Exploring the Role of Staphylococcus aureus in Inflammatory Diseases

 ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Virulence Factors of S. aureus
2.1. Secreted Virulence Factors
2.1.1. PFTs
- Hla, Hlb, and Hlg
- 2.
- α-Toxin
- 3.
- PVL
2.1.2. PSMs
- PSMα
- 2.
- PSMβ
- 3.
- PSMγ (δ Toxin)
2.1.3. Proteases
- ETs
- 2.
- Serine Protease-Like Proteins (Spls)
- 3.
- Staphopain B (SspB)
2.1.4. SAgs
- SEs
- 2.
- TSST-1
2.1.5. Secreted Enzymes (Exoenzymes) and Effectors
- EsxA and EsxB
- 2.
- Coa
- 3.
- Nuc and AdsA
- 4.
- Extracellular Adhesion Protein (Eap)
2.2. Surface Proteins of S. aureus (Cell Wall-Anchored (CWA) Proteins)
2.2.1. MSCRAMM
2.2.2. Staphylococcal Protein A (SpA)
2.3. PAMPs
2.3.1. Triacyl Lipopetides and Diacyl Lipoproteins
2.3.2. LTA
2.3.3. PGN
3. Different Inflammatory Cell Types Involved in S. aureus Pathogenesis
3.1. Keratinocytes/Epithelial Cells
3.2. Helper T Cells (Th Cells)
3.2.1. T Helper 1 (Th1) Cells
3.2.2. Th2 Cells
3.2.3. Th17 Cells
3.2.4. Tregs
3.2.5. Tfh Cells
3.2.6. Th9 Cells
3.3. ILCs
3.4. Macrophages
3.5. DCs
3.6. Mast Cells
3.7. Neutrophils
3.8. Eosinophils (Eos)
3.9. Basophils
3.10. B Cells
4. Types of Host Cell Death Caused by S. aureus
4.1. Pyroptosis
4.1.1. Activation of Inflammasomes Caused by S. aureus
4.1.2. Pyroptosis
4.2. Necroptosis
4.3. Apoptosis
4.4. Autophagy
4.5. Ferroptosis and Cuprotosis
5. Basic Types of Diseases Caused by S. aureus and Virulence Mechanisms
5.1. Skin Disease
5.1.1. AD
5.1.2. Psoriasis
5.2. Respiratory Diseases
5.2.1. Asthma
5.2.2. Chronic Rhinosinusitis
5.2.3. Pneumonia
5.2.4. CF
5.3. Food Poisoning
5.4. Autoimmune Diseases
5.5. Osteomyelitis
5.6. MRSA
5.7. DFI
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Shopsin, B.; Kreiswirth, B.N. Molecular epidemiology of methicillin-resistant Staphylococcus aureus. Emerg. Infect. Dis. 2001, 7, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol. 2015, 6, 1174. [Google Scholar] [CrossRef] [PubMed]
- Recsei, P.; Kreiswirth, B.; O’Reilly, M.; Schlievert, P.; Gruss, A.; Novick, R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol. Gen. Genet. 1986, 202, 58–61. [Google Scholar] [CrossRef]
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe 2017, 22, 667–677.e5. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.; Peters, A.T. Atopic dermatitis. Allergy Asthma Proc. 2019, 40, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Fujieda, S.; Imoto, Y.; Kato, Y.; Ninomiya, T.; Tokunaga, T.; Tsutsumiuchi, T.; Yoshida, K.; Kidoguchi, M.; Takabayashi, T. Eosinophilic chronic rhinosinusitis. Allergol. Int. 2019, 68, 403–412. [Google Scholar] [CrossRef]
- Pidwill, G.R.; Gibson, J.F.; Cole, J.; Renshaw, S.A.; Foster, S.J. The Role of Macrophages in Staphylococcus aureus Infection. Front. Immunol. 2020, 11, 620339. [Google Scholar] [CrossRef]
- Tatsuno, K.; Fujiyama, T.; Yamaguchi, H.; Waki, M.; Tokura, Y. TSLP Directly Interacts with Skin-Homing Th2 Cells Highly Expressing its Receptor to Enhance IL-4 Production in Atopic Dermatitis. J. Investig. Dermatol. 2015, 135, 3017–3024. [Google Scholar] [CrossRef]
- Geoghegan, J.A.; Irvine, A.D.; Foster, T.J. Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol. 2018, 26, 484–497. [Google Scholar] [CrossRef]
- Marchitto, M.C.; Dillen, C.A.; Liu, H.; Miller, R.J.; Archer, N.K.; Ortines, R.V.; Alphonse, M.P.; Marusina, A.I.; Merleev, A.A.; Wang, Y.; et al. Clonal Vγ6(+)Vδ4(+) T cells promote IL-17-mediated immunity against Staphylococcus aureus skin infection. Proc. Natl. Acad. Sci. USA 2019, 116, 10917–10926. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.U.; Bachert, C. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860.e6. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Huang, Q.; Liang, J.; Wang, J.; Zhang, J.; Yang, Y.; Ye, Q.; He, S.; Li, J.; Wu, Z.; et al. A review of the mechanisms of keratinocytes damage caused by Staphylococcus aureus infection in patients with atopic dermatitis. J. Leukoc. Biol. 2021, 110, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef]
- Ionin, B.; Hammamieh, R.; Shupp, J.W.; Das, R.; Pontzer, C.H.; Jett, M. Staphylococcal enterotoxin B causes differential expression of Rnd3 and RhoA in renal proximal tubule epithelial cells while inducing actin stress fiber assembly and apoptosis. Microb. Pathog. 2008, 45, 303–309. [Google Scholar] [CrossRef]
- Soe, Y.M.; Bedoui, S.; Stinear, T.P.; Hachani, A. Intracellular Staphylococcus aureus and host cell death pathways. Cell Microbiol. 2021, 23, e13317. [Google Scholar] [CrossRef]
- Arroyo, D.S.; Soria, J.A.; Gaviglio, E.A.; Garcia-Keller, C.; Cancela, L.M.; Rodriguez-Galan, M.C.; Wang, J.M.; Iribarren, P. Toll-like receptor 2 ligands promote microglial cell death by inducing autophagy. FASEB J. 2013, 27, 299–312. [Google Scholar] [CrossRef]
- Mempel, M.; Voelcker, V.; Köllisch, G.; Plank, C.; Rad, R.; Gerhard, M.; Schnopp, C.; Fraunberger, P.; Walli, A.K.; Ring, J.; et al. Toll-like receptor expression in human keratinocytes: Nuclear factor kappaB controlled gene activation by Staphylococcus aureus is toll-like receptor 2 but not toll-like receptor 4 or platelet activating factor receptor dependent. J. Investig. Dermatol. 2003, 121, 1389–1396. [Google Scholar] [CrossRef]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef]
- Rowe, S.E.; Wagner, N.J.; Li, L.; Beam, J.E.; Wilkinson, A.D.; Radlinski, L.C.; Zhang, Q.; Miao, E.A.; Conlon, B.P. Reactive oxygen species induce antibiotic tolerance during systemic Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 282–290. [Google Scholar] [CrossRef]
- Kumar, S.; Colpitts, S.L.; Ménoret, A.; Budelsky, A.L.; Lefrancois, L.; Vella, A.T. Rapid αβ T-cell responses orchestrate innate immunity in response to Staphylococcal enterotoxin A. Mucosal Immunol. 2013, 6, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Hattar, K.; Grandel, U.; Moeller, A.; Fink, L.; Iglhaut, J.; Hartung, T.; Morath, S.; Seeger, W.; Grimminger, F.; Sibelius, U. Lipoteichoic acid (LTA) from Staphylococcus aureus stimulates human neutrophil cytokine release by a CD14-dependent, Toll-like-receptor-independent mechanism: Autocrine role of tumor necrosis factor-[alpha] in mediating LTA-induced interleukin-8 generation. Crit. Care Med. 2006, 34, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yan, M.; Sugui, J.A.; Li, H.; Xu, C.; Joo, J.; Kwon-Chung, K.J.; Coleman, W.G.; Rodgers, G.P. Olfm4 deletion enhances defense against Staphylococcus aureus in chronic granulomatous disease. J. Clin. Investig. 2013, 123, 3751–3755. [Google Scholar] [CrossRef] [PubMed]
- Widaa, A.; Claro, T.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS ONE 2012, 7, e40586. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 2. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef]
- Askarian, F.; Wagner, T.; Johannessen, M.; Nizet, V. Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol. Rev. 2018, 42, 656–671. [Google Scholar] [CrossRef]
- Verma, P.; Gandhi, S.; Lata, K.; Chattopadhyay, K. Pore-forming toxins in infection and immunity. Biochem. Soc. Trans. 2021, 49, 455–465. [Google Scholar] [CrossRef]
- Otto, M. Phenol-soluble modulins. Int. J. Med. Microbiol. 2014, 304, 164–169. [Google Scholar] [CrossRef]
- Yagi, S.; Wakaki, N.; Ikeda, N.; Takagi, Y.; Uchida, H.; Kato, Y.; Minamino, M. Presence of staphylococcal exfoliative toxin A in sera of patients with atopic dermatitis. Clin. Exp. Allergy 2004, 34, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.; Harshman, S. Studies on the binding of staphylococcal 125I-labeled alpha-toxin to rabbit erythrocytes. Biochemistry 1976, 15, 2348–2355. [Google Scholar] [CrossRef]
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus Alpha-Toxin Disrupts Endothelial-Cell Tight Junctions via Acid Sphingomyelinase and Ceramide. Infect. Immun. 2018, 86, e00606-17. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.S.; Núñez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, M.E.; O’Brien, E.C.; O’Keeffe, K.M.; Vozza, E.G.; Leddy, N.; McLoughlin, R.M. Manipulation of Autophagy and Apoptosis Facilitates Intracellular Survival of Staphylococcus aureus in Human Neutrophils. Front. Immunol. 2020, 11, 565545. [Google Scholar] [CrossRef]
- Adler, A.; Temper, V.; Block, C.S.; Abramson, N.; Moses, A.E. Panton-Valentine leukocidin-producing Staphylococcus aureus. Emerg. Infect. Dis. 2006, 12, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, G.M. The hemolysins of Staphylococcus aureus. Bacteriol. Rev. 1975, 39, 317–344. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef]
- Lizak, M.; Yarovinsky, T.O. Phospholipid scramblase 1 mediates type i interferon-induced protection against staphylococcal α-toxin. Cell Host Microbe 2012, 11, 70–80. [Google Scholar] [CrossRef]
- Bhakdi, S.; Muhly, M.; Korom, S.; Hugo, F. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect. Immun. 1989, 57, 3512–3519. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, N.; Fuhrmann, M.; Tannert-Otto, S.; Grimminger, F.; Bhadki, S. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J. Exp. Med. 1993, 178, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef]
- Suttorp, N.; Seeger, W.; Dewein, E.; Bhakdi, S.; Roka, L. Staphylococcal alpha-toxin-induced PGI2 production in endothelial cells: Role of calcium. Am. J. Physiol. 1985, 248, C127–C134. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Rose, F.; Sibelius, U.; Meinhardt, M.; Pötzsch, B.; Spriestersbach, R.; Bhakdi, S.; Suttorp, N.; Seeger, W. Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J. Immunol. 1997, 159, 1909–1916. [Google Scholar]
