In Silico Evaluation of Sesquiterpenes and Benzoxazinoids Phytotoxins against Mpro, RNA Replicase and Spike Protein of SARS-CoV-2 by Molecular Dynamics. Inspired by Nature

 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Docking Studies
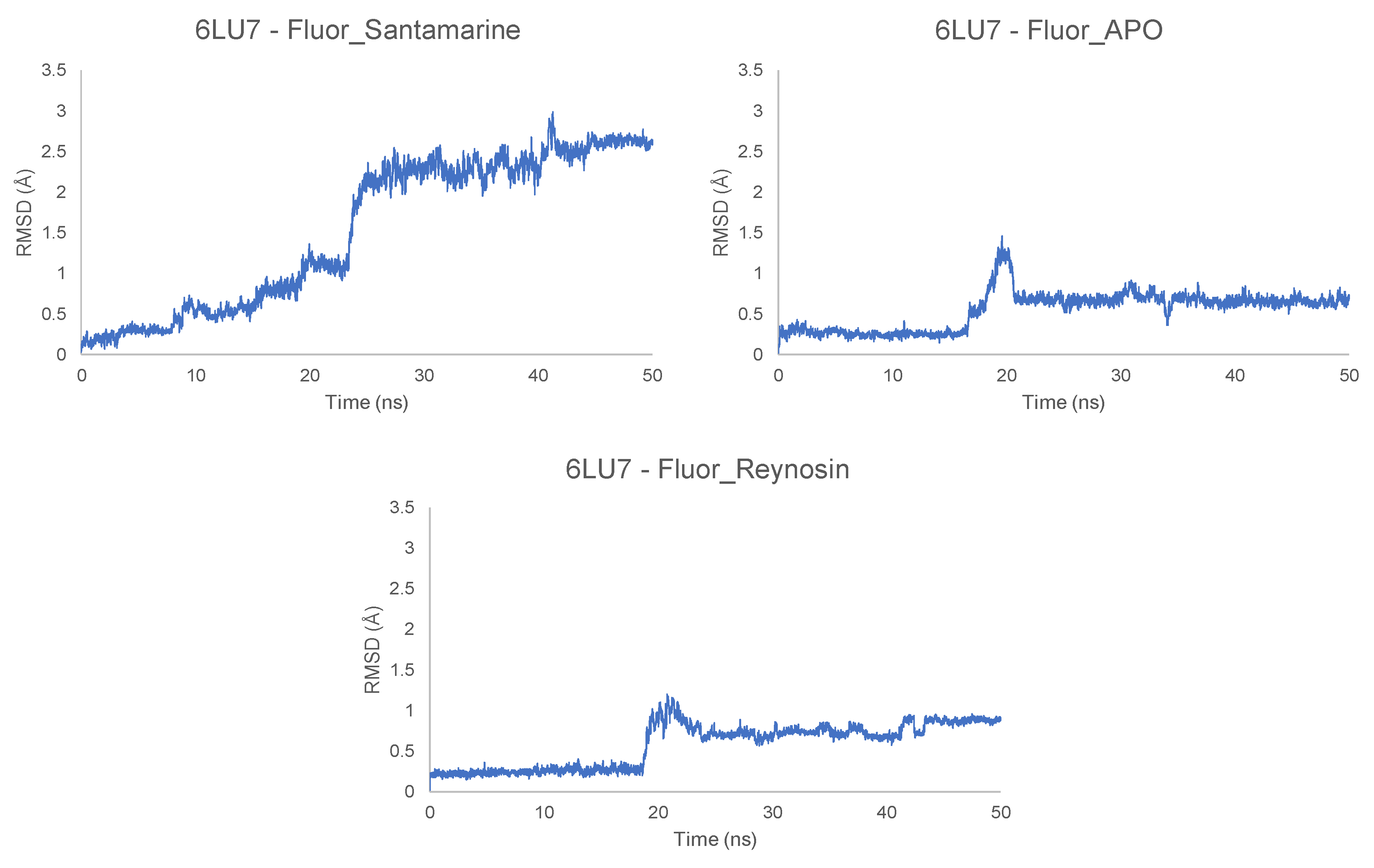
2.2. Molecular Dynamics Simulations
3. Conclusions
4. Materials and Methods
4.1. Molecular Docking Studies
4.2. Molecular Dynamics Simulation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Glass, W.G.; Subbarao, K.; Murphy, B.; Murphy, P.M. Mechanisms of Host Defense Following Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) Pulmonary Infection of Mice. J. Immunol. 2004, 173, 4030–4039. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Islam, M.S.; Wang, J.; Li, Y.; Chen, X. Traditional Chinese Medicine in the Treatment of Patients Infected with 2019-New Coronavirus (SARS-CoV-2): A Review and Perspective. Int. J. Biol. Sci. 2020, 16, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Farmanpour-Kalalagh, K.; Beyraghdar Kashkooli, A.; Babaei, A.; Rezaei, A.; van der Krol, A.R. Artemisinins in Combating Viral Infections Like SARS-CoV-2, Inflammation and Cancers and Options to Meet Increased Global Demand. Front. Plant Sci. 2022, 13, 780257. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.S.; Huang, Y.; Fidock, D.A.; Polyak, S.J.; Wagoner, J.; Towler, M.J.; Weathers, P.J. Artemisia Annua, L. Extracts Inhibit the in Vitro Replication of SARS-CoV-2 and Two of Its Variants. J. Ethnopharmacol. 2021, 274, 114016. [Google Scholar] [CrossRef]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential Inhibitors of Coronavirus 3-Chymotrypsin-like Protease (3CL pro ): An in Silico Screening of Alkaloids and Terpenoids from African Medicinal Plants. J. Biomol. Struct. Dyn. 2020, 39, 3396–3408. [Google Scholar] [CrossRef]
- Forrestall, K.L.; Burley, D.E.; Cash, M.K.; Pottie, I.R.; Darvesh, S. 2-Pyridone Natural Products as Inhibitors of SARS-CoV-2 Main Protease. Chem. Biol. Interact. 2021, 335, 109348. [Google Scholar] [CrossRef]
- Narkhede, R.R.; Pise, A.v.; Cheke, R.S.; Shinde, S.D. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect 2020, 10, 297–306. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Protein Data Bank. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3. Available online: https://www.rcsb.org/structure/6LU7 (accessed on 23 August 2022). [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Protein Data Bank. Crystal Structure of SARS-CoV-2 Spike Receptor-Binding Domain Bound with ACE2. Available online: https://www.rcsb.org/structure/6M0J (accessed on 23 August 2022). [CrossRef]
