Characterization of Two Dehydrogenases from Gluconobacter oxydans Involved in the Transformation of Patulin to Ascladiol



Abstract
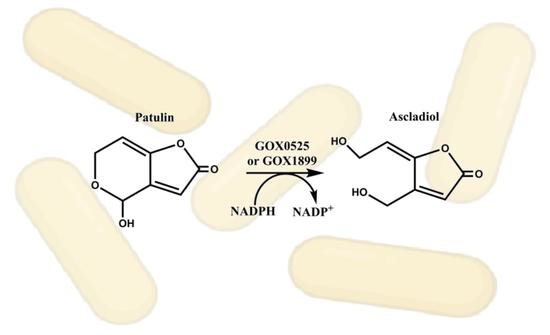
1. Introduction
2. Results
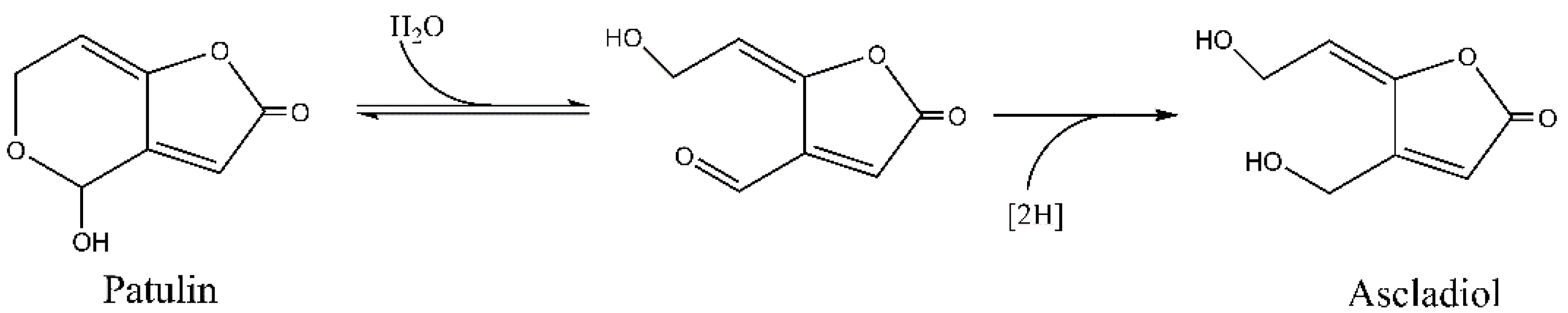
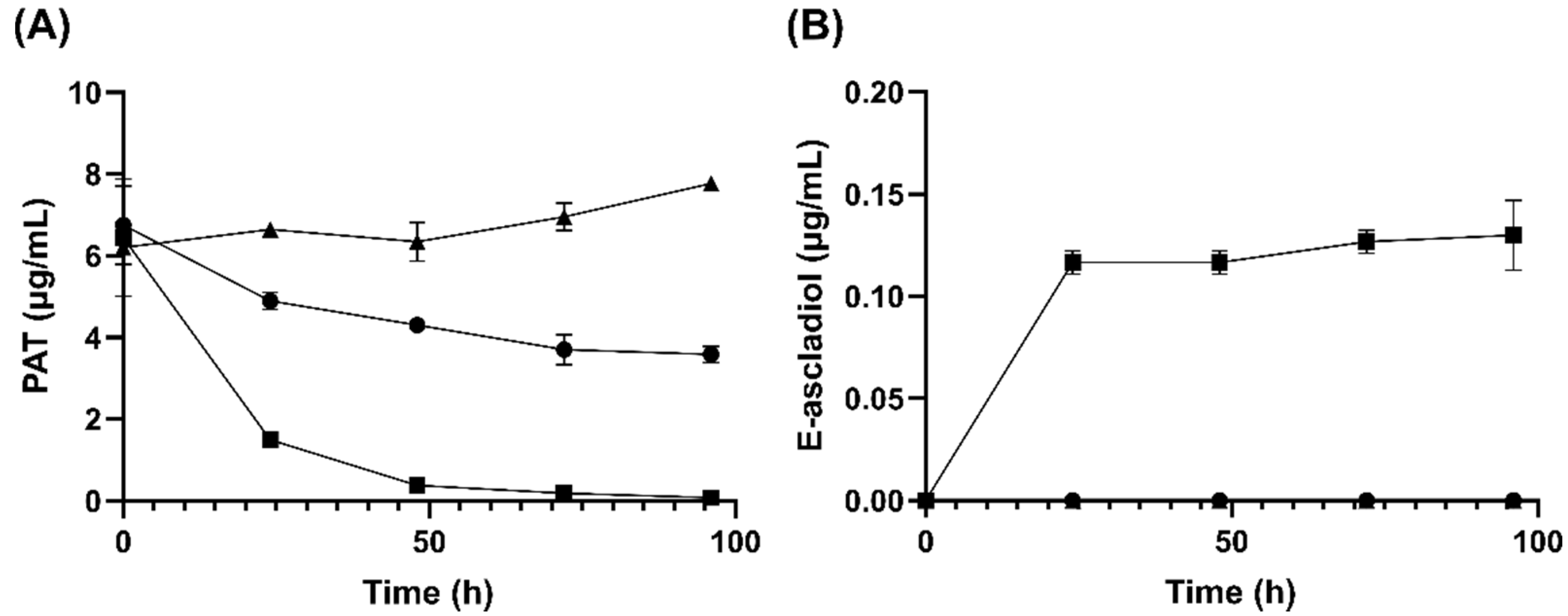
2.1. Analysis of Patulin Transformation by G. oxydans 621
2.2. Protein Identification
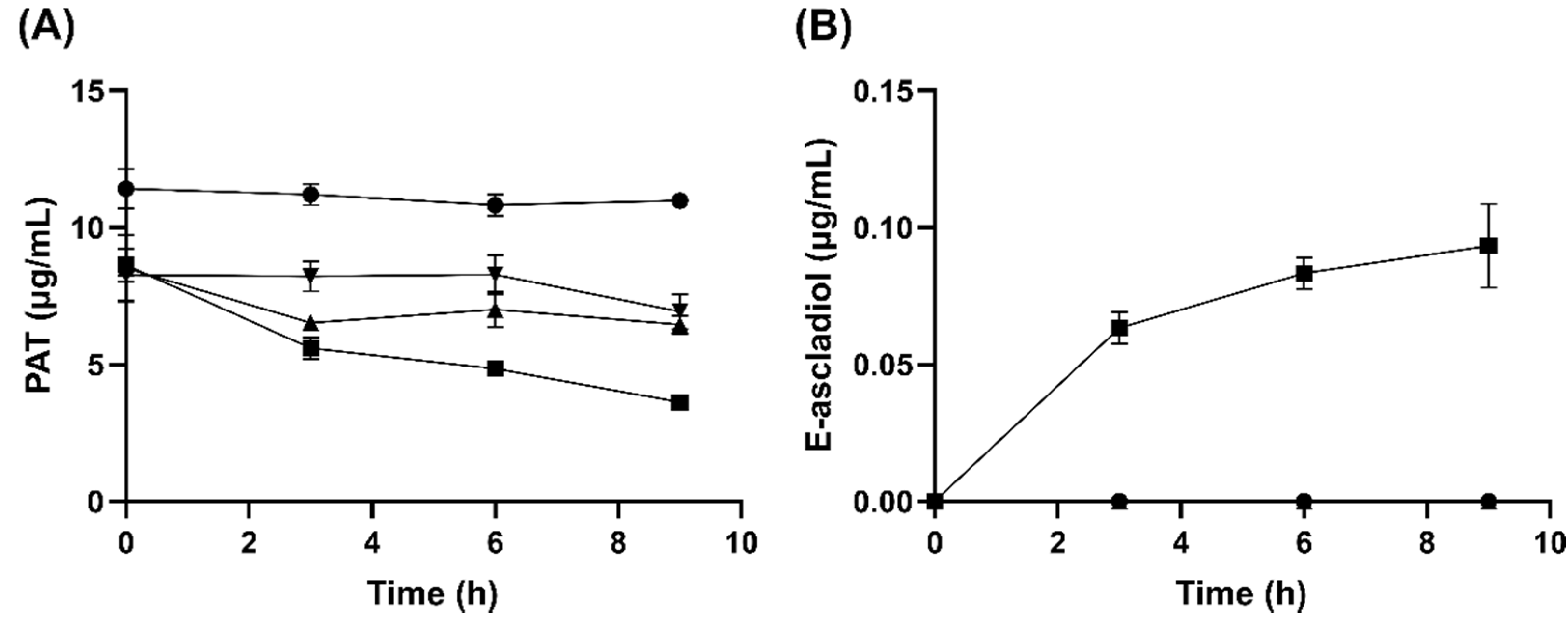
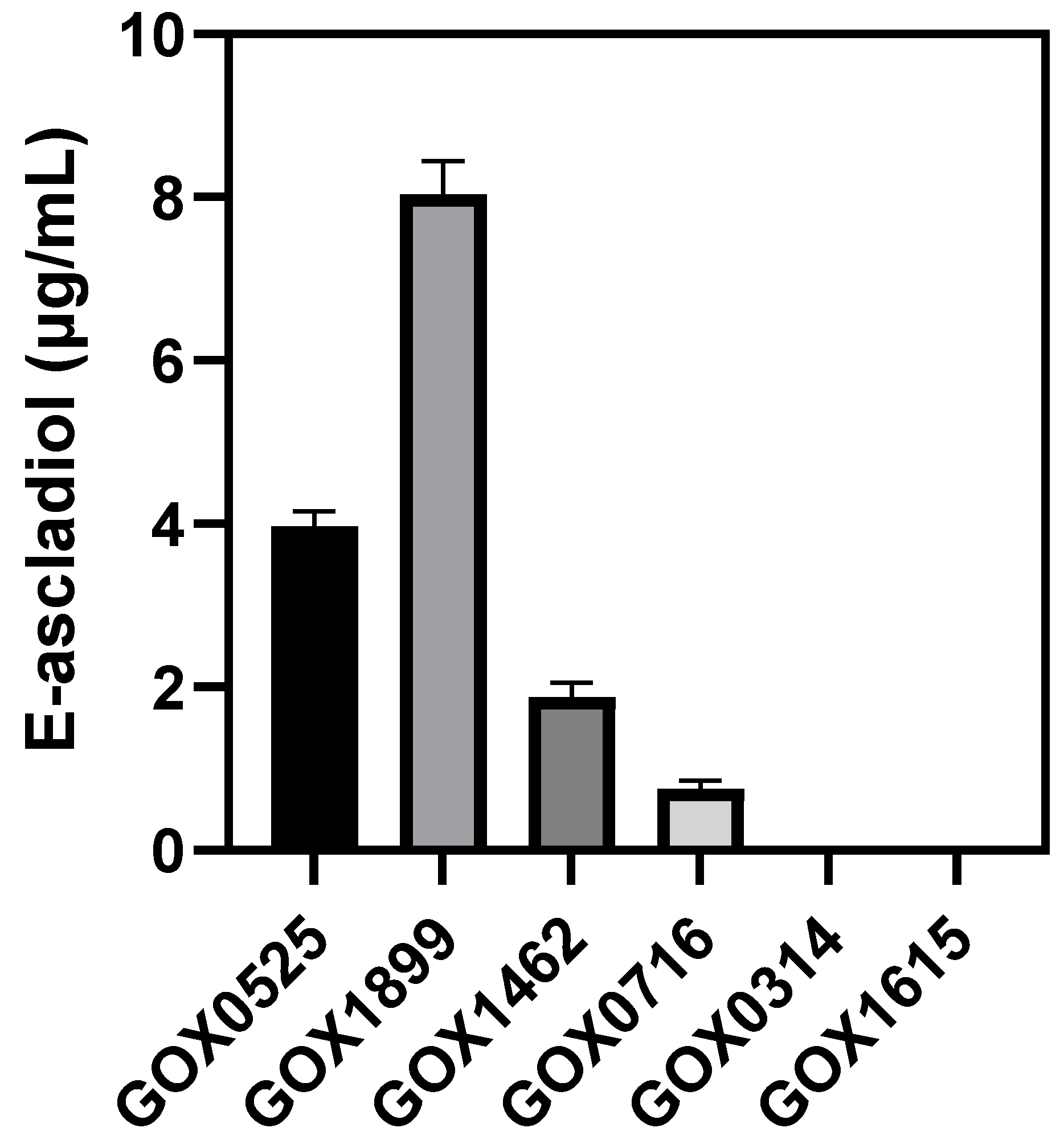
2.3. Analysis of Ascladiol Formation by Whole Recombinant E. coli Cells
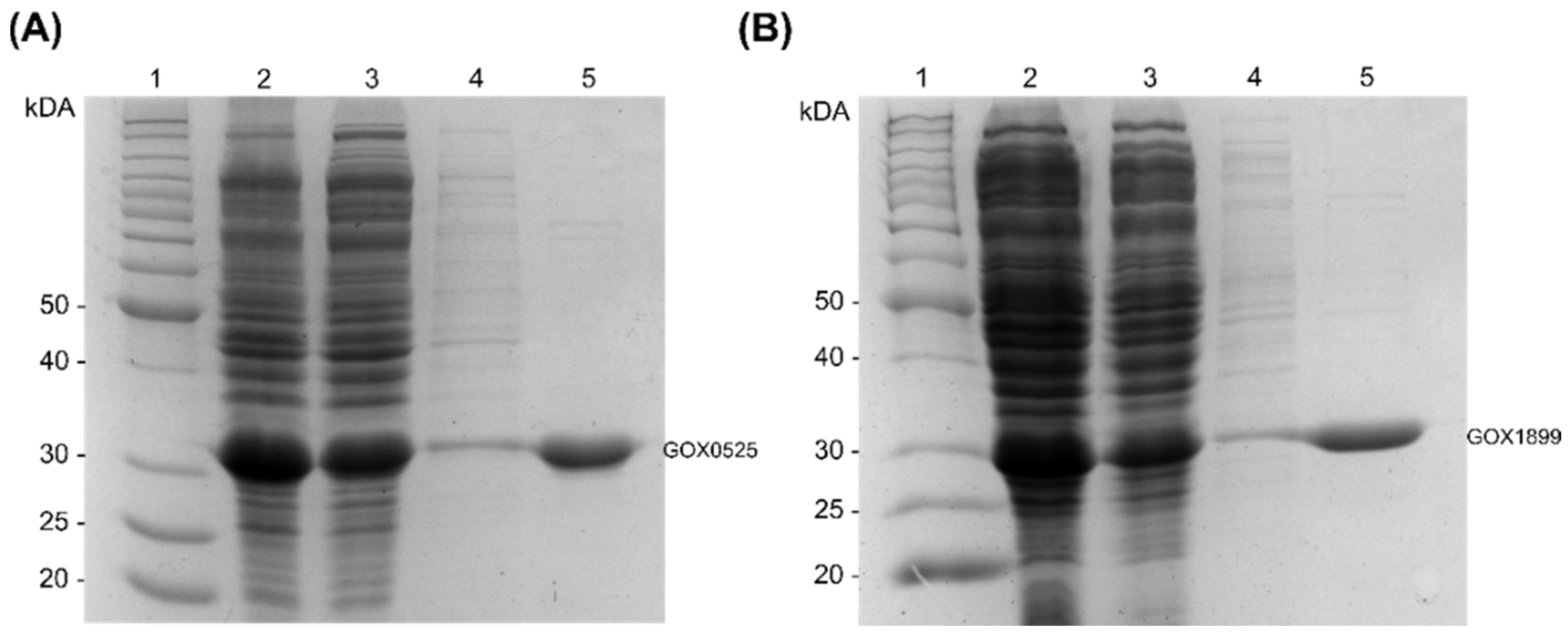
2.4. Purification of Recombinant Enzymes That Transform Patulin to E-Ascladiol
2.5. Kinetic Analysis
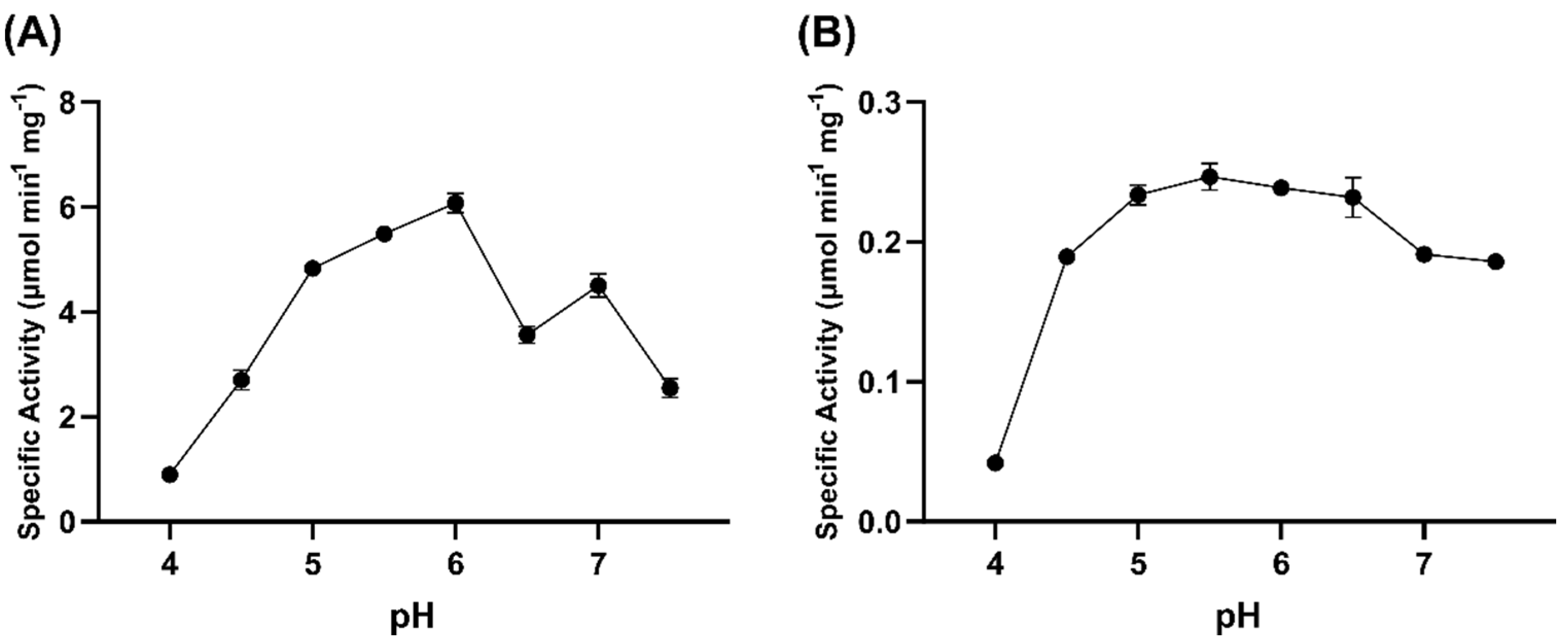
2.6. pH Dependence of GOX0525 and GOX1899 Activity
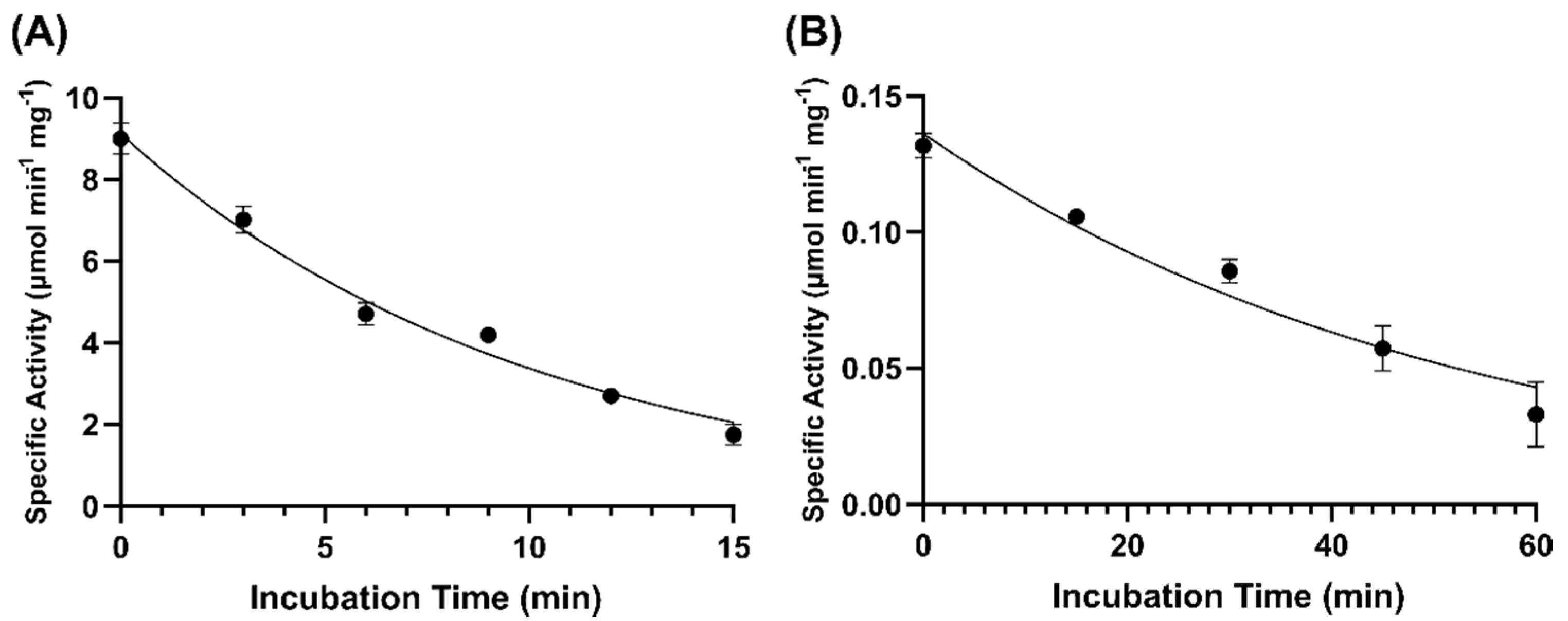
2.7. Thermostability
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Chemicals
4.3. High-Performance Liquid Chromatography and Mass Spectrometry
4.4. Screening for Whole G. oxydans Cells for Patulin Transformation Activity