- Iacovache, I.; Bischofberger, M.; van der Goot, F.G. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 2010, 20, 241–246. [Google Scholar] [CrossRef]
- Breuer, K.; Wittmann, M.; Kempe, K.; Kapp, A.; Mai, U.; Dittrich-Breiholz, O.; Kracht, M.; Mrabet-Dahbi, S.; Werfel, T. Alpha-toxin is produced by skin colonizing Staphylococcus aureus and induces a T helper type 1 response in atopic dermatitis. Clin. Exp. Allergy 2005, 35, 1088–1095. [Google Scholar] [CrossRef]
- Kasraie, S.; Niebuhr, M.; Kopfnagel, V.; Dittrich-Breiholz, O.; Kracht, M.; Werfel, T. Macrophages from patients with atopic dermatitis show a reduced CXCL10 expression in response to staphylococcal α-toxin. Allergy 2012, 67, 41–49. [Google Scholar] [CrossRef]
- Schlievert, P.M.; Roller, R.J.; Kilgore, S.H.; Villarreal, M.; Klingelhutz, A.J.; Leung, D.Y.M. Staphylococcal TSST-1 Association with Eczema Herpeticum in Humans. mSphere 2021, 6, e00608-21. [Google Scholar] [CrossRef]
- Okano, M.; Fujiwara, T.; Kariya, S.; Higaki, T.; Haruna, T.; Matsushita, O.; Noda, Y.; Makihara, S.; Kanai, K.; Noyama, Y.; et al. Cellular responses to Staphylococcus aureus alpha-toxin in chronic rhinosinusitis with nasal polyps. Allergol. Int. 2014, 63, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Noh, S.Y.; Park, O.J.; Yun, C.H.; Han, S.H. Staphylococcus aureus induces IL-8 expression through its lipoproteins in the human intestinal epithelial cell, Caco-2. Cytokine 2015, 75, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.K.; Vikström, E.; Magnusson, K.E.; Vécsey-Semjén, B.; Colque-Navarro, P.; Möllby, R. The Staphylococcus aureus alpha-toxin perturbs the barrier function in Caco-2 epithelial cell monolayers by altering junctional integrity. Infect. Immun. 2012, 80, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Gillet, Y.; Issartel, B.; Vanhems, P.; Fournet, J.C.; Lina, G.; Bes, M.; Vandenesch, F.; Piémont, Y.; Brousse, N.; Floret, D.; et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 2002, 359, 753–759. [Google Scholar] [CrossRef]
- Finck-Barbançon, V.; Duportail, G.; Meunier, O.; Colin, D.A. Pore formation by a two-component leukocidin from Staphylococcus aureus within the membrane of human polymorphonuclear leukocytes. Biochim. Biophys. Acta 1993, 1182, 275–282. [Google Scholar] [CrossRef]
- Yanai, M.; Rocha, M.A.; Matolek, A.Z.; Chintalacharuvu, A.; Taira, Y.; Chintalacharuvu, K.; Beenhouwer, D.O. Separately or combined, LukG/LukH is functionally unique compared to other staphylococcal bicomponent leukotoxins. PLoS ONE 2014, 9, e89308. [Google Scholar] [CrossRef]
- Melehani, J.H.; James, D.B.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog. 2015, 11, e1004970. [Google Scholar] [CrossRef]
- Holzinger, D.; Gieldon, L.; Mysore, V.; Nippe, N.; Taxman, D.J.; Duncan, J.A.; Broglie, P.M.; Marketon, K.; Austermann, J.; Vogl, T.; et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Biol. 2012, 92, 1069–1081. [Google Scholar] [CrossRef]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef]
- Staali, L.; Monteil, H.; Colin, D.A. The staphylococcal pore-forming leukotoxins open Ca2+ channels in the membrane of human polymorphonuclear neutrophils. J. Membr. Biol. 1998, 162, 209–216. [Google Scholar] [CrossRef]
- Noda, M.; Kato, I.; Hirayama, T.; Matsuda, F. Mode of action of staphylococcal leukocidin: Effects of the S and F components on the activities of membrane-associated enzymes of rabbit polymorphonuclear leukocytes. Infect. Immun. 1982, 35, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.H.; Deo, P.; Yeung, A.T.Y.; Kostoulias, X.P.; Jeon, Y.; Gao, M.L.; Seidi, A.; Olivier, F.A.B.; Sridhar, S.; Nethercott, C.; et al. Targeting NLRP3 and Staphylococcal pore-forming toxin receptors in human-induced pluripotent stem cell-derived macrophages. J. Leukoc. Biol. 2020, 108, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Shallcross, L.J.; Fragaszy, E.; Johnson, A.M.; Hayward, A.C. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 43–54. [Google Scholar] [CrossRef]
- Genestier, A.L.; Michallet, M.C.; Prévost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M.; et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, T.; Zou, X.; Wu, S.; Zhu, J. Panton-valentine leucocidin carrying Staphylococcus aureus causing necrotizing pneumonia inactivates the JAK/STAT signaling pathway and increases the expression of inflammatory cytokines. Infect. Genet. Evol. 2020, 86, 104582. [Google Scholar] [CrossRef]
- Gray, B.; Hall, P.; Gresham, H. Targeting agr- and agr-Like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensors 2013, 13, 5130–5166. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, C.; Ma, B.; Xue, X.; Li, Z.; Chen, Z.; Li, F.; Zhou, S.; Luo, X.; Hou, Z. Inhibiting PSMα-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis. 2018, 9, 362. [Google Scholar] [CrossRef]
- Hanzelmann, D.; Joo, H.S.; Franz-Wachtel, M.; Hertlein, T.; Stevanovic, S.; Macek, B.; Wolz, C.; Götz, F.; Otto, M.; Kretschmer, D.; et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat. Commun. 2016, 7, 12304. [Google Scholar] [CrossRef]
- Patrick, G.J.; Liu, H.; Alphonse, M.P.; Dikeman, D.A.; Youn, C.; Otterson, J.C.; Wang, Y.; Ravipati, A.; Mazhar, M.; Denny, G.; et al. Epicutaneous Staphylococcus aureus induces IL-36 to enhance IgE production and ensuing allergic disease. J. Clin. Investig. 2021, 131, e143334. [Google Scholar] [CrossRef]
- McKevitt, A.I.; Bjornson, G.L.; Mauracher, C.A.; Scheifele, D.W. Amino acid sequence of a deltalike toxin from Staphylococcus epidermidis. Infect. Immun. 1990, 58, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, D.; Gleske, A.K.; Rautenberg, M.; Wang, R.; Köberle, M.; Bohn, E.; Schöneberg, T.; Rabiet, M.J.; Boulay, F.; Klebanoff, S.J.; et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 2010, 7, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Rautenberg, M.; Joo, H.S.; Otto, M.; Peschel, A. Neutrophil responses to staphylococcal pathogens and commensals via the formyl peptide receptor 2 relates to phenol-soluble modulin release and virulence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef]
- Kapral, F.A. Staphylococcus aureus delta toxin as an enterotoxin. Ciba. Found. Symp. 1985, 112, 215–229. [Google Scholar] [CrossRef]
- Ullah, A.; Khan, A.; Al-Harrasi, A.; Ullah, K.; Shabbir, A. Three-Dimensional Structure Characterization and Inhibition Study of Exfoliative Toxin D from Staphylococcus aureus. Front. Pharmacol. 2022, 13, 800970. [Google Scholar] [CrossRef]
- Imanishi, I.; Nicolas, A.; Caetano, A.B.; Castro, T.L.P.; Tartaglia, N.R.; Mariutti, R.; Guédon, E.; Even, S.; Berkova, N.; Arni, R.K.; et al. Exfoliative toxin E, a new Staphylococcus aureus virulence factor with host-specific activity. Sci. Rep. 2019, 9, 16336. [Google Scholar] [CrossRef]
- Stentzel, S.; Teufelberger, A.; Nordengrün, M.; Kolata, J.; Schmidt, F.; van Crombruggen, K.; Michalik, S.; Kumpfmüller, J.; Tischer, S.; Schweder, T.; et al. Staphylococcal serine protease-like proteins are pacemakers of allergic airway reactions to Staphylococcus aureus. J. Allergy Clin. Immunol. 2017, 139, 492–500.e8. [Google Scholar] [CrossRef]
- Melish, M.E.; Glasgow, L.A. Staphylococcal scalded skin syndrome: The expanded clinical syndrome. J. Pediatr. 1971, 78, 958–967. [Google Scholar] [CrossRef]
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef]
- Nishifuji, K.; Sugai, M.; Amagai, M. Staphylococcal exfoliative toxins: “molecular scissors” of bacteria that attack the cutaneous defense barrier in mammals. J. Dermatol. Sci. 2008, 49, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M.; Yamaguchi, T.; Hanakawa, Y.; Nishifuji, K.; Sugai, M.; Stanley, J.R. Staphylococcal exfoliative toxin B specifically cleaves desmoglein 1. J. Investig. Dermatol. 2002, 118, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Nordengrün, M.; Abdurrahman, G.; Treffon, J.; Wächter, H.; Kahl, B.C.; Bröker, B.M. Allergic Reactions to Serine Protease-Like Proteins of Staphylococcus aureus. Front. Immunol. 2021, 12, 651060. [Google Scholar] [CrossRef] [PubMed]
- Smagur, J.; Guzik, K.; Magiera, L.; Bzowska, M.; Gruca, M.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. A new pathway of staphylococcal pathogenesis: Apoptosis-like death induced by Staphopain B in human neutrophils and monocytes. J. Innate Immun. 2009, 1, 98–108. [Google Scholar] [CrossRef]
- Kulig, P.; Zabel, B.A.; Dubin, G.; Allen, S.J.; Ohyama, T.; Potempa, J.; Handel, T.M.; Butcher, E.C.; Cichy, J. Staphylococcus aureus-derived staphopain B, a potent cysteine protease activator of plasma chemerin. J. Immunol. 2007, 178, 3713–3720. [Google Scholar] [CrossRef]
- Bergdoll, M.S.; Crass, B.A.; Reiser, R.F.; Robbins, R.N.; Davis, J.P. A new staphylococcal enterotoxin, enterotoxin F, associated with toxic-shock-syndrome Staphylococcus aureus isolates. Lancet 1981, 1, 1017–1021. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Calus, L.; Derycke, L.; Dullaers, M.; Van Zele, T.; De Ruyck, N.; Pérez-Novo, C.; Holtappels, G.; De Vos, G.; Lambrecht, B.N.; Bachert, C.; et al. IL-21 Is Increased in Nasal Polyposis and after Stimulation with Staphylococcus aureus Enterotoxin B. Int. Arch. Allergy Immunol. 2017, 174, 161–169. [Google Scholar] [CrossRef]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef]