- Litter, D.; Gully, B.; Colson, R.; Rossjohn, J. Crystal Structure of the SARS-CoV-2 Non-structural Protein 9, Nsp9. iScience 2020, 23, 101258–101265. [Google Scholar] [CrossRef]
- Protein Data Bank. The Crystal Structure of Nsp9 RNA Binding Protein of SARS CoV-2. Available online: https://www.rcsb.org/structure/6W4B (accessed on 23 August 2022). [CrossRef]
- Moriya, S.; Miyazawa, K.; Kawaguchi, T.; Che, X.F.; Tomoda, A. Involvement of Endoplasmic Reticulum Stress-Mediated CHOP (GADD153) Induction in the Cytotoxicity of 2-Aminophenoxazine-3-One in Cancer Cells. Int. J. Oncol. 2011, 39, 981–988. [Google Scholar] [CrossRef] [PubMed]
- babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G.G. Application of Sesquiterpene Lactone: A New Promising Way for Cancer Therapy Based on Anticancer Activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Russo, A.; Perri, M.; Cione, E.; di Gioia, M.L.; Nardi, M.; Cristina Caroleo, M. Biochemical and Chemical Characterization of Cynara Cardunculus, L. Extract and Its Potential Use as Co-Adjuvant Therapy of Chronic Myeloid Leukemia. J. Ethnopharmacol. 2017, 202, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.-R.; Wu, Y.-S.; Chang, C.-W.; Lien, T.-W.; Chen, W.-C.; Tan, U.-K.; Hsu, J.T.A.; Hsieh, H.-P. Synthesis and Anti-Viral Activity of a Series of Sesquiterpene Lactones and Analogues in the Subgenomic HCV Replicon System. Bioorg. Med. Chem. 2006, 14, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Elsebai, M.F.; Koutsoudakis, G.; Saludes, V.; Pérez-Vilaró, G.; Turpeinen, A.; Mattila, S.; Pirttilä, A.M.; Fontaine-Vive, F.; Mehiri, M.; Meyerhans, A.; et al. Pan-Genotypic Hepatitis C Virus Inhibition by Natural Products Derived from the Wild Egyptian Artichoke. J. Virol. 2016, 90, 1918–1930. [Google Scholar] [CrossRef]
- Hayashi, K.; Hayashi, T.; Tomoda, A. Phenoxazine Derivatives Inactivate Human Cytomegalovirus, Herpes Simplex Virus-1, and Herpes Simplex Virus-2 In Vitro. J. Pharmacol. Sci. 2008, 106, 369–375. [Google Scholar] [CrossRef]
- Hayashi, K.; Hayashi, T.; Miyazawa, K.; Tomoda, A. Phenoxazine Derivatives Suppress the Infections Caused by Herpes Simplex Virus Type-1 and Herpes Simplex Virus Type-2 Intravaginally Inoculated Into Mice. J. Pharmacol. Sci. 2010, 114, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Touret, F.; de Lamballerie, X. Of Chloroquine and COVID-19. Antivir. Res. 2020, 177, 104762. [Google Scholar] [CrossRef]