4.5. Evaluating Patulin Transformation Activity in G. oxydans Cell Lysates
4.6. Anion Exchange Chromatography and Analysis of Fractions for Patulin Transformation Activity
4.7. Identification of Proteins in Anion Exchange Chromatography Fractions by Mass-Spectrometry
4.8. Gene Cloning
4.9. Recombinant E. coli Patulin Transformation Assay
4.10. Expression and Purification of Recombinant G. oxydans Genes in E. coli LOBSTR-BL21 (DE3)
4.11. Assessment of Enzyme Purity and Concentration
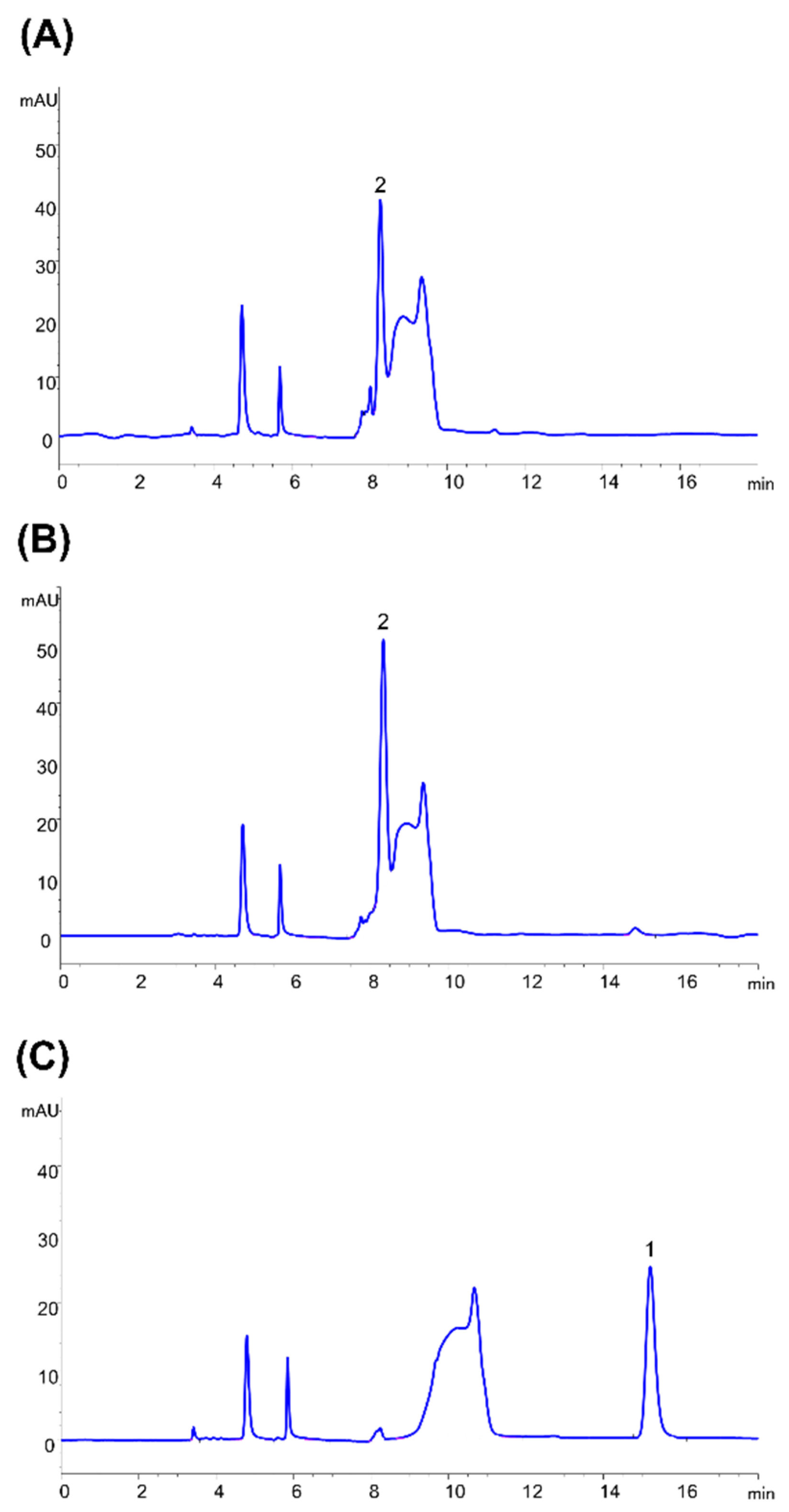
4.12. Enzyme Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, M.S.; Dutton, M.F.; Harding, K. Production and Degradation of Patulin by Paecilomyces Species, a Common Contaminant of Silage. J. Sci. Food Agric. 1979, 30, 229–232. [Google Scholar] [CrossRef]
- Lovett, J.; Thompson, R.G. Patulin Producution by Species of Aspergillus and Penicillium at 1.7, 7.2 and 12.8 C. J. Food Prot. 1978, 41, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.L.; Beuchat, L.R.; Worthington, R.E. Patulin Production by Byssochlamys Spp. in Fruit Juices. J. Food Sci. 1977, 34, 791–796. [Google Scholar] [CrossRef]