- Van Zele, T.; Gevaert, P.; Watelet, J.B.; Claeys, G.; Holtappels, G.; Claeys, C.; van Cauwenberge, P.; Bachert, C. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J. Allergy Clin. Immunol. 2004, 114, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Orfali, R.L.; da Silva Oliveira, L.M.; de Lima, J.F.; de Carvalho, G.C.; Ramos, Y.A.L.; Pereira, N.Z.; Pereira, N.V.; Zaniboni, M.C.; Sotto, M.N.; da Silva Duarte, A.J.; et al. Staphylococcus aureus enterotoxins modulate IL-22-secreting cells in adults with atopic dermatitis. Sci. Rep. 2018, 8, 6665. [Google Scholar] [CrossRef] [PubMed]
- Hellings, P.W.; Hens, G.; Meyts, I.; Bullens, D.; Vanoirbeek, J.; Gevaert, P.; Jorissen, M.; Ceuppens, J.L.; Bachert, C. Aggravation of bronchial eosinophilia in mice by nasal and bronchial exposure to Staphylococcus aureus enterotoxin B. Clin. Exp. Allergy 2006, 36, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, Y.; Wang, D.; Zhao, Z.; Jeffry, J.; Zeng, L.; Zou, Z.; Chen, H.; Tao, A. Synergistic activation of Src, ERK and STAT pathways in PBMCs for Staphylococcal enterotoxin A induced production of cytokines and chemokines. Asian Pac. J. Allergy Immunol. 2020, 38, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Smith, F.; Ho, J.; Croft, N.M.; Domizio, P.; Price, E.; Sanderson, I.R.; Meadows, N.J. Staphylococcal enterotoxins G and I, a cause of severe but reversible neonatal enteropathy. Clin. Gastroenterol. Hepatol. 2008, 6, 251–254. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Ali, T.; Alkasir, R.; Yin, J.; Liu, G.; Han, B. Staphylococcal enterotoxin H induced apoptosis of bovine mammary epithelial cells in vitro. Toxins 2014, 6, 3552–3567. [Google Scholar] [CrossRef]
- Hou, F.; Peng, L.; Jiang, J.; Chen, T.; Xu, D.; Huang, Q.; Ye, C.; Peng, Y.; Hu, D.L.; Fang, R. ATP Facilitates Staphylococcal Enterotoxin O Induced Neutrophil IL-1β Secretion via NLRP3 Inflammasome Dependent Pathways. Front. Immunol. 2021, 12, 649235. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, J.; Yang, D.; Tang, C.; Chen, J. Staphylococcal enterotoxin M induced inflammation and impairment of bovine mammary epithelial cells. J. Dairy Sci. 2020, 103, 8350–8359. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, A.; Tang, J.; Tang, C.; Chen, J. Identification and measurement of staphylococcal enterotoxin M from Staphylococcus aureus isolate associated with staphylococcal food poisoning. Lett. Appl. Microbiol. 2017, 65, 27–34. [Google Scholar] [CrossRef]
- Hu, D.L.; Ono, H.K.; Isayama, S.; Okada, R.; Okamura, M.; Lei, L.C.; Liu, Z.S.; Zhang, X.C.; Liu, M.Y.; Cui, J.C.; et al. Biological characteristics of staphylococcal enterotoxin Q and its potential risk for food poisoning. J. Appl. Microbiol. 2017, 122, 1672–1679. [Google Scholar] [CrossRef]
- Cruciani, M.; Etna, M.P.; Camilli, R.; Giacomini, E.; Percario, Z.A.; Severa, M.; Sandini, S.; Rizzo, F.; Brandi, V.; Balsamo, G.; et al. Staphylococcus aureus Esx Factors Control Human Dendritic Cell Functions Conditioning Th1/Th17 Response. Front. Cell. Infect. Microbiol. 2017, 7, 330. [Google Scholar] [CrossRef] [PubMed]
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 2014, 82, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Zhu, Y.L.; Li, J.; Shi, J.; He, X.Q.; Ding, J.; Xu, Y.Q. Staphylococcal protein A, Panton-Valentine leukocidin and coagulase aggravate the bone loss and bone destruction in osteomyelitis. Cell Physiol. Biochem. 2013, 32, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef]
- Winstel, V.; Schneewind, O.; Missiakas, D. Staphylococcus aureus Exploits the Host Apoptotic Pathway To Persist during Infection. mBio 2019, 10, e02270-19. [Google Scholar] [CrossRef]
- Wang, H.; von Rohrscheidt, J.; Roehrbein, J.; Peters, T.; Sindrilaru, A.; Kess, D.; Preissner, K.T.; Scharffetter-Kochanek, K. Extracellular adherence protein of Staphylococcus aureus suppresses disease by inhibiting T-cell recruitment in a mouse model of psoriasis. J. Investig. Dermatol. 2010, 130, 743–754. [Google Scholar] [CrossRef]
- Foster, T.J. The MSCRAMM Family of Cell-Wall-Anchored Surface Proteins of Gram-Positive Cocci. Trends Microbiol. 2019, 27, 927–941. [Google Scholar] [CrossRef]
- Pazos, M.; Peters, K. Peptidoglycan. Subcell. Biochem. 2019, 92, 127–168. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Surface Proteins of Staphylococcus aureus. Microbiol. Spectr. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.M.; Speziale, P.; Patti, J.M.; Höök, M. MSCRAMM--targeted vaccines and immunotherapy for staphylococcal infection. Curr. Opin. Drug Discov. Dev. 2004, 7, 223–227. [Google Scholar]
- Fox, P.G.; Schiavetti, F.; Rappuoli, R.; McLoughlin, R.M.; Bagnoli, F. Staphylococcal Protein A Induces Leukocyte Necrosis by Complexing with Human Immunoglobulins. mBio 2021, 12, e00899-21. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Sa, G.; Chattopadhyay, S.; Ray, P.K. Protein A-induced apoptosis of cancer cells is effected by soluble immune mediators. Cancer Immunol. Immunother. 2002, 51, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene 2000, 19, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Claro, T.; Widaa, A.; McDonnell, C.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology 2013, 159, 147–154. [Google Scholar] [CrossRef]
- Claro, T.; Widaa, A.; O’Seaghdha, M.; Miajlovic, H.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS ONE 2011, 6, e18748. [Google Scholar] [CrossRef]
- Al Kindi, A.; Williams, H.; Matsuda, K.; Alkahtani, A.M.; Saville, C.; Bennett, H.; Alshammari, Y.; Tan, S.Y.; O’Neill, C.; Tanaka, A.; et al. Staphylococcus aureus second immunoglobulin-binding protein drives atopic dermatitis via IL-33. J. Allergy Clin. Immunol. 2021, 147, 1354–1368.e3. [Google Scholar] [CrossRef]
- Garcovich, S.; Maurelli, M.; Gisondi, P.; Peris, K.; Yosipovitch, G.; Girolomoni, G. Pruritus as a Distinctive Feature of Type 2 Inflammation. Vaccines 2021, 9, 303. [Google Scholar] [CrossRef]
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Takeuchi, O.; Akira, S. Recognition of lipopeptides by Toll-like receptors. J. Endotoxin. Res. 2002, 8, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Stoll, H.; Dengjel, J.; Nerz, C.; Götz, F. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect. Immun. 2005, 73, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e14. [Google Scholar] [CrossRef] [PubMed]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus Lipoteichoic Acid Initiates a TSLP-Basophil-IL4 Axis in the Skin. J. Investig. Dermatol. 2020, 140, 915–917.e2. [Google Scholar] [CrossRef] [PubMed]
- Misawa, Y.; Kelley, K.A.; Wang, X.; Wang, L.; Park, W.B.; Birtel, J.; Saslowsky, D.; Lee, J.C. Staphylococcus aureus Colonization of the Mouse Gastrointestinal Tract Is Modulated by Wall Teichoic Acid, Capsule, and Surface Proteins. PLoS Pathog. 2015, 11, e1005061. [Google Scholar] [CrossRef] [PubMed]
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297. [Google Scholar] [CrossRef]
- Covas, G.; Vaz, F.; Henriques, G.; Pinho, M.G.; Filipe, S.R. Analysis of Cell Wall Teichoic Acids in Staphylococcus aureus. Methods Mol. Biol. 2016, 1440, 201–213. [Google Scholar] [CrossRef]
- Matsui, K.; Tofukuji, S.; Ikeda, R. CCL17 production by mouse langerhans cells stimulated with Staphylococcus aureus cell wall components. Biol. Pharm. Bull. 2015, 38, 317–320. [Google Scholar] [CrossRef]
- Matsui, K.; Wirotesangthong, M.; Nishikawa, A. Peptidoglycan from Staphylococcus aureus induces IL-4 production from murine spleen cells via an IL-18-dependent mechanism. Int. Arch. Allergy Immunol. 2008, 146, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Supajatura, V.; Ushio, H.; Nakao, A.; Akira, S.; Okumura, K.; Ra, C.; Ogawa, H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Investig. 2002, 109, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Biggs, T.C.; Abadalkareem, R.S.; Hayes, S.M.; Holding, R.E.; Lau, L.C.; Harries, P.G.; Allan, R.N.; Pender, S.L.F.; Walls, A.F.; Salib, R.J. Staphylococcus aureus internalisation enhances bacterial survival through modulation of host immune responses and mast cell activation. Allergy 2021, 76, 1893–1896. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Tang, C.H.; Chen, J.H.; Chuang, J.Y.; Huang, S.M.; Tan, T.W.; Lai, C.H.; Lu, D.Y. Peptidoglycan induces interleukin-6 expression through the TLR2 receptor, JNK, c-Jun, and AP-1 pathways in microglia. J. Cell. Physiol. 2011, 226, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.J.; Chang, C.K.; Chen, M.C.; Chen, B.C.; Ma, H.P.; Hong, C.Y.; Lin, C.H. Apoptosis signal-regulating kinase 1 in peptidoglycan-induced COX-2 expression in macrophages. J. Leukoc. Biol. 2010, 87, 1069–1082. [Google Scholar] [CrossRef]
- Wang, D.; Xiao, P.L.; Duan, H.X.; Zhou, M.; Liu, J.; Li, W.; Luo, K.L.; Chen, J.J.; Hu, J.Y. Peptidoglycans promotes human leukemic THP-1 cell apoptosis and differentiation. Asian Pac. J. Cancer Prev. 2012, 13, 6409–6413. [Google Scholar] [CrossRef]
- Namazi, M.R. Paradoxical exacerbation of psoriasis in AIDS: Proposed explanations including the potential roles of substance P and gram-negative bacteria. Autoimmunity 2004, 37, 67–71. [Google Scholar] [CrossRef]
- Ruíz-González, V.; Cancino-Diaz, J.C.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E. Keratinocytes treated with peptidoglycan from Staphylococcus aureus produce vascular endothelial growth factor, and its expression is amplified by the subsequent production of interleukin-13. Int. J. Dermatol. 2009, 48, 846–854. [Google Scholar] [CrossRef]
- Wu, H.M.; Wang, J.; Zhang, B.; Fang, L.; Xu, K.; Liu, R.Y. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci. 2016, 161, 51–59. [Google Scholar] [CrossRef]