- Colson, P.; Rolain, J.M.; Raoult, D. Chloroquine for the 2019 Novel Coronavirus SARS-CoV-2. Int. J. Antimicrob. Agents 2020, 55, 105923. [Google Scholar] [CrossRef]
- Echeverría-Esnal, D.; Martin-Ontiyuelo, C.; Navarrete-Rouco, M.E.; De-Antonio Cuscó, M.; Ferrández, O.; Horcajada, J.P.; Grau, S. Azithromycin in the Treatment of COVID-19: A Review. Expert Rev. Anti. Infect. Ther. 2021, 19, 147–163. [Google Scholar] [CrossRef]
- Cala, A.; Zorrilla, J.G.; Rial, C.; Molinillo, J.M.G.; Varela, R.M.; MacÍas, F.A. Easy Access to Alkoxy, Amino, Carbamoyl, Hydroxy, and Thiol Derivatives of Sesquiterpene Lactones and Evaluation of Their Bioactivity on Parasitic Weeds. J. Agric. Food Chem. 2019, 67, 10764–10773. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef]
- Lu, I.-L.; Mahindroo, N.; Liang, P.-H.; Peng, Y.-H.; Kuo, C.-J.; Tsai, K.-C.; Hsieh, H.-P.; Chao, Y.-S.; Wu, S.-Y. Structure-Based Drug Design and Structural Biology Study of Novel Nonpeptide Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Main Protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef]
- Xu, Z.; Peng, C.; Shi, Y.; Zhu, Z.; Mu, K.; Wang, X.; Zhu, W. Nelfinavir Was Predicted to Be a Potential Inhibitor of 2019-NCov Main Protease by an Integrative Approach Combining Homology Modelling, Molecular Docking and Binding Free Energy Calculation. BioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a Less Toxic Derivative of Chloroquine, Is Effective in Inhibiting SARS-CoV-2 Infection in Vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine Is a Potent Inhibitor of SARS Coronavirus Infection and Spread. Virol. J. 2005, 2, 1–10. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Rial, C.; Novaes, P.; Varela, R.M.; Molinillo, J.M.G.; Macias, F.A. Phytotoxicity of Cardoon (Cynara Cardunculus) Allelochemicals on Standard Target Species and Weeds. J. Agric. Food Chem. 2014, 62, 6699–6706. [Google Scholar] [CrossRef]
- Schulz, M.; Marocco, A.; Tabaglio, V.; Macias, F.A.; Molinillo, J.M.G. Benzoxazinoids in Rye Allelopathy—From Discovery to Application in Sustainable Weed Control and Organic Farming. J. Chem. Ecol. 2013, 39, 154–174. [Google Scholar] [CrossRef]
- Zhang, M.-Q.; Wilkinson, B. Drug Discovery beyond the ‘Rule-of-Five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME Evaluation in Drug Discovery. 7. Prediction of Oral Absorption by Correlation and Classification. J. Chem. Inf. Model. 2007, 47, 208–218. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute—USA Chemical Indetifier Resolver. Available online: https://cactus.nci.nih.gov/chemical/structure (accessed on 23 August 2022).