- Roland, J.O.; Beuchat, L.R. Biomass and Patulin Production by Byssochlamys nivea in Apple Juice as Affected by Sorbate, Benzoate, SO2 and Temperature. J. Food Sci. 1984, 49, 402–406. [Google Scholar] [CrossRef]
- Fliege, R.; Metzler, M. Electrophilic Properties of Patulin. Adduct Structures and Reaction Pathways with 4-Bromothiophenol and Other Model Nucleophiles. Chem. Res. Toxicol. 2000, 13, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Bullerman, L.B.; Olivigni, F.J. Mycotoxin Producing-Potential of Molds Isolated From Cheddar Cheese. J. Food Sci. 1974, 39, 1166–1168. [Google Scholar] [CrossRef]
- Harwig, J.; Blanchfield, B.J.; Scott, P.M. Patulin Production by Penicillium roqueforti Thom from Grape. Can. Inst. Food Sci. Technol. J. 1978, 11, 149–151. [Google Scholar] [CrossRef]
- Reiss, J. Prevention of the Formation of Mycotoxins in Whole Wheat Bread by Citric Acid and Lactic Acid (Mycotoxins in Foodstuffs. IX). Experientia 1976, 32, 168–169. [Google Scholar] [CrossRef]
- Rychlik, M.; Schieberle, P. Quantification of the Mycotoxin Patulin by a Stable Isotope Dilution Assay. J. Agric. Food Chem. 1999, 47, 3749–3755. [Google Scholar] [CrossRef]
- Scott, P.M.; Miles, W.F.; Toft, P.; Dubé, J.G. Occurrence of Patulin in Apple Juice. J. Agric. Food Chem. 1972, 20, 450–451. [Google Scholar] [CrossRef]
- Zouaoui, N.; Sbaii, N.; Bacha, H.; Abid-Essefi, S. Occurrence of Patulin in Various Fruit Juice Marketed in Tunisia. Food Control 2015, 51, 356–360. [Google Scholar] [CrossRef]
- de Sylos, C.M.; Rodriguez-Amaya, D.B. Incidence of Patulin in Fruits and Fruit Juices Marketed in Campinas, Brazil. Food Addit. Contam. 1999, 16, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Gökmen, V.; Acar, J. Incidence of Patulin in Apple Juice Concentrates Produced in Turkey. J. Chromatogr. A 1998, 815, 99–102. [Google Scholar] [CrossRef]
- Leggott, N.L.; Shephard, G.S. Patulin in South African Commercial Apple Products. Food Control 2001, 12, 73–76. [Google Scholar] [CrossRef]
- Ritieni, A. Patulin in Italian Commercial Apple Products. J. Agric. Food Chem. 2003, 51, 6086–6090. [Google Scholar] [CrossRef]
- Tangni, E.K.; Theys, R.; Mignolet, E.; Maudoux, M.; Michelet, J.Y.; Larondelle, Y. Patulin in Domestic and Imported Apple-Based Drinks in Belgium: Occurrence and Exposure Assessment. Food Addit. Contam. 2003, 20, 482–489. [Google Scholar] [CrossRef]
- Puel, O.; Galtier, P.; Oswald, I.P. Biosynthesis and Toxicological Effects of Patulin. Toxins 2010, 2, 613–631. [Google Scholar] [CrossRef]
- European Commission Commission Regulation (EC). No. 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs. Off. J. Eur. Union 2006, 364, 7–17. [Google Scholar]
- FDA. CPG Sec.510.150 Apple Juice, Apple Juice Concentrates and Apple Juice Products—Adulteration with Patulin. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cpg-sec510150-apple-juice-apple-juice-concentrates-and-apple-juice-products-adulteration-patulin (accessed on 19 October 2019).