- Müller, S.; Wolf, A.J.; Iliev, I.D.; Berg, B.L.; Underhill, D.M.; Liu, G.Y. Poorly Cross-Linked Peptidoglycan in MRSA Due to mecA Induction Activates the Inflammasome and Exacerbates Immunopathology. Cell Host Microbe 2015, 18, 604–612. [Google Scholar] [CrossRef]
- Zhu, F.; Zhou, Y.; Jiang, C.; Zhang, X. Role of JAK-STAT signaling in maturation of phagosomes containing Staphylococcus aureus. Sci. Rep. 2015, 5, 14854. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Willing, S.E.; Kim, H.K.; Schneewind, O.; Missiakas, D. Peptidoglycan Contribution to the B Cell Superantigen Activity of Staphylococcal Protein A. mBio 2021, 12, e00039-21. [Google Scholar] [CrossRef] [PubMed]
- Soong, G.; Chun, J.; Parker, D.; Prince, A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J. Infect. Dis. 2012, 205, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Jänicke, R.U. α-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the i.intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 2001, 155, 637–648. [Google Scholar] [CrossRef]
- Mestre, M.B.; Colombo, M.I. Staphylococcus aureus promotes autophagy by decreasing intracellular cAMP levels. Autophagy 2012, 8, 1865–1867. [Google Scholar] [CrossRef]
- Mestre, M.B.; Colombo, M.I. cAMP and EPAC are key players in the regulation of the signal transduction pathway involved in the α-hemolysin autophagic response. PLoS Pathog. 2012, 8, e1002664. [Google Scholar] [CrossRef]
- McFadden, J.P.; Noble, W.C.; Camp, R.D. Superantigenic exotoxin-secreting potential of staphylococci isolated from atopic eczematous skin. Br. J. Dermatol. 1993, 128, 631–632. [Google Scholar] [CrossRef]
- Porichis, F.; Morou, A.; Baritaki, S.; Spandidos, D.A.; Krambovitis, E. Activation-induced cell death signalling in CD4+ T cells by staphylococcal enterotoxin A. Toxicol. Lett. 2008, 176, 77–84. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, S.M.; Kim, J.M.; Oh, B.M.; Kim, J.Y.; Jung, H.J.; Lim, H.J.; Kim, B.S.; Lee, W.J.; Lee, S.J.; et al. Potential Immunoinflammatory Role of Staphylococcal Enterotoxin A in Atopic Dermatitis: Immunohistopathological Analysis and in vitro Assay. Ann. Dermatol. 2013, 25, 173–180. [Google Scholar] [CrossRef]
- Maina, E.K.; Hu, D.L.; Tsuji, T.; Omoe, K.; Nakane, A. Staphylococcal enterotoxin A has potent superantigenic and emetic activities but not diarrheagenic activity. Int. J. Med. Microbiol. 2012, 302, 88–95. [Google Scholar] [CrossRef]
- Bardoel, B.W.; Vos, R.; Bouman, T.; Aerts, P.C.; Bestebroer, J.; Huizinga, E.G.; Brondijk, T.H.; van Strijp, J.A.; de Haas, C.J. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med. 2012, 90, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, M.; Iwamoto, K.; Niitsu, Y.; Matsushima, A.; Yanase, Y.; Hisatsune, J.; Sugai, M.; Hide, M. Staphylococcus aureus from atopic dermatitis skin accumulates in the lysosomes of keratinocytes with induction of IL-1α secretion via TLR9. Allergy 2019, 74, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sweren, E.; Liu, H.; Wier, E.; Alphonse, M.P.; Chen, R.; Islam, N.; Li, A.; Xue, Y.; Chen, J.; et al. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe 2021, 29, 777–791.e6. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Liu, B.; Tai, Y.; Achanta, S.; Kaelberer, M.M.; Caceres, A.I.; Shao, X.; Fang, J.; Jordt, S.E. IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc. Natl. Acad. Sci. USA 2016, 113, E7572–E7579. [Google Scholar] [CrossRef]
- Vu, A.T.; Baba, T.; Chen, X.; Le, T.A.; Kinoshita, H.; Xie, Y.; Kamijo, S.; Hiramatsu, K.; Ikeda, S.; Ogawa, H.; et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J. Allergy Clin. Immunol. 2010, 126, 985–993.e3. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Thé, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef]
- Son, E.D.; Kim, H.J.; Park, T.; Shin, K.; Bae, I.H.; Lim, K.M.; Cho, E.G.; Lee, T.R. Staphylococcus aureus inhibits terminal differentiation of normal human keratinocytes by stimulating interleukin-6 secretion. J. Dermatol. Sci. 2014, 74, 64–71. [Google Scholar] [CrossRef]
- Williams, M.R.; Nakatsuji, T.; Sanford, J.A.; Vrbanac, A.F.; Gallo, R.L. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Investig. Dermatol. 2017, 137, 377–384. [Google Scholar] [CrossRef]
- Lan, F.; Zhang, N.; Holtappels, G.; De Ruyck, N.; Krysko, O.; Van Crombruggen, K.; Braun, H.; Johnston, S.L.; Papadopoulos, N.G.; Zhang, L.; et al. Staphylococcus aureus Induces a Mucosal Type 2 Immune Response via Epithelial Cell-derived Cytokines. Am. J. Respir. Crit Care Med. 2018, 198, 452–463. [Google Scholar] [CrossRef]
- Hardman, C.S.; Chen, Y.L.; Salimi, M.; Nahler, J.; Corridoni, D.; Jagielowicz, M.; Fonseka, C.L.; Johnson, D.; Repapi, E.; Cousins, D.J.; et al. IL-6 effector function of group 2 innate lymphoid cells (ILC2) is NOD2 dependent. Sci. Immunol. 2021, 6, eabe5084. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. T-cell subsets (Th1 versus Th2). Ann. Allergy Asthma Immunol. 2000, 85, 9–18; quiz 18, 21. [Google Scholar] [CrossRef]
- Pérez Novo, C.A.; Jedrzejczak-Czechowicz, M.; Lewandowska-Polak, A.; Claeys, C.; Holtappels, G.; Van Cauwenberge, P.; Kowalski, M.L.; Bachert, C. T cell inflammatory response, Foxp3 and TNFRS18-L regulation of peripheral blood mononuclear cells from patients with nasal polyps-asthma after staphylococcal superantigen stimulation. Clin. Exp. Allergy 2010, 40, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Orciani, M.; Campanati, A.; Caffarini, M.; Ganzetti, G.; Consales, V.; Lucarini, G.; Offidani, A.; Di Primio, R. T helper (Th)1, Th17 and Th2 imbalance in mesenchymal stem cells of adult patients with atopic dermatitis: At the origin of the problem. Br. J. Dermatol. 2017, 176, 1569–1576. [Google Scholar] [CrossRef]
- Tada, Y.; Asahina, A.; Takekoshi, T.; Kishimoto, E.; Mitsui, H.; Saeki, H.; Komine, M.; Tamaki, K. Interleukin 12 production by monocytes from patients with psoriasis and its inhibition by ciclosporin A. Br. J. Dermatol. 2006, 154, 1180–1183. [Google Scholar] [CrossRef]
- Stetson, D.B.; Voehringer, D.; Grogan, J.L.; Xu, M.; Reinhardt, R.L.; Scheu, S.; Kelly, B.L.; Locksley, R.M. Th2 cells: Orchestrating barrier immunity. Adv. Immunol. 2004, 83, 163–189. [Google Scholar] [CrossRef]
- Kamijo, H.; Miyagaki, T.; Hayashi, Y.; Akatsuka, T.; Watanabe-Otobe, S.; Oka, T.; Shishido-Takahashi, N.; Suga, H.; Sugaya, M.; Sato, S. Increased IL-26 Expression Promotes T Helper Type 17- and T Helper Type 2-Associated Cytokine Production by Keratinocytes in Atopic Dermatitis. J. Investig. Dermatol. 2020, 140, 636–644.e2. [Google Scholar] [CrossRef]
- Chang, H.W.; Yan, D.; Singh, R.; Liu, J.; Lu, X.; Ucmak, D.; Lee, K.; Afifi, L.; Fadrosh, D.; Leech, J.; et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 2018, 6, 154. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J. Investig. Dermatol. 2014, 134, 2114–2121. [Google Scholar] [CrossRef]
- Ou, L.S.; Goleva, E.; Hall, C.; Leung, D.Y. T regulatory cells in atopic dermatitis and subversion of their activity by superantigens. J. Allergy Clin. Immunol. 2004, 113, 756–763. [Google Scholar] [CrossRef]
- Laouini, D.; Kawamoto, S.; Yalcindag, A.; Bryce, P.; Mizoguchi, E.; Oettgen, H.; Geha, R.S. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J. Allergy Clin. Immunol. 2003, 112, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.A.; Ochkur, S.I.; Lee, N.A.; Lee, J.J. Eosinophils and asthma. Curr. Allergy Asthma Rep. 2007, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.A.; McGirt, L.Y.; Beck, L.A. Biomarkers of Th2 polarity are predictive of staphylococcal colonization in subjects with atopic dermatitis. Br. J. Dermatol. 2009, 160, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Dubin, C.; Del Duca, E.; Guttman-Yassky, E. The IL-4, IL-13 and IL-31 pathways in atopic dermatitis. Expert Rev. Clin. Immunol. 2021, 17, 835–852. [Google Scholar] [CrossRef]
- Iwaszko, M.; Biały, S.; Bogunia-Kubik, K. Significance of Interleukin (IL)-4 and IL-13 in Inflammatory Arthritis. Cells 2021, 10, 3000. [Google Scholar] [CrossRef]
- Kimura, A.; Kishimoto, T. Th17 cells in inflammation. Int. Immunopharmacol 2011, 11, 319–322. [Google Scholar] [CrossRef]
- Tokura, Y. Th17 cells and skin diseases. Nihon Rinsho Meneki Gakkai Kaishi 2012, 35, 388–392. [Google Scholar] [CrossRef][Green Version]
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J. Investig. Dermatol. 2008, 128, 2625–2630. [Google Scholar] [CrossRef]
- Orfali, R.L.; Yoshikawa, F.S.Y.; Oliveira, L.; Pereira, N.Z.; de Lima, J.F.; Ramos YÁ, L.; Duarte, A.; Sato, M.N.; Aoki, V. Staphylococcal enterotoxins modulate the effector CD4(+) T cell response by reshaping the gene expression profile in adults with atopic dermatitis. Sci. Rep. 2019, 9, 13082. [Google Scholar] [CrossRef]
- Sugaya, M. The Role of Th17-Related Cytokines in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 1314. [Google Scholar] [CrossRef]