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ΔG (Kcal/mol) | ||
|---|---|---|---|
| Main Protease | RNA Replicase | Spike Protein | |
| Azithromycin | −1.20 ± 0.47 | −0.76 ± 0.88 | −4.64 ± 0.78 |
| Hydroxychloroquine | −3.45 ± 0.16 | −2.67 ± 0.81 | −4.29 ± 0.76 |
| Favipiravir | −3.21 ± 0.16 | −3.58 ± 0.24 | −3.93 ± 0.42 |
| Artemisinin | −6.25 ± 0.23 | −6.07 ± 0.07 | −5.96 ± 0.19 |
| Cynaropicrin | −3.49 ± 0.07 | −4.02 ± 0.25 | −4.19 ± 0.28 |
| Met-4F-Benzo | −5.32 ± 0.64 | −5.71 ± 0.93 | −5.81 ± 0.48 |
| Fluor-Cynaro | −3.97 ± 1.01 | −5.56 ± 0.73 | −8.13 ± 1.08 |
| Costunolide | −6.11 ± 0.41 | −5.77 ± 0.20 | −6.15 ± 0.20 |
| DHC | −6.08 ± 0.30 | −5.57 ± 0.34 | −6.00 ± 0.28 |
| Reynosin | −5.54 ± 0.48 | −5.92 ± 0.41 | −6.11 ± 0.18 |
| Santamarine | −5.81 ± 0.59 | −5.97 ± 0.48 | −6.12 ± 0.31 |
| Fluor-Reynosin | −7.37 ± 0.50 | −7.10 ± 0.93 | −7.89 ± 0.77 |
| Fluor-Santamarine | −7.77 ± 0.77 | −6.35 ± 0.54 | −7.68 ± 0.74 |
| Alanto | −5.82 ± 0.22 | −6.57 ± 0.26 | −6.46 ± 0.17 |
| Alpha-Cyclo | −5.99 ± 0.37 | −5.95 ± 0.36 | −6.20 ± 0.25 |
| Beta-Cyclo | −5.98 ± 0.49 | −6.02 ± 0.52 | −6.20 ± 0.29 |
| 3-DeBra | −6.36 ± 0.37 | −6.04 ± 0.37 | −6.19 ± 0.25 |
| Compounds | ΔG (Kcal/mol) | ||
|---|---|---|---|
| Main Protease | RNA Replicase | Spike Protein | |
| Azithromycin | −1.20 ± 0.47 | −0.76 ± 0.88 | −4.64 ± 0.78 |
| Hydroxychloroquine | −3.45 ± 0.16 | −2.67 ± 0.81 | −4.29 ± 0.76 |
| Favipiravir | −3.21 ± 0.16 | −3.58 ± 0.24 | −3.93 ± 0.42 |
| Artemisinin | −6.25 ± 0.23 | −6.07 ± 0.07 | −5.96 ± 0.19 |
| Met-4F-Benzo | −3.49 ± 0.08 | −4.02 ± 0.25 | −4.19 ± 0.28 |
| APO | −5.13 ± 0.31 | −5.93 ± 0.66 | −5.52 ± 0.24 |
| DisOH | −4.84 ± 0.69 | −4.74 ± 0.84 | −4.66 ± 0.59 |
| DisNH2 | −4.48 ± 0.22 | −4.68 ± 0.44 | −4.88 ± 0.38 |
| Fluor-APO | −6.01 ± 0.53 | −6.08 ± 0.41 | −7.79 ± 0.88 |
| Fluor-DisOH | −5.71 ± 1.36 | −4.67 ± 0.69 | −5.01 ± 0.86 |
| Fluor-DisNH | −4.45 ± 1.31 | −5.77 ± 0.55 | −5.91 ± 0.93 |
| DIBOAa | −4.05 ± 0.28 | −4.05 ± 0.34 | −4.94 ± 0.33 |
| DIBOAb | −3.90 ± 0.17 | −4.50 ± 0.49 | −4.33 ± 0.28 |
| DIMBOAa | −3.93 ± 0.17 | −4.03 ± 0.38 | −4.61 ± 0.30 |
| DIMBOAb | −3.91 ± 0.13 | −3.71 ± 0.47 | −4.53 ± 0.36 |
| DDIBOA | −4.12 ± 0.17 | −4.19 ± 0.19 | −4.41 ± 0.27 |
| 6Cl-DDIBOA | −4.42 ± 0.11 | −4.28 ± 0.24 | −4.77 ± 0.21 |
| 6F-DDIBOA | −4.07 ± 0.28 | −4.30 ± 0.36 | −4.28 ± 0.19 |
| 6F-DDIBOA | −4.46 ± 0.22 | −4.57 ± 0.38 | −4.57 ± 0.18 |
| (A). Lipinski’s Rules for standard compounds tested. | ||||