- Health Canada List of Contaminants and Other Adulterating Substances in Foods. Available online: https://www.canada.ca/en/health-canada/services/food-nutrition/food-safety/chemical-contaminants/contaminants-adulterating-substances-foods.html (accessed on 19 October 2019).
- Zhong, L.; Carere, J.; Lu, Z.; Lu, F.; Zhou, T. Patulin in Apples and Apple-Based Food Products: The Burdens and the Mitigation Strategies. Toxins 2018, 10, 475. [Google Scholar] [CrossRef]
- Ioi, J.D.; Zhou, T.; Tsao, R.; Marcone, M.F. Mitigation of Patulin in Fresh and Processed Foods and Beverages. Toxins 2017, 9, 157. [Google Scholar] [CrossRef]
- Xing, M.; Li, B.; Chen, Y.; Tian, S. Ribonucleoside Diphosphate Reductase Plays an Important Role in Patulin Degradation by Enterobacter cloacae Subsp. dissolvens. J. Agric. Food Chem. 2020, 68, 5232–5240. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zheng, X.; Yang, Q.; Zhang, H.; Apaliya, M.T.; Dhanasekaran, S.; Zhang, X.; Zhao, L.; Li, J.; Jiang, Z. S-Adenosylmethionine-Dependent Methyltransferase Helps Pichia caribbica Degrade Patulin. J. Agric. Food Chem. 2019, 67, 11758–11768. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, X.; Zhang, F.; Meng, X.; Liu, B. Biodegradation of the Mycotoxin Patulin in Apple Juice by Orotate Phosphoribosyltransferase from Rhodotorula mucilaginosa. Food Control 2019, 100, 158–164. [Google Scholar] [CrossRef]
- Liu, B.; Peng, X.; Meng, X. Effective Biodegradation of Mycotoxin Patulin by Porcine Pancreatic Lipase. Front. Microbiol. 2018, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.O.; Long, M.T. Fate of Patulin in the Presence of the Yeast Saccharomyces cerevisiae. Food Addit. Contam. 2002, 19, 387–399. [Google Scholar] [CrossRef]
- Dong, X.; Jiang, W.; Li, C.; Ma, N.; Xu, Y.; Meng, X. Patulin Biodegradation by Marine Yeast Kodameae ohmeri. Food Addit. Contam.—Part A 2015, 32, 352–360. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, H.-M.; Wang, X.; Li, B.-Q.; Long, M.-Y.; Tian, S. Biodegradation Mechanisms of Patulin in Candida guilliermondii: An ITRAQ-Based Proteomic Analysis. Toxins 2017, 9, 48. [Google Scholar] [CrossRef]
- Ianiri, G.; Idnurm, A.; Wright, S.A.I.; Durán-Patrón, R.; Mannina, L.; Ferracane, R.; Ritieni, A.; Castoria, R. Searching for Genes Responsible for Patulin Degradation in a Biocontrol Yeast Provides Insight into the Basis for Resistance to This. Appl. Environ. Microbiol. 2013, 79, 3101–3115. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Shimamoto, T.; Yamada, Y.; Gaucher, G.M. Patulin Biosynthesis: Enzymatic and Nonezymatic Transformations of the Mycotoxin (E)-Ascladiol. Appl. Environ. Microbiol. 1983, 45, 1939–1942. [Google Scholar] [CrossRef]
- Tannous, J.; Snini, S.P.; el Khoury, R.; Canlet, C.; Pinton, P.; Lippi, Y.; Alassane-Kpembi, I.; Gauthier, T.; el Khoury, A.; Atoui, A.; et al. Patulin Transformation Products and Last Intermediates in Its Biosynthetic Pathway, E- and Z-Ascladiol, Are Not Toxic to Human Cells. Arch. Toxicol. 2017, 91, 2455–2467. [Google Scholar] [CrossRef]
- Xing, M.; Chen, Y.; Li, B.; Tian, S. Characterization of a Short-Chain Dehydrogenase/Reductase and Its Function in Patulin Biodegradation in Apple Juice. Food Chem. 2021, 348, 129046. [Google Scholar] [CrossRef] [PubMed]
- Ricelli, A.; Baruzzi, F.; Solfrizzo, M.; Morea, M.; Fanizzi, F.P. Biotransformation of Patulin by Gluconobacter oxydans. Appl. Environ. Microbiol. 2007, 73, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Bevardi, M.; Frece, J.; Mesarek, D.; Bošnir, J.; Mrvčić, J.; Delaš, F.; Markov, K. Antifungal and Antipatulin Activity of Gluconobacter oxydans Isolated from Apple Surface. Arh. Hig. Za Rada I Toksikol. 2013, 64, 279–284. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Z.; Yuan, Y.; Cai, R.; Niu, C.; Yue, T. Identification of Key Factors Involved in the Biosorption of Patulin by Inactivated Lactic Acid Bacteria (LAB) Cells. PLoS ONE 2015, 10, e0143431. [Google Scholar] [CrossRef]