- Liu, H.; Archer, N.K.; Dillen, C.A.; Wang, Y.; Ashbaugh, A.G.; Ortines, R.V.; Kao, T.; Lee, S.K.; Cai, S.S.; Miller, R.J.; et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe 2017, 22, 653–666.e5. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Bishayi, B. Role of Th17 and Treg cells in septic arthritis and the impact of the Th17/Treg-derived cytokines in the pathogenesis of S. aureus induced septic arthritis in mice. Microb. Pathog. 2017, 113, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Dey, I.; Bishayi, B. Role of different Th17 and Treg downstream signalling pathways in the pathogenesis of Staphylococcus aureus infection induced septic arthritis in mice. Exp. Mol. Pathol. 2020, 116, 104485. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Dey, R.; Bishayi, B. Dual neutralization of TNFR-2 and MMP-2 regulates the severity of induced septic arthritis correlating alteration in the level of interferon gamma and interleukin-10 in terms of TNFR2 blocking. Immunol. Res. 2018, 66, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Dey, R.; Sawoo, R.; Bishayi, B. Neutralization of IL-17 and treatment with IL-2 protects septic arthritis by regulating free radical production and antioxidant enzymes in Th17 and Tregs: An immunomodulatory TLR2 versus TNFR response. Cell Immunol. 2021, 370, 104441. [Google Scholar] [CrossRef]
- Saito, S.; Quadery, A.F. Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens 2018, 7, 64. [Google Scholar] [CrossRef]
- Taylor, A.L.; Llewelyn, M.J. Superantigen-induced proliferation of human CD4+CD25-T cells is followed by a switch to a functional regulatory phenotype. J. Immunol. 2010, 185, 6591–6598. [Google Scholar] [CrossRef]
- Seneschal, J.; Clark, R.A.; Gehad, A.; Baecher-Allan, C.M.; Kupper, T.S. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 2012, 36, 873–884. [Google Scholar] [CrossRef]
- van Dalen, R.; De La Cruz Diaz, J.S.; Rumpret, M.; Fuchsberger, F.F.; van Teijlingen, N.H.; Hanske, J.; Rademacher, C.; Geijtenbeek, T.B.H.; van Strijp, J.A.G.; Weidenmaier, C.; et al. Langerhans Cells Sense Staphylococcus aureus Wall Teichoic Acid through Langerin To Induce Inflammatory Responses. mBio 2019, 10, e00330-19. [Google Scholar] [CrossRef]
- Ma, L.; Xue, H.B.; Guan, X.H.; Shu, C.M.; Wang, F.; Zhang, J.H.; An, R.Z. The Imbalance of Th17 cells and CD4(+) CD25(high) Foxp3(+) Treg cells in patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1079–1086. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef]
- Moser, B. CXCR5, the Defining Marker for Follicular B Helper T (TFH) Cells. Front. Immunol. 2015, 6, 296. [Google Scholar] [CrossRef]
- Bossaller, L.; Burger, J.; Draeger, R.; Grimbacher, B.; Knoth, R.; Plebani, A.; Durandy, A.; Baumann, U.; Schlesier, M.; Welcher, A.A.; et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J. Immunol. 2006, 177, 4927–4932. [Google Scholar] [CrossRef]
- Bennett, F.; Luxenberg, D.; Ling, V.; Wang, I.M.; Marquette, K.; Lowe, D.; Khan, N.; Veldman, G.; Jacobs, K.A.; Valge-Archer, V.E.; et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 2003, 170, 711–718. [Google Scholar] [CrossRef]
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039. [Google Scholar] [CrossRef]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef]
- Dupont, C.D.; Scully, I.L.; Zimnisky, R.M.; Monian, B.; Rossitto, C.P.; O’Connell, E.B.; Jansen, K.U.; Anderson, A.S.; Love, J.C. Two Vaccines for Staphylococcus aureus Induce a B-Cell-Mediated Immune Response. mSphere 2018, 3, e00217-18. [Google Scholar] [CrossRef]
- Chang, H.C.; Sehra, S.; Goswami, R.; Yao, W.; Yu, Q.; Stritesky, G.L.; Jabeen, R.; McKinley, C.; Ahyi, A.N.; Han, L.; et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010, 11, 527–534. [Google Scholar] [CrossRef]
- Soussi-Gounni, A.; Kontolemos, M.; Hamid, Q. Role of IL-9 in the pathophysiology of allergic diseases. J. Allergy Clin. Immunol. 2001, 107, 575–582. [Google Scholar] [CrossRef]
- Jäger, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 2009, 183, 7169–7177. [Google Scholar] [CrossRef]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Ghaedi, M.; Takei, F. Innate lymphoid cell development. J. Allergy Clin. Immunol. 2021, 147, 1549–1560. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef]
- Wang, X.; Eagen, W.J.; Lee, J.C. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 3174–3184. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Heinrichs, D.E. Macrophage-driven nutrient delivery to phagosomal Staphylococcus aureus supports bacterial growth. EMBO Rep. 2020, 21, e50348. [Google Scholar] [CrossRef]
- Grayczyk, J.P.; Alonzo, F., 3rd. Staphylococcus aureus Lipoic Acid Synthesis Limits Macrophage Reactive Oxygen and Nitrogen Species Production To Promote Survival during Infection. Infect. Immun. 2019, 87, e00344-19. [Google Scholar] [CrossRef]
- Iwamoto, K.; Nümm, T.J.; Koch, S.; Herrmann, N.; Leib, N.; Bieber, T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward TLR2 activation. Allergy 2018, 73, 2205–2213. [Google Scholar] [CrossRef]
- Asahina, A.; Tamaki, K. Role of Langerhans cells in cutaneous protective immunity: Is the reappraisal necessary? J. Dermatol. Sci. 2006, 44, 1–9. [Google Scholar] [CrossRef]
- Berends, E.T.M.; Zheng, X.; Zwack, E.E.; Ménager, M.M.; Cammer, M.; Shopsin, B.; Torres, V.J. Staphylococcus aureus Impairs the Function of and Kills Human Dendritic Cells via the LukAB Toxin. mBio 2019, 10, e01918-18. [Google Scholar] [CrossRef]
- Matsui, K.; Kanai, S.; Ikuta, M.; Horikawa, S. Mast Cells Stimulated with Peptidoglycan from Staphylococcus aureus Augment the Development of Th1 Cells. J. Pharm. Pharm. Sci. 2018, 21, 296–304. [Google Scholar] [CrossRef]
- Hayes, S.M.; Biggs, T.C.; Goldie, S.P.; Harries, P.G.; Walls, A.F.; Allan, R.N.; Pender, S.L.F.; Salib, R.J. Staphylococcus aureus internalization in mast cells in nasal polyps: Characterization of interactions and potential mechanisms. J. Allergy Clin. Immunol. 2020, 145, 147–159. [Google Scholar] [CrossRef]
- Bachert, C.; Humbert, M.; Hanania, N.A.; Zhang, N.; Holgate, S.; Buhl, R.; Bröker, B.M. Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: Current knowledge. Eur. Respir. J. 2020, 55, 1901592. [Google Scholar] [CrossRef]
- Liu, C.; Yang, L.; Han, Y.; Ouyang, W.; Yin, W.; Xu, F. Mast cells participate in regulation of lung-gut axis during Staphylococcus aureus pneumonia. Cell Prolif. 2019, 52, e12565. [Google Scholar] [CrossRef]
- Lehman, H.K.; Segal, B.H. The role of neutrophils in host defense and disease. J. Allergy Clin. Immunol. 2020, 145, 1535–1544. [Google Scholar] [CrossRef]
- Gough, P.; Ganesan, S.; Datta, S.K. IL-20 Signaling in Activated Human Neutrophils Inhibits Neutrophil Migration and Function. J. Immunol. 2017, 198, 4373–4382. [Google Scholar] [CrossRef]
- Cho, J.S.; Guo, Y.; Ramos, R.I.; Hebroni, F.; Plaisier, S.B.; Xuan, C.; Granick, J.L.; Matsushima, H.; Takashima, A.; Iwakura, Y.; et al. Neutrophil-derived IL-1β is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012, 8, e1003047. [Google Scholar] [CrossRef]
- Lutalo, P.M.; D’Cruz, D.P. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J. Autoimmun. 2014, 48–49, 94–98. [Google Scholar] [CrossRef]
- Ravin, K.A.; Loy, M. The Eosinophil in Infection. Clin. Rev. Allergy Immunol. 2016, 50, 214–227. [Google Scholar] [CrossRef]
- Gangwar, R.S.; Levi-Schaffer, F. sCD48 is anti-inflammatory in Staphylococcus aureus Enterotoxin B-induced eosinophilic inflammation. Allergy 2016, 71, 829–839. [Google Scholar] [CrossRef]
- Prince, L.R.; Graham, K.J.; Connolly, J.; Anwar, S.; Ridley, R.; Sabroe, I.; Foster, S.J.; Whyte, M.K. Staphylococcus aureus induces eosinophil cell death mediated by α-hemolysin. PLoS ONE 2012, 7, e31506. [Google Scholar] [CrossRef]
- Kay, A.B. TH2-type cytokines in asthma. Ann. N. Y. Acad. Sci. 1996, 796, 1–8. [Google Scholar] [CrossRef]
- Holgate, S.T. Innate and adaptive immune responses in asthma. Nat. Med. 2012, 18, 673–683. [Google Scholar] [CrossRef]
- Jiao, D.; Wong, C.K.; Qiu, H.N.; Dong, J.; Cai, Z.; Chu, M.; Hon, K.L.; Tsang, M.S.; Lam, C.W. NOD2 and TLR2 ligands trigger the activation of basophils and eosinophils by interacting with dermal fibroblasts in atopic dermatitis-like skin inflammation. Cell Mol. Immunol. 2016, 13, 535–550. [Google Scholar] [CrossRef]
- Leyva-Castillo, J.M.; Das, M.; Kane, J.; Strakosha, M.; Singh, S.; Wong, D.S.H.; Horswill, A.R.; Karasuyama, H.; Brombacher, F.; Miller, L.S.; et al. Basophil-derived IL-4 promotes cutaneous Staphylococcus aureus infection. JCI Insight 2021, 6, e149953. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, X.; Rao, X. Apoptosis induced by Staphylococcus aureus toxins. Microbiol. Res. 2017, 205, 19–24. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, S.K.; Baik, J.E.; Ko, E.B.; Ahn, K.B.; Yun, C.H.; Han, S.H. Staphylococcal LTA antagonizes the B cell-mitogenic potential of LPS. Sci. Rep. 2018, 8, 1496. [Google Scholar] [CrossRef]
- Place, D.E.; Lee, S.; Kanneganti, T.D. PANoptosis in microbial infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Hara, H. The inflammasome and its regulation. Crit. Rev. Immunol. 2014, 34, 41–80. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Champagne, C.; Morizot, A.; Lapointe, J.M.; Saleh, M. The Inflammatory Caspases-1 and -11 Mediate the Pathogenesis of Dermatitis in Sharpin-Deficient Mice. J. Immunol. 2015, 195, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhou, F. Inflammasomes in Common Immune-Related Skin Diseases. Front. Immunol. 2020, 11, 882. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef]
- Tilburgs, T.; Meissner, T.B.; Ferreira, L.M.R.; Mulder, A.; Musunuru, K.; Ye, J.; Strominger, J.L. NLRP2 is a suppressor of NF-κB signaling and HLA-C expression in human trophoblasts. Biol. Reprod. 2017, 96, 831–842. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef]
- Li, R.; Zhu, S. NLRP6 inflammasome. Mol. Asp. Med. 2020, 76, 100859. [Google Scholar] [CrossRef]