| No | Standard | Molecular Formula | Lipinski’s rule of 5 | |
| Properties | Value | |||
| 1 | 6-fluoropyrazine-2-carboxamide (Favipiravir) | C5H4FN3O | M.W. (≤500 amu) | 141.11 |
| cLog P (≤5) | –0.50873 | |||
| H-bond donors (≤5) | 1 | |||
| H-bond acceptors (≤10) | 5 | |||
| Violations | 0 | |||
| 2 | Hydroxychloroquine | C18H26ClN3O | M.W. (≤500 amu) | 335.88 |
| cLog P (≤5) | 4.11588 | |||
| H-bond donors (≤5) | 2 | |||
| H-bond acceptors (≤10) | 4 | |||
| Violations | 0 | |||
| 3 | Artemisinin | C15H22O5 | M.W. (≤500 amu) | 282.34 |
| cLog P (≤5) | 2.71630 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 5 | |||
| Violations | 0 | |||
| 4 | Azithromycin | C38H72N2O12 | M.W. (≤500 amu) | 749.00 |
| cLog P (≤5) | 2.63825 | |||
| H-bond donors (≤5) | 5 | |||
| H-bond acceptors (≤10) | 14 | |||
| Violations | 2 | |||
| (B). Lipinski’s Rules for the most relevant sesquiterpenoid compounds tested. | ||||
| No | Sesquiterpenoids | Molecular Formula | Lipinski’s rule of 5 | |
| Properties | Value | |||
| 1 | Cynaropicrin | C19H22O6 | M.W. (≤500 amu) | 346.38 |
| cLog P (≤5) | 0.045825 | |||
| H-bond donors (≤5) | 2 | |||
| H-bond acceptors (≤10) | 6 | |||
| Violations | 0 | |||
| 2 | 3,3’-di(4’-fluorobenzoyloxy)cynaropicrin (Fluor-Cynaro) | C33H28F2O8 | M.W. (≤500 amu) | 590.58 |
| cLog P (≤5) | 5.82662 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 10 | |||
| Violations | 2 | |||
| 3 | Costunolide | C15H20O2 | M.W. (≤500 amu) | 232.32 |
| cLog P (≤5) | 3.79 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 4 | Dehydrocostuslactone (DHC) | C15H18O2 | M.W. (≤500 amu) | 230.31 |
| cLog P (≤5) | 2.786 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 5 | Reynosin | C15H20O3 | M.W. (≤500 amu) | 248.32 |
| cLog P (≤5) | 1.183 | |||
| H-bond donors (≤5) | 1 | |||
| H-bond acceptors (≤10) | 3 | |||
| Violations | 0 | |||
| 6 | 1-(4’-fluorobenzoyloxy)reynosin (Fluor-Reynosin) | C22H23FO4 | M.W. (≤500 amu) | 370.42 |
| cLog P (≤5) | 4.201 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 5 | |||
| Violations | 0 | |||
| 7 | Santamarine | C15H20O3 | M.W. (≤500 amu) | 248.32 |
| cLog P (≤5) | 1.183 | |||
| H-bond donors (≤5) | 1 | |||
| H-bond acceptors (≤10) | 3 | |||
| Violations | 0 | |||
| 8 | 1-(4-fluorobenzoyloxy)santamarine (Fluor-Santamarine) | C22H23FO4 | M.W. (≤500 amu) | 370.42 |
| cLog P (≤5) | 4.201 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 5 | |||
| Violations | 0 | |||