- Prust, C.; Hoffmeister, M.; Liesegang, H.; Wiezer, A.; Fricke, W.F.; Ehrenreich, A.; Gottschalk, G.; Deppenmeier, U. Complete Genome Sequence of the Acetic Acid Bacterium Gluconobacter oxydans. Nat. Biotechnol. 2005, 23, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Lee, S.; Kwon, O.-S.; Park, S.-Y.; Lee, S.-J.; Park, B.-J.; Kim, K.-J. Redox-Switch Modulation of Human SSADH by Dynamic Catalytic Loop. EMBO J. 2009, 28, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Argos, P. NADP-Dependent Enzymes. I: Conserved Stereochemistry of Cofactor Binding. Proteins 1997, 28, 10–28. [Google Scholar] [CrossRef]
- Chen, R.; Liu, X.; Lin, J.; Wei, D. A Genomic Search Approach to Identify Carbonyl Reductases in Gluconobacter oxydans for Enantioselective Reduction of Ketones. Biosci. Biotechnol. Biochem. 2014, 78, 1350–1356. [Google Scholar] [CrossRef]
- Macauley, S.; Mcneil, B.; Harvey, L.M. The Genus Gluconobacter and Its Applications in Biotechnology. Crit. Rev. Biotechnol. 2001, 21, 1–25. [Google Scholar] [CrossRef]
- de Ley, J.; Swings, J. Family VI, Actinobacteriacaea. Genus II, Gluconobacter. In Bergey’s Manual of Systematic Bacteriology; Whitman, W.B., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; pp. 275–278. [Google Scholar]
- Deppenmeier, U.; Hoffmeister, M.; Prust, C. Biochemistry and Biotechnological Applications of Gluconobacter Strains. Appl. Microbiol. Biotechnol. 2002, 60, 233–242. [Google Scholar] [CrossRef]
- Shanbhag, A.P. FabG: From a Core to Circumstantial Catalyst. Biotechnol. Lett. 2019, 41, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, X.; Wang, J.; Lin, J.; Wei, D. Cloning, Expression, and Characterization of an Anti-Prelog Stereospecific Carbonyl Reductase from Gluconobacter oxydans DSM2343. Enzym. Microb. Technol. 2015, 70, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, P.; Deppenmeier, U. Analysis of Aldehyde Reductases from Gluconobacter oxydans 621H. Appl. Microbiol. Biotechnol. 2010, 85, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Jörnvall, H.; Persson, B.; Krook, M.; Atrian, S.; Roser Gonzàilez-Duarte, R.; Jeffery, J.; Ghosh, D. Short-Chain Dehydrogenases/Reductases (SDR). Biochemistry 1995, 34, 6003–6013. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Kallberg, Y.; Oppermann, U.; Jörnvall, H. Coenzyme-Based Functional Assignments of Short-Chain Dehydrogenases/Reductases (SDRs). Chem.-Biol. Interact. 2003, 143–144, 271–278. [Google Scholar] [CrossRef]
- Jörnvall, H.; von Bahr-Lindström, H.; Jany, K.-D.; Ulmer, W.; Fröschle, M. Extended Superfamily of Short Alcohol-Polyol-Sugar Dehydrogenases: Structural Similarities between Glucose and Ribitol Dehydrogenases. FEBS Lett. 1984, 165, 190–196. [Google Scholar] [CrossRef]
- Wierenga, R.K.; de Maeyer, M.C.H.; Hol, W.G.J. Interaction of Pyrophosphate Moeieties with α-Helixes in Dinucleotide Binding Proteins. Biochemistry 1985, 24, 1346–1357. [Google Scholar] [CrossRef]
- Rossman, M.G.; Liljas, A.; Branden, C.-I.; Banaszak, L.J. Evolutionary and Structural Relationships among Dehydrogenases. In The Enzymes; Boyer, P.D., Ed.; Academic Press: New York, NY, USA, 1975; Volume 11, pp. 61–102. [Google Scholar]
- Kallberg, Y.; Oppermann, U.; Jörnvall, H.; Persson, B. Short-Chain Dehydrogenases/Reductases (SDRs). Coenzyme-Based Functional Assignments in Completed Genomes. Eur. J. Biochem. 2002, 269, 4409–4417. [Google Scholar] [CrossRef]
- Wu, J.T.; Wu, L.H.; Knight, J.A. Stability of NADPH: Effect of Various Factors on the Kinetics of Degradation. Clin. Chem. 1986, 32, 314. [Google Scholar] [CrossRef]