- Radian, A.D.; de Almeida, L.; Dorfleutner, A.; Stehlik, C. NLRP7 and related inflammasome activating pattern recognition receptors and their function in host defense and disease. Microbes Infect. 2013, 15, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Tuladhar, S.; Kanneganti, T.D. NLRP12 in innate immunity and inflammation. Mol. Asp. Med. 2020, 76, 100887. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Wang, B.; Tian, Y.; Yin, Q. AIM2 Inflammasome Assembly and Signaling. Adv. Exp. Med. Biol. 2019, 1172, 143–155. [Google Scholar] [CrossRef]
- Song, Y.; Wu, X.; Xu, Y.; Zhu, J.; Li, J.; Zou, Z.; Chen, L.; Zhang, B.; Hua, C.; Rui, H.; et al. HPV E7 inhibits cell pyroptosis by promoting TRIM21-mediated degradation and ubiquitination of the IFI16 inflammasome. Int. J. Biol. Sci. 2020, 16, 2924–2937. [Google Scholar] [CrossRef] [PubMed]
- Loeven, N.A.; Medici, N.P.; Bliska, J.B. The pyrin inflammasome in host-microbe interactions. Curr. Opin. Microbiol. 2020, 54, 77–86. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Stappers, M.H.; Thys, Y.; Oosting, M.; Plantinga, T.S.; Ioana, M.; Reimnitz, P.; Mouton, J.W.; Netea, M.G.; Joosten, L.A.; Gyssens, I.C. TLR1, TLR2, and TLR6 gene polymorphisms are associated with increased susceptibility to complicated skin and skin structure infections. J. Infect. Dis. 2014, 210, 311–318. [Google Scholar] [CrossRef][Green Version]
- Niebuhr, M.; Baumert, K.; Heratizadeh, A.; Satzger, I.; Werfel, T. Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy 2014, 69, 1058–1067. [Google Scholar] [CrossRef]
- Schuepbach-Mallepell, S.; Philippe, V.; Brüggen, M.C.; Watanabe, H.; Roques, S.; Baldeschi, C.; Gaide, O. Antagonistic effect of the inflammasome on thymic stromal lymphopoietin expression in the skin. J. Allergy Clin. Immunol. 2013, 132, 1348–1357. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, M.; Geng, N.; Du, Y.; Li, Z.; Gao, X.; Han, B.; Liu, J.; Liu, Y. Staphylococcus aureus mediates pyroptosis in bovine mammary epithelial cell via activation of NLRP3 inflammasome. Vet. Res. 2022, 53, 10. [Google Scholar] [CrossRef]
- Accarias, S.; Lugo-Villarino, G.; Foucras, G.; Neyrolles, O.; Boullier, S.; Tabouret, G. Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. Eur. J. Immunol. 2015, 45, 794–806. [Google Scholar] [CrossRef]
- Yao, R.; Chen, Y.; Hao, H.; Guo, Z.; Cheng, X.; Ma, Y.; Ji, Q.; Yang, X.; Wang, Y.; Li, X.; et al. Pathogenic effects of inhibition of mTORC1/STAT3 axis facilitates Staphylococcus aureus-induced pyroptosis in human macrophages. Cell Commun. Signal. 2020, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yousefi, S.; Simon, H.U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 2018, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Chen, F.; Xie, N.; Cao, M.Y.; Chen, P.; Lou, Q.; Zhao, Y.; He, C.; Zhang, S.; Song, X.; et al. c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis. 2018, 9, 1140. [Google Scholar] [CrossRef]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 2020, 5, 141–153. [Google Scholar] [CrossRef]
- Xu, S.X.; McCormick, J.K. Staphylococcal superantigens in colonization and disease. Front. Cell Infect. Microbiol. 2012, 2, 52. [Google Scholar] [CrossRef]
- Ayala, A.; Wesche-Soldato, D.E.; Perl, M.; Lomas-Neira, J.L.; Swan, R.; Chung, C.S. Blockade of apoptosis as a rational therapeutic strategy for the treatment of sepsis. Novartis Found. Symp. 2007, 280, 37–49; discussion 49–52, 160–164. [Google Scholar] [CrossRef]
- Dziarski, R.; Gupta, D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: A reevaluation. Infect. Immun. 2005, 73, 5212–5216. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.S.; Jones-Nelson, O.; Hotz, M.; Cheng, L.; Miller, L.S.; Suzich, J.; Stover, C.K.; Sellman, B.R. S. aureus blocks efferocytosis of neutrophils by macrophages through the activity of its virulence factor alpha toxin. Sci. Rep. 2016, 6, 35466. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef]
- López de Armentia, M.M.; Amaya, C.; Colombo, M.I. Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles. Cells 2016, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fan, Z.; Han, H. Autophagy in Staphylococcus aureus Infection. Front. Cell. Infect. Microbiol. 2021, 11, 750222. [Google Scholar] [CrossRef]
- Schnaith, A.; Kashkar, H.; Leggio, S.A.; Addicks, K.; Krönke, M.; Krut, O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 2007, 282, 2695–2706. [Google Scholar] [CrossRef]
- Fang, L.; Wu, H.M.; Ding, P.S.; Liu, R.Y. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated RAW264.7 cells. Cell. Signal. 2014, 26, 806–814. [Google Scholar] [CrossRef]
- Li, S.; Fang, L.; Wang, J.; Liu, R. TLR2 modulates Staphylococcus aureus-induced inflammatory response and autophagy in macrophages through PI3K signaling pathway. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2017, 33, 1160–1164. [Google Scholar]
- Geng, N.; Wang, X.; Yu, X.; Wang, R.; Zhu, Y.; Zhang, M.; Liu, J.; Liu, Y. Staphylococcus aureus Avoids Autophagy Clearance of Bovine Mammary Epithelial Cells by Impairing Lysosomal Function. Front. Immunol. 2020, 11, 746. [Google Scholar] [CrossRef]
- O’Keeffe, K.M.; Wilk, M.M.; Leech, J.M.; Murphy, A.G.; Laabei, M.; Monk, I.R.; Massey, R.C.; Lindsay, J.A.; Foster, T.J.; Geoghegan, J.A.; et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus aureus Bloodstream Infection. Infect. Immun. 2015, 83, 3445–3457. [Google Scholar] [CrossRef]
- Soong, G.; Paulino, F.; Wachtel, S.; Parker, D.; Wickersham, M.; Zhang, D.; Brown, A.; Lauren, C.; Dowd, M.; West, E.; et al. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. mBio 2015, 6, e00289-15. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic granulomatous disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2017, 41, 139–157. [Google Scholar] [CrossRef]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kahlson, M.A.; Dixon, S.J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, I.; Neumayer, D.; Herrmann, M. Senescence of staphylococci: Using functional genomics to unravel the roles of ClpC ATPase during late stationary phase. Int. J. Med. Microbiol. 2010, 300, 130–136. [Google Scholar] [CrossRef]
- Chatterjee, I.; Becker, P.; Grundmeier, M.; Bischoff, M.; Somerville, G.A.; Peters, G.; Sinha, B.; Harraghy, N.; Proctor, R.A.; Herrmann, M. Staphylococcus aureus ClpC is required for stress resistance, aconitase activity, growth recovery, and death. J. Bacteriol. 2005, 187, 4488–4496. [Google Scholar] [CrossRef]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef]
- Kim, H.S.; Yosipovitch, G. The Skin Microbiota and Itch: Is There a Link? J. Clin. Med. 2020, 9, 1190. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.; Perricone, C.; Olivieri, G.; Cipriano, E.; Spinelli, F.R.; Valesini, G.; Conti, F. Staphylococcus aureus Nasal Carriage and Autoimmune Diseases: From Pathogenic Mechanisms to Disease Susceptibility and Phenotype. Int. J. Mol. Sci. 2019, 20, 5624. [Google Scholar] [CrossRef] [PubMed]
- Hülpüsch, C.; Tremmel, K.; Hammel, G.; Bhattacharyya, M.; de Tomassi, A.; Nussbaumer, T.; Neumann, A.U.; Reiger, M.; Traidl-Hoffmann, C. Skin pH-dependent Staphylococcus aureus abundance as predictor for increasing atopic dermatitis severity. Allergy 2020, 75, 2888–2898. [Google Scholar] [CrossRef]
- Strickland, I.; Hauk, P.J.; Trumble, A.E.; Picker, L.J.; Leung, D.Y. Evidence for superantigen involvement in skin homing of T cells in atopic dermatitis. J. Investig. Dermatol. 1999, 112, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Seiti Yamada Yoshikawa, F.; Feitosa de Lima, J.; Notomi Sato, M.; Álefe Leuzzi Ramos, Y.; Aoki, V.; Leao Orfali, R. Exploring the Role of Staphylococcus Aureus Toxins in Atopic Dermatitis. Toxins 2019, 11, 321. [Google Scholar] [CrossRef]
- Ng, C.Y.; Huang, Y.H.; Chu, C.F.; Wu, T.C.; Liu, S.H. Risks for Staphylococcus aureus colonization in patients with psoriasis: A systematic review and meta-analysis. Br. J. Dermatol. 2017, 177, 967–977. [Google Scholar] [CrossRef]
- Baker, B.S.; Laman, J.D.; Powles, A.; van der Fits, L.; Voerman, J.S.; Melief, M.J.; Fry, L. Peptidoglycan and peptidoglycan-specific Th1 cells in psoriatic skin lesions. J. Pathol. 2006, 209, 174–181. [Google Scholar] [CrossRef]
- Yilmaz, V.; Yentür, S.P.; Saruhan-Direskeneli, G. IL-12 and IL-10 polymorphisms and their effects on cytokine production. Cytokine 2005, 30, 188–194. [Google Scholar] [CrossRef]
- Schünke, H.; Göbel, U.; Dikic, I.; Pasparakis, M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat. Commun. 2021, 12, 5912. [Google Scholar] [CrossRef]
- Teufelberger, A.R.; Bröker, B.M.; Krysko, D.V.; Bachert, C.; Krysko, O. Staphylococcus aureus Orchestrates Type 2 Airway Diseases. Trends Mol. Med. 2019, 25, 696–707. [Google Scholar] [CrossRef]
- Krysko, O.; Teufelberger, A.; Van Nevel, S.; Krysko, D.V.; Bachert, C. Protease/antiprotease network in allergy: The role of Staphylococcus aureus protease-like proteins. Allergy 2019, 74, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Poddighe, D.; Vangelista, L. Staphylococcus aureus Infection and Persistence in Chronic Rhinosinusitis: Focus on Leukocidin ED. Toxins 2020, 12, 678. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Li, M.; Chen, K.; Zhu, H.; Tang, M.; Zhou, C.; Zheng, Y.; Wen, J.; Han, M.; Zhang, J.; et al. Necroptosis Underlies Neutrophilic Inflammation Associated with the Chronic Rhinosinusitis with Nasal Polyps (CRSwNP). J. Inflamm. Res. 2021, 14, 3969–3983. [Google Scholar] [CrossRef]