| 9 | Alantolactone (Alanto) | C15H20O2 | M.W. (≤500 amu) | 232.32 |
| cLog P (≤5) | 3.27 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 10 | β-cyclocostunolide (Beta-Cyclo) | C15H20O2 | M.W. (≤500 amu) | 232.32 |
| cLog P (≤5) | 3.27 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 11 | α-cyclocostunolide (Alpha-Cyclo) | C15H20O2 | M.W. (≤500 amu) | 232.32 |
| cLog P (≤5) | 3.27 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 12 | 3-deoxybrachylaenolide (3-DeBra) | C15H16O3 | M.W. (≤500 amu) | 244.29 |
| cLog P (≤5) | 1.024 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 3 | |||
| Violations | 0 | |||
| (C). Lipinski’s Rules for the most relevant aminophenoxazinoids tested. | ||||
| No | Benzoxazinoids | Molecular Formula | Lipinski’s rule of 5 | |
| Properties | Value | |||
| 13 | 2-amino-3H-phenoxazin-3-one (APO) | C12H8N2O2 | M.W. (≤500 amu) | 212.21 |
| cLog P (≤5) | 1.13575 | |||
| H-bond donors (≤5) | 1 | |||
| H-bond acceptors (≤10) | 4 | |||
| Violations | 0 | |||
| 14 | 4-fluoro-N-(3-oxo-3H-phenoxazin-2-yl)benzamide (Fluor-APO) | C19H11FN2O3 | M.W. (≤500 amu) | 334.31 |
| cLog P (≤5) | 2.97045 | |||
| H-bond donors (≤5) | 1 | |||
| H-bond acceptors (≤10) | 6 | |||
| Violations | 0 | |||
| 15 | 2,2′-disulfanediyldiphenol (DisOH) | C12H10O2S2 | M.W. (≤500 amu) | 250.33 |
| cLog P (≤5) | 3.0194 | |||
| H-bond donors (≤5) | 2 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 16 | disulfanediylbis(2,1-phenylene) bis(4-fluorobenzoate) (Fluor-DisOH) | C26H16F2O4S2 | M.W. (≤500 amu) | 494.53 |
| cLog P (≤5) | 7.2229 | |||
| H-bond donors (≤5) | 0 | |||
| H-bond acceptors (≤10) | 6 | |||
| Violations | 1 | |||
| 17 | 2,2′-dithiodianiline (DisNH2) | C12H12N2S2 | M.W. (≤500 amu) | 248.36 |
| cLog P (≤5) | 2.736 | |||
| H-bond donors (≤5) | 2 | |||
| H-bond acceptors (≤10) | 2 | |||
| Violations | 0 | |||
| 18 | N,N′-(disulfanediylbis(2,1-phenylene))bis(4-fluorobenzamide) (Fluor-DisNH) | C26H18F2N2O2S2 | M.W. (≤500 amu) | 492.56 |
| cLog P (≤5) | 5.06192 | |||
| H-bond donors (≤5) | 2 | |||
| H-bond acceptors (≤10) | 4 | |||
| Violations | 1 | |||
| Protein–LIG Energy (kJ/mol) Lennard–Jones | ||||||||
| 6M0J | 6LU7 | 6W4B | ||||||
| Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin |
| −8.846 ± 4.279 | −123.866 ± 3.611 | −113.915 ± 2.960 | −105.696 ± 2.446 | −68.819 ± 3.583 | −126.618 ± 2.007 | −54.026 ± 2.800 | −118.928 ± 1.982 | −125.207 ± 10.865 |