- Sydenham, E.W.; Vismer, H.F.; Walter, F.O.; Brown, N.; der Westhuizen, V.; Rheeder, J.P. Reduction of Patulin in Apple Juice Samples—Influence of Initial Processing. Food Control 1995, 6, 195–200. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Inglis, K.A.; Urch, J.E.; Müller, S.; van Aalten, D.M.F.; Fairlamb, A.H. Kinetic, Inhibition and Structural Studies on 3-Oxoacyl-ACP Reductase from Plasmodium falciparum, a Key Enzyme in Fatty Acid Biosynthesis. Biochem. J. 2006, 393, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- di Costanzo, L.; Gomez, G.A.; Christianson, D.W. Crystal Structure of Lactaldehyde Dehydrogenase from Escherichia coli and Inferences Regarding Substrate and Cofactor Specificity. J. Mol. Biol. 2007, 366, 481–493. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fisher, M.; Kroon, J.T.; Martindale, W.; Stuitje, A.R.; Slabas, A.R.; Rafferty, J.B. The X-Ray Structure of Brassica napus β-Keto Acyl Carrier Protein Reductase and Its Implications for Substrate Binding and Catalysis. Structure 2000, 8, 339–347. [Google Scholar] [CrossRef]
- Price, A.C.; Zhang, Y.M.; Rock, C.O.; White, S.W. Structure of β-Ketoacyl-[Acyl Carrier Protein] Reductase from Escherichia coli: Negative Cooperativity and Its Structural Basis. Biochemistry 2001, 40, 12772–12781. [Google Scholar] [CrossRef] [PubMed]
- Valencia, E.; Larroy, C.; Ochoa, W.F.; Parés, X.; Fita, I.; Biosca, J.A. Apo and Holo Structures of an NADP(H)-Dependent Cinnamyl Alcohol Dehydrogenase from Saccharomyces cerevisiae. J. Mol. Biol. 2004, 341, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Ehrensberger, A.H.; Wilson, D.K. Structural and Catalytic Diversity in the Two Family 11 Aldo-Keto Reductases. J. Mol. Biol. 2004, 337, 661–673. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
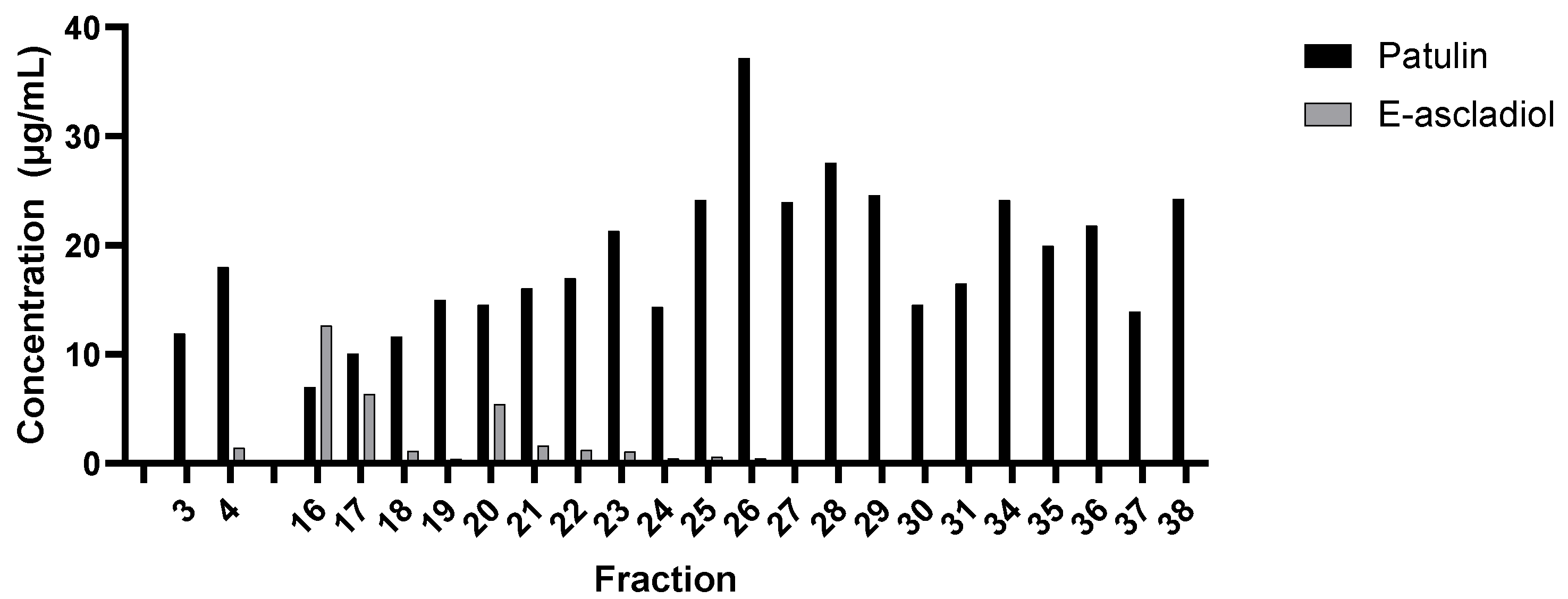{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer | Sequence |
|---|---|---|
| GOX0525 | Forward | GGC GGA ATT CAT GAC CCA CAG AGT AGC GCT C |
| Reverse | GGG CAA GCT TCA GGC GAC GAG ACC GCC | |
| GOX1899 | Forward | GCG GCA TAT GAC CAC GGA CAA CAT CG’ |
| Reverse | GGG CAA GCT TAA AAC TCC TGT CTG GTC GG | |
| GOX1462 | Forward | CGC CCA TAT GCA GTA TCG TCA GCT TGG |
| Reverse | GGG CAA GCT TCA TTT TTT CGT TTC AAG GGC CTC | |
| GOX0716 | Forward | CGC TCA TAT GGC CGA TCA CAG CAT C |
| Reverse | CGC CAA GCT TAC TTC GTC GTG TAC CCT CC | |
| GOX0314 | Forward | CCG CCA TAT GTT CGC CAT GCA GCT C |
| Reverse | CCG CAA GCT TCA CGG CAG CAA TAC GGC | |
| GOX1615 | Forward | GGG AGA ATT CAG TCC CGT GCC GG |
| Reverse | CGC TCT CGA GTC AGT CCC GTG CCG GG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, E.T.S.; Zhu, Y.; Li, X.-Z.; Zhou, T.; Seah, S.Y.K. Characterization of Two Dehydrogenases from Gluconobacter oxydans Involved in the Transformation of Patulin to Ascladiol. Toxins 2022, 14, 423. https://doi.org/10.3390/toxins14070423
Chan ETS, Zhu Y, Li X-Z, Zhou T, Seah SYK. Characterization of Two Dehydrogenases from Gluconobacter oxydans Involved in the Transformation of Patulin to Ascladiol. Toxins. 2022; 14(7):423. https://doi.org/10.3390/toxins14070423
Chicago/Turabian StyleChan, Edicon T. S., Yan Zhu, Xiu-Zhen Li, Ting Zhou, and Stephen Y. K. Seah. 2022. "Characterization of Two Dehydrogenases from Gluconobacter oxydans Involved in the Transformation of Patulin to Ascladiol" Toxins 14, no. 7: 423. https://doi.org/10.3390/toxins14070423
APA StyleChan, E. T. S., Zhu, Y., Li, X.-Z., Zhou, T., & Seah, S. Y. K. (2022). Characterization of Two Dehydrogenases from Gluconobacter oxydans Involved in the Transformation of Patulin to Ascladiol. Toxins, 14(7), 423. https://doi.org/10.3390/toxins14070423