- Wang, B.F.; Cao, P.P.; Wang, Z.C.; Li, Z.Y.; Wang, Z.Z.; Ma, J.; Liao, B.; Deng, Y.K.; Long, X.B.; Xu, K.; et al. Interferon-γ-induced insufficient autophagy contributes to p62-dependent apoptosis of epithelial cells in chronic rhinosinusitis with nasal polyps. Allergy 2017, 72, 1384–1397. [Google Scholar] [CrossRef]
- Morgene, M.F.; Botelho-Nevers, E.; Grattard, F.; Pillet, S.; Berthelot, P.; Pozzetto, B.; Verhoeven, P.O. Staphylococcus aureus colonization and non-influenza respiratory viruses: Interactions and synergism mechanisms. Virulence 2018, 9, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Bianconi, I.; Baldan, R.; De Simone, M.; Riva, C.; Sipione, B.; Rossi, G.; Cirillo, D.M.; Bragonzi, A. Staphylococcus aureus Impacts Pseudomonas aeruginosa Chronic Respiratory Disease in Murine Models. J. Infect. Dis. 2018, 217, 933–942. [Google Scholar] [CrossRef]
- Stelzner, K.; Boyny, A.; Hertlein, T.; Sroka, A.; Moldovan, A.; Paprotka, K.; Kessie, D.; Mehling, H.; Potempa, J.; Ohlsen, K.; et al. Intracellular Staphylococcus aureus employs the cysteine protease staphopain A to induce host cell death in epithelial cells. PLoS Pathog. 2021, 17, e1009874. [Google Scholar] [CrossRef]
- Goerke, C.; Wolz, C. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int. J. Med. Microbiol. 2010, 300, 520–525. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.L.; Settembre, C.; Gavina, M.; Raia, V.; Ballabio, A.; Maiuri, L. Cystic fibrosis: A disorder with defective autophagy. Autophagy 2011, 7, 104–106. [Google Scholar] [CrossRef]
- Edwards, L.A.; O’Neill, C.; Furman, M.A.; Hicks, S.; Torrente, F.; Pérez-Machado, M.; Wellington, E.M.; Phillips, A.D.; Murch, S.H. Enterotoxin-producing staphylococci cause intestinal inflammation by a combination of direct epithelial cytopathy and superantigen-mediated T-cell activation. Inflamm. Bowel Dis. 2012, 18, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Stelzner, K.; Winkler, A.C.; Liang, C.; Boyny, A.; Ade, C.P.; Dandekar, T.; Fraunholz, M.J.; Rudel, T. Intracellular Staphylococcus aureus Perturbs the Host Cell Ca(2+) Homeostasis To Promote Cell Death. mBio 2020, 11, e02250-20. [Google Scholar] [CrossRef] [PubMed]
- Argudín, M.; Mendoza, M.C.; Rodicio, M.R. Food poisoning and Staphylococcus aureus enterotoxins. Toxins 2010, 2, 1751–1773. [Google Scholar] [CrossRef]
- Fisher, E.L.; Otto, M.; Cheung, G.Y.C. Basis of Virulence in Enterotoxin-Mediated Staphylococcal Food Poisoning. Front. Microbiol. 2018, 9, 436. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.D. The morphologic consequences of acute exogenous (Staphylococcic) gastroenteritis on the gastric mucosa. Gastroenterology 1951, 19, 462–475. [Google Scholar] [CrossRef]
- Alber, G.; Scheuber, P.H.; Reck, B.; Sailer-Kramer, B.; Hartmann, A.; Hammer, D.K. Role of substance P in immediate-type skin reactions induced by staphylococcal enterotoxin B in unsensitized monkeys. J. Allergy Clin. Immunol. 1989, 84, 880–885. [Google Scholar] [CrossRef]
- Jett, M.; Brinkley, W.; Neill, R.; Gemski, P.; Hunt, R. Staphylococcus aureus enterotoxin B challenge of monkeys: Correlation of plasma levels of arachidonic acid cascade products with occurrence of illness. Infect. Immun. 1990, 58, 3494–3499. [Google Scholar] [CrossRef]
- Scheuber, P.H.; Golecki, J.R.; Kickhöfen, B.; Scheel, D.; Beck, G.; Hammer, D.K. Skin reactivity of unsensitized monkeys upon challenge with staphylococcal enterotoxin B: A new approach for investigating the site of toxin action. Infect. Immun. 1985, 50, 869–876. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Blake, P.A. Staphylococcal food poisoning in the United States. New facts and old misconceptions. JAMA 1984, 251, 487–489. [Google Scholar] [CrossRef]
- Pérez-Bosque, A.; Moretó, M. A rat model of mild intestinal inflammation induced by Staphylococcus aureus enterotoxin B. Proc. Nutr. Soc. 2010, 69, 447–453. [Google Scholar] [CrossRef]
- Kadariya, J.; Smith, T.C.; Thapaliya, D. Staphylococcus aureus and staphylococcal food-borne disease: An ongoing challenge in public health. BioMed Res. Int. 2014, 2014, 827965. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.; Fujikawa, H. Effects of Lactic Acid and Salt on Enterotoxin A Production and Growth of Staphylococcus aureus. J. Food Sci. 2019, 84, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Medina, C.B.; Barron, B.J.; Karvelyte, L.; Aaes, T.L.; Lambertz, I.; Perry, J.S.A.; Mehrotra, P.; Gonçalves, A.; Lemeire, K.; et al. Microbes exploit death-induced nutrient release by gut epithelial cells. Nature 2021, 596, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Ooi, J.D.; Jiang, J.H.; Eggenhuizen, P.J.; Chua, L.L.; van Timmeren, M.; Loh, K.L.; O’Sullivan, K.M.; Gan, P.Y.; Zhong, Y.; Tsyganov, K.; et al. A plasmid-encoded peptide from Staphylococcus aureus induces anti-myeloperoxidase nephritogenic autoimmunity. Nat. Commun. 2019, 10, 3392. [Google Scholar] [CrossRef]
- Farinelli, G.; Jofra Hernandez, R.; Rossi, A.; Ranucci, S.; Sanvito, F.; Migliavacca, M.; Brombin, C.; Pramov, A.; Di Serio, C.; Bovolenta, C.; et al. Lentiviral Vector Gene Therapy Protects XCGD Mice from Acute Staphylococcus aureus Pneumonia and Inflammatory Response. Mol. Ther. 2016, 24, 1873–1880. [Google Scholar] [CrossRef]
- Harfi, I.; D’Hondt, S.; Sariban, E. iPLA(2) Activation Mediates Granular Exocytosis and Corrects Microbicidal Defects in ROS-Deficient and CGD Human Neutrophils. J. Clin. Immunol. 2019, 39, 486–493. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Cao, Y.; Mo, A.; Peng, Q. Potentials of nanotechnology in treatment of methicillin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2021, 213, 113056. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef]
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [Google Scholar] [CrossRef]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane Microdomain Disassembly Inhibits MRSA Antibiotic Resistance. Cell 2017, 171, 1354–1367.e20. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, prevalence, and management of MRSA bacteremia across patient populations-a review of recent developments in MRSA management and treatment. Crit. Care 2017, 21, 211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, X.; Xiu, H.; Zhang, Z.; Zhang, K.; Cai, J.; Cai, Z.; Chen, Z.; Zhang, Z.; Cui, W.; et al. The attenuation of Th1 and Th17 responses via autophagy protects against methicillin-resistant Staphylococcus aureus-induced sepsis. Microbes Infect. 2021, 23, 104833. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Ulrich, A.; Suliman, H.B.; Piantadosi, C.A. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic. Biol. Med. 2015, 78, 179–189. [Google Scholar] [CrossRef]
- Myles, I.A.; Fontecilla, N.M.; Valdez, P.A.; Vithayathil, P.J.; Naik, S.; Belkaid, Y.; Ouyang, W.; Datta, S.K. Signaling via the IL-20 receptor inhibits cutaneous production of IL-1β and IL-17A to promote infection with methicillin-resistant Staphylococcus aureus. Nat. Immunol. 2013, 14, 804–811. [Google Scholar] [CrossRef]
- Singh, N.; Armstrong, D.G.; Lipsky, B.A. Preventing foot ulcers in patients with diabetes. JAMA 2005, 293, 217–228. [Google Scholar] [CrossRef]
- Gemechu, F.W.; Seemant, F.; Curley, C.A. Diabetic foot infections. Am. Fam. Physician 2013, 88, 177–184. [Google Scholar]
- Lavery, L.A.; Fontaine, J.L.; Bhavan, K.; Kim, P.J.; Williams, J.R.; Hunt, N.A. Risk factors for methicillin-resistant Staphylococcus aureus in diabetic foot infections. Diabet. Foot Ankle 2014, 5, 23575. [Google Scholar] [CrossRef][Green Version]
- Taha, A.B. Relationship and susceptibility profile of Staphylococcus aureus infection diabetic foot ulcers with Staphylococcus aureus nasal carriage. Foot 2013, 23, 11–16. [Google Scholar] [CrossRef]
- Lipsky, B.A.; Berendt, A.R.; Deery, H.G.; Embil, J.M.; Joseph, W.S.; Karchmer, A.W.; LeFrock, J.L.; Lew, D.P.; Mader, J.T.; Norden, C.; et al. Diagnosis and treatment of diabetic foot infections. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2004, 39, 885–910. [Google Scholar] [CrossRef]
- Sotto, A.; Lina, G.; Richard, J.L.; Combescure, C.; Bourg, G.; Vidal, L.; Jourdan, N.; Etienne, J.; Lavigne, J.P. Virulence potential of Staphylococcus aureus strains isolated from diabetic foot ulcers: A new paradigm. Diabetes Care 2008, 31, 2318–2324. [Google Scholar] [CrossRef] [PubMed]
- Munro, P.; Benchetrit, M.; Nahori, M.A.; Stefani, C.; Clément, R.; Michiels, J.F.; Landraud, L.; Dussurget, O.; Lemichez, E. The Staphylococcus aureus epidermal cell differentiation inhibitor toxin promotes formation of infection foci in a mouse model of bacteremia. Infect. Immun. 2010, 78, 3404–3411. [Google Scholar] [CrossRef] [PubMed]
- Messad, N.; Landraud, L.; Canivet, B.; Lina, G.; Richard, J.L.; Sotto, A.; Lavigne, J.P.; Lemichez, E. Distribution of edin in Staphylococcus aureus isolated from diabetic foot ulcers. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2013, 19, 875–880. [Google Scholar] [CrossRef]
- Bader, M.S. Diabetic foot infection. Am. Fam. Physician 2008, 78, 71–79. [Google Scholar] [PubMed]
- Uçkay, I.; Kressmann, B.; Malacarne, S.; Toumanova, A.; Jaafar, J.; Lew, D.; Lipsky, B.A. A randomized, controlled study to investigate the efficacy and safety of a topical gentamicin-collagen sponge in combination with systemic antibiotic therapy in diabetic patients with a moderate or severe foot ulcer infection. BMC Infect. Dis. 2018, 18, 361. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Marinelli, F.; Marcone, G.L. Old and New Glycopeptide Antibiotics: Action and Resistance. Antibiotics. 2014, 3, 572–594. [Google Scholar] [CrossRef]