| Protein–LIG Total Number of H-Bonds along 50 ns | ||||||||
| 6M0J | 6LU7 | 6W4B | ||||||
| Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin |
| 355 | 1267 | 1674 | 3600 | 2490 | 3708 | 1293 | 3571 | 2385 |
| Protein–LIG Average Number of H-Bonds per ns | ||||||||
| 6M0J | 6LU7 | 6W4B | ||||||
| Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin |
| 0.07 | 0.25 | 0.33 | 0.72 | 0.50 | 0.74 | 0.26 | 0.91 | 0.48 |
| Protein–LIG Average Distance of H-Bonds (nm) | ||||||||
| 6M0J | 6LU7 | 6W4B | ||||||
| Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin |
| 0.2925 | 0.3075 | 0.2825 | 0.2875 | 0.2975 | 0.3125 | 0.3275 | 0.2875 | 0.2825 |
| Protein–LIG Lifetime of H-Bonds (ps) | ||||||||
| 6M0J | 6LU7 | 6W4B | ||||||
| Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin | Fluor-APO | Fluor-Santamarine | Fluor-Reynosin |
| 19.63 | 18.08 | 14.03 | 65.95 | 24.34 | 19.81 | 27.57 | 74.94 | 75.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejías, F.J.R.; Durán, A.G.; Chinchilla, N.; Varela, R.M.; Álvarez, J.A.; Molinillo, J.M.G.; García-Cozar, F.; Macías, F.A. In Silico Evaluation of Sesquiterpenes and Benzoxazinoids Phytotoxins against Mpro, RNA Replicase and Spike Protein of SARS-CoV-2 by Molecular Dynamics. Inspired by Nature. Toxins 2022, 14, 599. https://doi.org/10.3390/toxins14090599
Mejías FJR, Durán AG, Chinchilla N, Varela RM, Álvarez JA, Molinillo JMG, García-Cozar F, Macías FA. In Silico Evaluation of Sesquiterpenes and Benzoxazinoids Phytotoxins against Mpro, RNA Replicase and Spike Protein of SARS-CoV-2 by Molecular Dynamics. Inspired by Nature. Toxins. 2022; 14(9):599. https://doi.org/10.3390/toxins14090599
Chicago/Turabian StyleMejías, Francisco J. R., Alexandra G. Durán, Nuria Chinchilla, Rosa M. Varela, José A. Álvarez, José M. G. Molinillo, Francisco García-Cozar, and Francisco A. Macías. 2022. "In Silico Evaluation of Sesquiterpenes and Benzoxazinoids Phytotoxins against Mpro, RNA Replicase and Spike Protein of SARS-CoV-2 by Molecular Dynamics. Inspired by Nature" Toxins 14, no. 9: 599. https://doi.org/10.3390/toxins14090599
APA StyleMejías, F. J. R., Durán, A. G., Chinchilla, N., Varela, R. M., Álvarez, J. A., Molinillo, J. M. G., García-Cozar, F., & Macías, F. A. (2022). In Silico Evaluation of Sesquiterpenes and Benzoxazinoids Phytotoxins against Mpro, RNA Replicase and Spike Protein of SARS-CoV-2 by Molecular Dynamics. Inspired by Nature. Toxins, 14(9), 599. https://doi.org/10.3390/toxins14090599