Iota-Toxin. Toxins. 2023; 15(12):695. https://doi.org/10.3390/toxins15120695
Chicago/Turabian StyleNagahama, Masahiro, Masaya Takehara, Soshi Seike, and Yoshihiko Sakaguchi. 2023. "Cellular Uptake and Cytotoxicity of Clostridium perfringens Iota-Toxin" Toxins 15, no. 12: 695. https://doi.org/10.3390/toxins15120695
APA StyleNagahama, M., Takehara, M., Seike, S., & Sakaguchi, Y. (2023). Cellular Uptake and Cytotoxicity of Clostridium perfringens Iota-Toxin. Toxins, 15(12), 695. https://doi.org/10.3390/toxins15120695