- Mottola, C.; Matias, C.S.; Mendes, J.J.; Melo-Cristino, J.; Tavares, L.; Cavaco-Silva, P.; Oliveira, M. Susceptibility patterns of Staphylococcus aureus biofilms in diabetic foot infections. BMC Microbiol. 2016, 16, 119. [Google Scholar] [CrossRef]
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta 2016, 1858, 1044–1060. [Google Scholar] [CrossRef]
- Santos, R.; Gomes, D.; Macedo, H.; Barros, D.; Tibério, C.; Veiga, A.S.; Tavares, L.; Castanho, M.; Oliveira, M. Guar gum as a new antimicrobial peptide delivery system against diabetic foot ulcers Staphylococcus aureus isolates. J. Med. Microbiol. 2016, 65, 1092–1099. [Google Scholar] [CrossRef]
- Touzel, R.E.; Sutton, J.M.; Wand, M.E. Establishment of a multi-species biofilm model to evaluate chlorhexidine efficacy. J. Hosp. Infect. 2016, 92, 154–160. [Google Scholar] [CrossRef]
- Bonez, P.C.; Dos Santos Alves, C.F.; Dalmolin, T.V.; Agertt, V.A.; Mizdal, C.R.; Flores Vda, C.; Marques, J.B.; Santos, R.C.; Anraku de Campos, M.M. Chlorhexidine activity against bacterial biofilms. Am. J. Infect. Control 2013, 41, e119–e122. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ruza, D.; Cunha, E.; Tavares, L.; Oliveira, M. Diabetic foot infections: Application of a nisin-biogel to complement the activity of conventional antibiotics and antiseptics against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0220000. [Google Scholar] [CrossRef] [PubMed]
- Zenão, S.; Aires, A.; Dias, C.; Saavedra, M.J.; Fernandes, C. Antibacterial potential of Urtica dioica and Lavandula angustifolia extracts against methicillin resistant Staphylococcus aureus isolated from diabetic foot ulcers. J. Herb. Med. 2017, 10, 53–58. [Google Scholar] [CrossRef]




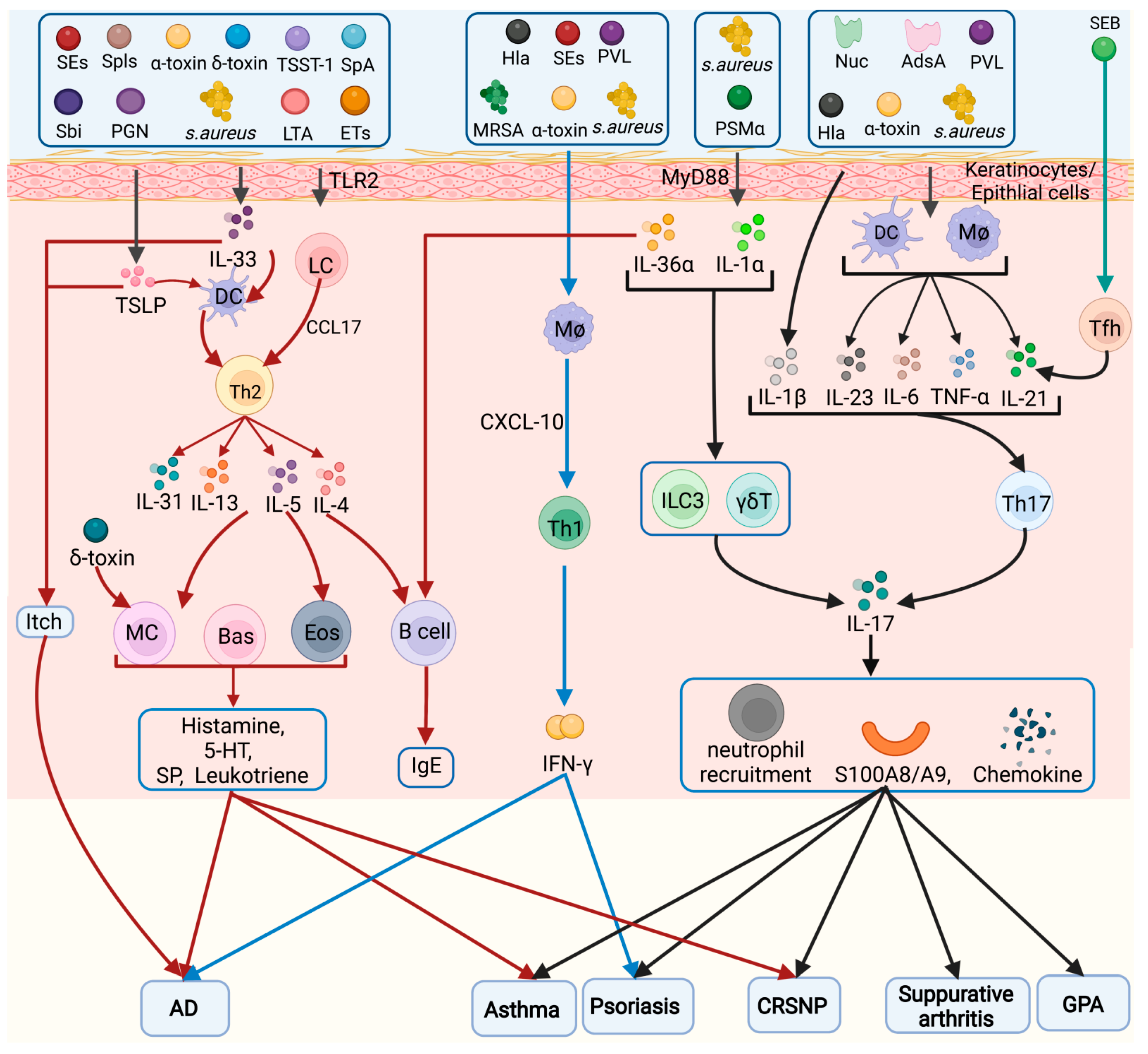{kind=link}
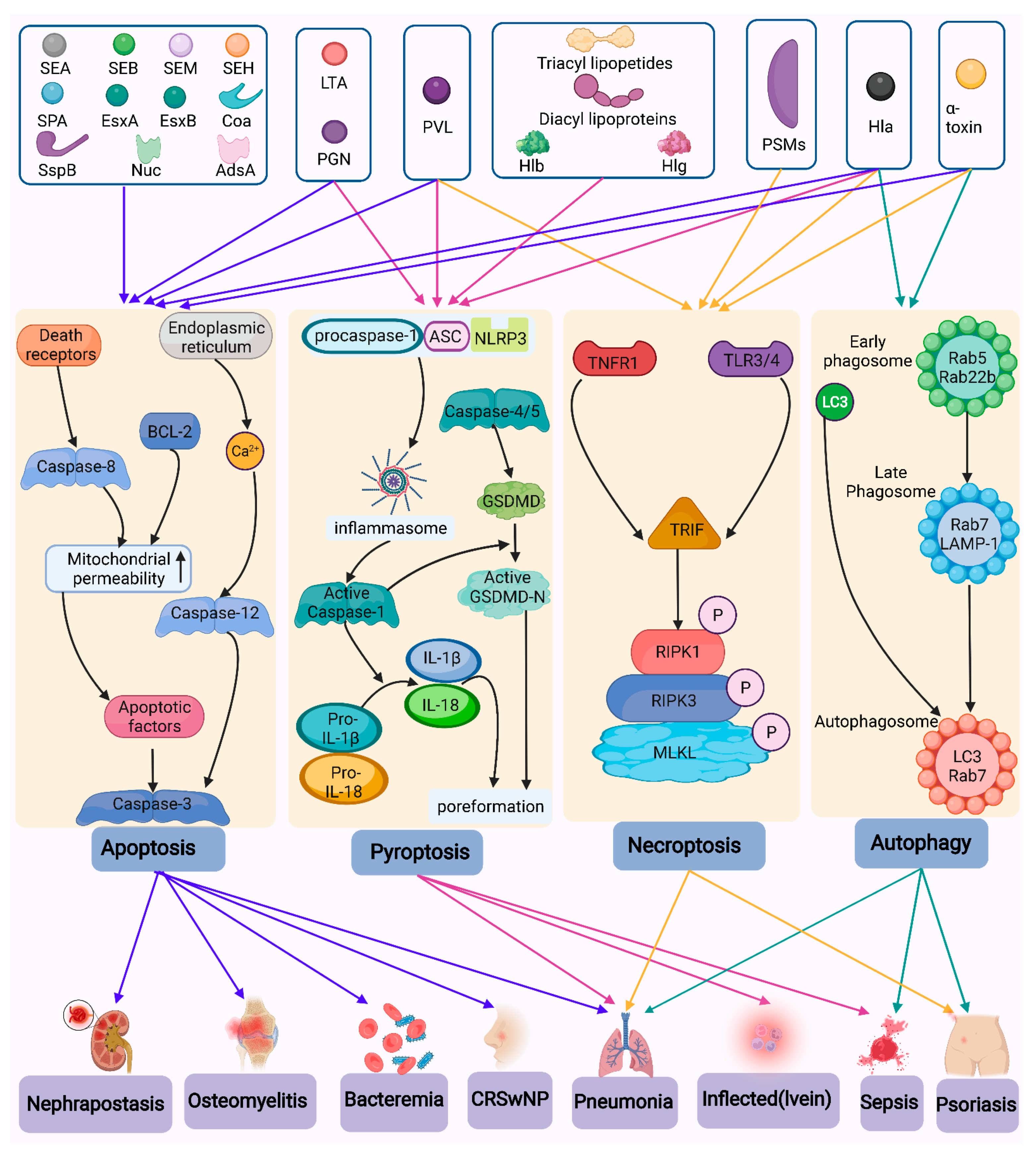{kind=link}
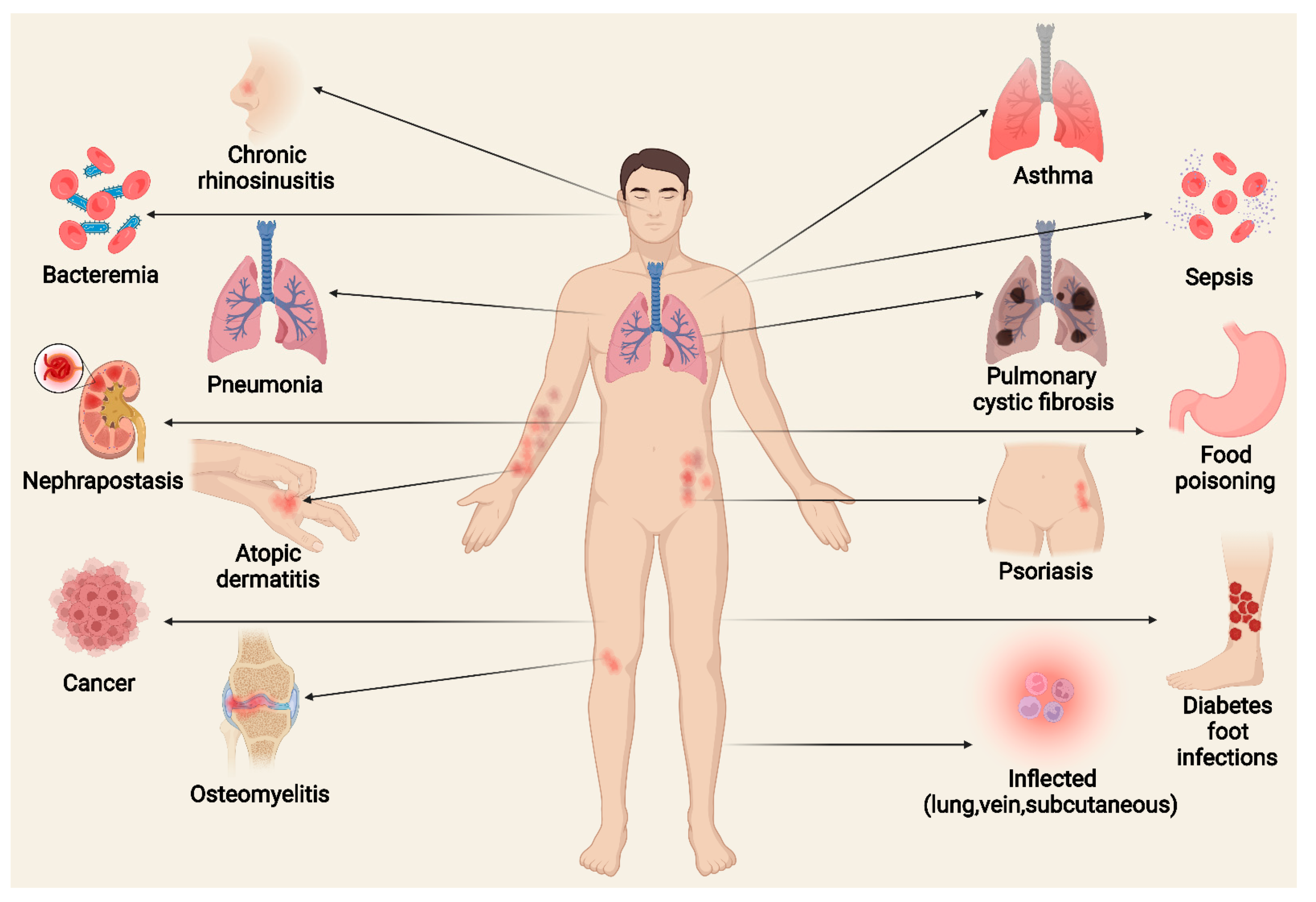{kind=link}
| Abbreviation | Full Name | Abbreviation | Full Name |
|---|---|---|---|
| 5-HT | 5-hydroxytryptamine | MMPs | Matrix metal proteinases |
| AAV | Associated vasculitis | MPO | Myeloperoxidase |
| Atopic dermatitis | MRSA | Methicillin-resistant S. aureus | |
| AdsA | Adenosine synthase A | MSCRAM | Mucin matrix molecules |
| Agr | Auxotrophic gene regulator | MSCRAMM | Microbial surface components recognizing adhesive matrix molecules |
| AMP | Antimicrobial peptide | NETs | Neutrophil extracellular traps |
| AMs | Alveolar macrophages | NF-κB | Nuclear factor kappa B |
| ANCA | Antineutrophil cytoplasmic antibody | NK | Natural killer cells |
| BCL-6 | B-cell lymphoma-6 | NO | Nitric oxide |
| BMECs | Bovine mammary epithelial cells | Nuc | Nuclease |
| CF | Cystic fibrosis | PA | Protein A |
| CFTR | Conductance regulator | PAMPs | Pathogen-associated molecular patterns |
| CGD | Chronic granulomatous disease | PBP2a | Penicillin-binding protein |
| Coa | Coagulase | PCD | Programmed cell death |
| CpG | Cytosine–phosphate–guanine | PD-1 | Programmed death 1 |
| CRSwNP | Chronic rhinosinusitis with nasal polyposis | PFTs | Pore-forming toxins |
| CWA | Cell wall-anchored | PGN | Peptidoglycan |
| CXCR5 | CXC chemokine receptor 5 | PI3K | Phosphatidylinositol 3-kinase |
| DAMPs | Damage-associated molecular patterns | PIP | Phosphatidylinositol phosphate |
| DCs | Dendritic cells | PMN | Polymorphonuclear leukocytes |
| DFI | Diabetic foot infection | PRRs | Pattern recognition receptors |
| Dsg1 | Desmoglein 1 | PSMs | Phenol-soluble modulins |
| Eap | Extracellular adhesion protein | PVL | Panton–Valentine leukocidin |
| ECP | Eosinophil cationic protein | RIPK1 | Receptor-interacting protein kinase 1 |
| EDIN | Epidermal differentiation inhibitor | ROS | Reactive oxygen specie |
| eDNA | Extracellular DNA | S. aureus | Staphylococcus aureus |
| EPO | Eosinophil peroxidase | SAgs | Superantigens |
| ESS | Early-secretion antigen-6 secretion system | Sbi | Second immunoglobulin-binding protein |
| ETs | Exfoliative toxins | SEs | Staphyloccocal enterotoxins |
| FPR2 | Formylpeptide receptor 2 | Socs3 | Suppressor of cytokine signaling 3 |
| GC | Germinal center | SpA | Staphylococcal protein A |
| G-CSF | Granulocyte colony-stimulating factor | Spls | Serine protease-like proteins |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor | SspB | Staphopain B |
| GPA | Granulomatous polyangiitis | SSSS | Staphylococcal scalded skin syndrome |
| Hla | α-hemolysin | TCA | Tricarboxylic acid |
| Hlb | β-hemolysin | Tem | Memory T |
| Hlg | γ-hemolysin | TER | Transmembrane electrical |
| I-CAM | Intercellular adhesion molecules | Tfh | Follicular helper T |
| IFN-γ | Interferon γ | TGF-β | Transforming growth factor β |
| lgE | Immunoglobulin-E | Th | T helper |
| IL | Interleukin | TLR | Toll-like receptor |
| ILCs | Innate lymphoid cells | TNF | Tumor necrosis factor |
| iPLA2 | Inhibition of calcium-dependent phospholipase A2 | TNFR1 | Tumor necrosis factor receptor 1 |
| JNK | Jun N-terminal kinase | Treg | Regulatory T |
| LCs | Langerhans cells | TSST-1 | Toxic shock syndrome toxin 1 |
| LTA | Lipoteichoic acid | V-CAM | Vascular cell adhesion molecules |
| MDP | Muramyl dipeptide | VEGF | Vascular endothelial growth factor |
| MLKL | Mixed-spectrum kinase-like protein |
| SEs | Functions |
|---|---|
| SEA | Anti-protein hydrolase, supercarcinogenic and emetic toxin, causes food poisoning, but no diarrheal activity |
| SEB | Increase in the number of T lymphocytes, as well as the concentration of pro-inflammatory cytokines and inflammatory mediators in the intestinal mucosa; increase mucosal permeability and intestinal secretion |
| SEC | Supercarcinogenic and emetic toxin, but no diarrheal activity |
| SEG | Reversible destruction of intestinal cell ultrastructure |
| SEH | Causes food poisoning |
| SEI | Reversibly disrupts enterocyte ultrastructure and exhibits weak emetic activity |
| SEM | Strong emetic potential |
| SEO | Has emetic activity |
| SEQ | Significantly stable for heat treatment and digestive enzyme degradation, and exhibits significant supercarcinogenic and emetic activity |
| Classification | Virulence Factors | Cell Types | Cell Death Mechanisms | Diseases | Refs. | |
|---|---|---|---|---|---|---|
| Secreted Virulence factors | PFT | Hla | Keratinocytes Macrophages | Pyroptosis Necroptosis Autophagy Apoptosis | [43,143,144,145,146] | |
| Hlb | Pyroptosis | [34] | ||||
| Hlg | Pyroptosis | [35] | ||||
| α-Toxin | Th1, Th2 Neutrophils Macrophages | Apoptosis Necroptosis Autophagy | AD GPA Pneumonia Chronic sinusitis sepsis | [47,48,49,50,51,52,53] | ||
| PVL | DCs Macrophages | Apoptosis | Pneumonia | [58,62,63,64,65,66] | ||
| PSMs | PSMα | Keratinocytes Neutrophils | Necroptosis | AD Pneumonia | [5,67,68,69,70] | |
| PSMβ | Neutrophil | [73] | ||||
| PSMγ (δ toxin) | Mast cells Keratinocytes | AD Diarrhea | [74,75,147] | |||
| SAgs | SEA | B cells CD4+ T cells Keratinocytes Eosinophils | Apoptosis | AD Asthma Food poisoning | [13,91,92,148,149] | |
| SEB | Th1, Th2 Eosinophils Proximal renal tubular epithelial cells | Apoptosis | Chronic sinusitis AD Asthma | [15,92,93,149] | ||
| SEC | B cells | AD Food poisoning | [13,150] | |||
| SEG | Food poisoning | [95] | ||||
| SEH | Bovine mammary gland epithelial cells | Apoptosis | Food poisoning | [96] | ||
| SEI | Food poisoning | [95,97] | ||||
| SEM | Apoptosis | Food poisoning | [98,99] | |||
| SEO | Food poisoning | [97] | ||||
| SEQ | Food poisoning | [100] | ||||
| TSST-1 | B cells Keratinocytes | AD | [13,50] | |||
| Proteases | ETs | AD | [31] | |||
| Spls | T cells | Asthma SSSS | [78] [76] | |||
| SspB | Macrophages | Apoptosis | [85] | |||
| Secreted Enzymes (proteins) | EsxA, EsxB | Apoptosis | [101,102] | |||
| Coa | Apoptosis | [104] | ||||
| Eap | T cells Lung epithelial cells | Psoriasis CF | [109] | |||
| Nuc | Neutrophils Macrophages | Apoptosis | Bacteraemia Nephrapostasis | [108] | ||
| AdsA | Neutrophils Macrophages | Apoptosis | Bacteraemia Nephrapostasis | [107] | ||
| Cell wall components | Surface Proteins (CWA) | Sbi | Keratinocytes | AD | [119] | |
| SpA | Tumor cells | Apoptosis | [115,116,117,118] | |||
| PAMPs | Triacyl lipopetides | Pyroptosis | Sepsis | [69,124] | ||
| Diacyl lipoproteins | Pyroptosis | Sepsis | [69,151] | |||
| LTA | Neutrophils Macrophages B cell | Pyroptosis | Food poisoning | [23,125,126,127] | ||
| PGN | LCs Mast cells Macrophages Macrophages | Autophagy | AD Sepsis Pneumonia Psoriasis | [17,130,131,132,133,140,141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Zhang, J.; He, Y.; Lv, Z.; Liang, Z.; Chen, J.; Li, P.; Liu, J.; Yang, H.; Tao, A.; et al. Exploring the Role of Staphylococcus aureus in Inflammatory Diseases. Toxins 2022, 14, 464. https://doi.org/10.3390/toxins14070464
Chen H, Zhang J, He Y, Lv Z, Liang Z, Chen J, Li P, Liu J, Yang H, Tao A, et al. Exploring the Role of Staphylococcus aureus in Inflammatory Diseases. Toxins. 2022; 14(7):464. https://doi.org/10.3390/toxins14070464
Chicago/Turabian StyleChen, Huanquan, Junyan Zhang, Ying He, Zhuoyi Lv, Zhengtong Liang, Jianze Chen, Peishan Li, Jiawei Liu, Hongchen Yang, Ailin Tao, and et al. 2022. "Exploring the Role of Staphylococcus aureus in Inflammatory Diseases" Toxins 14, no. 7: 464. https://doi.org/10.3390/toxins14070464
APA StyleChen, H., Zhang, J., He, Y., Lv, Z., Liang, Z., Chen, J., Li, P., Liu, J., Yang, H., Tao, A., & Liu, X. (2022). Exploring the Role of Staphylococcus aureus in Inflammatory Diseases. Toxins, 14(7), 464. https://doi.org/10.3390/toxins14070464