Computer-Aided Analysis of West Sub-Saharan Africa Snakes Venom towards the Design of Epitope-Based Poly-Specific Antivenoms

 ,
,  , and
, and
Abstract
:1. Introduction
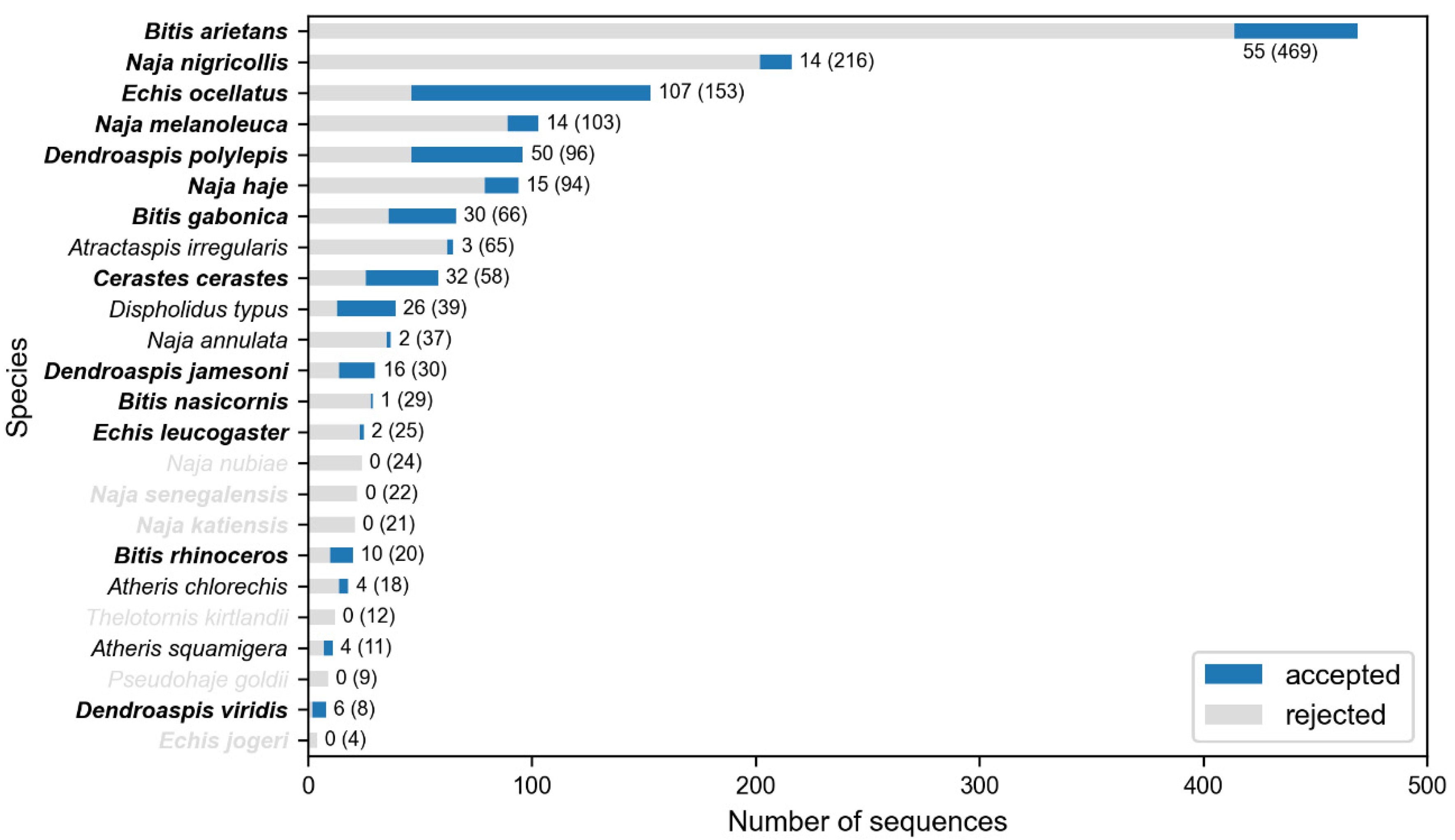
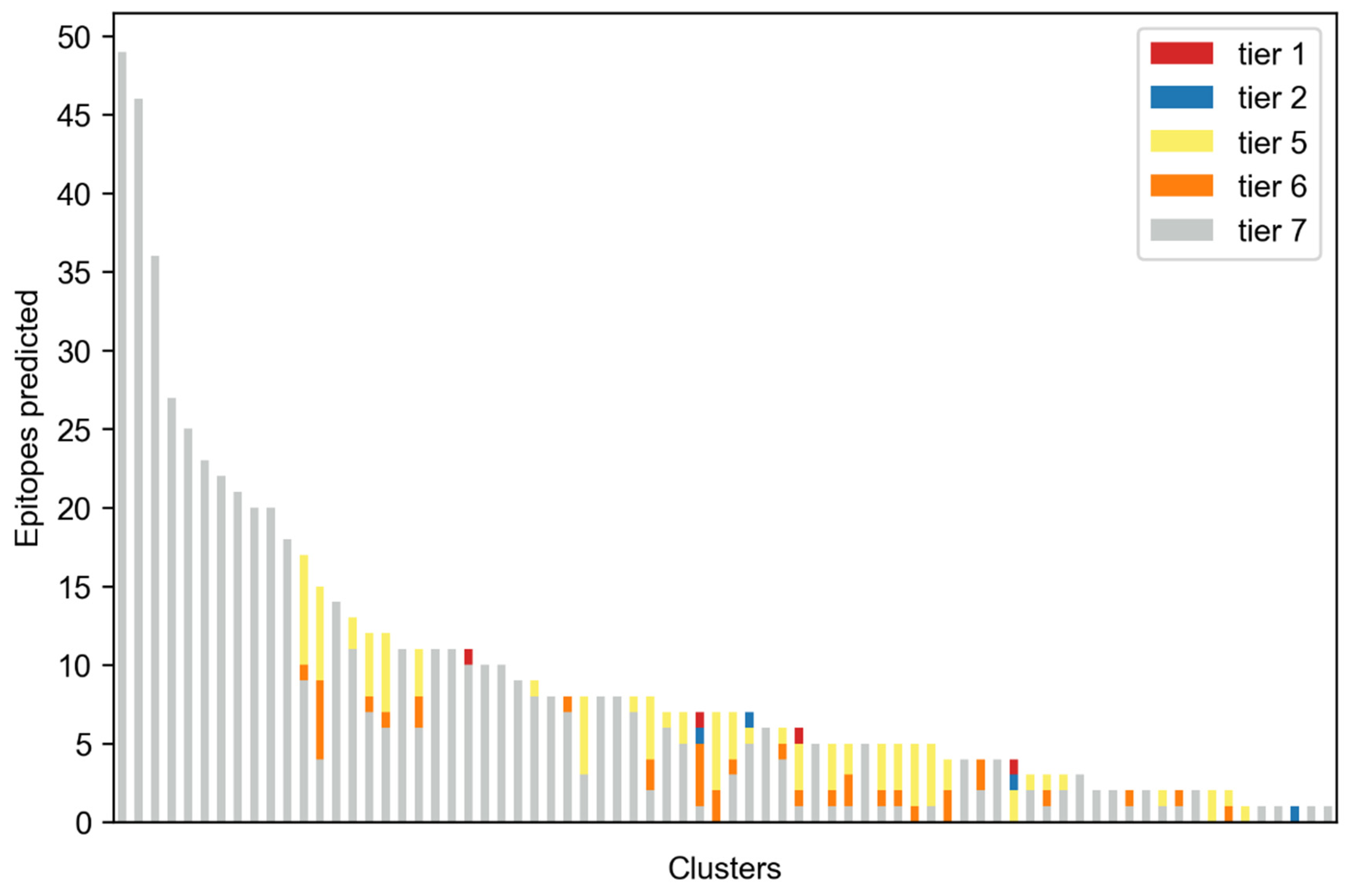
2. Results
3. Discussion
4. Methods
4.1. Collection of Protein Sequences from West Sub-Saharan Snakes
4.2. Clustering, Filtering and Multiple Sequence Alignments (MSAs)
4.3. Conservation Analysis
4.4. PDB BLASTP and Modeling
4.5. B-Cell Epitopes Prediction and Features for Epitopes Characterization and Triage
4.6. Epitope Scoring and Selection
- For predicted epitopes in which their best tier value (Table 2) was between 1 and 4 inclusive, three points; for tiers 5 and 6, two points; and for tier 7, one point.
- If a predicted epitope covered two species, one extra point was given. If it covered three or more, two extra points were given.
- If the cluster depth (number of peptide sequences within a given cluster) was higher than 10, two extra points were given. If it was between 5 and 10, one extra point was given.
- If any predicted epitope was found inside an IEDB epitope or the other way around, three extra points were given.
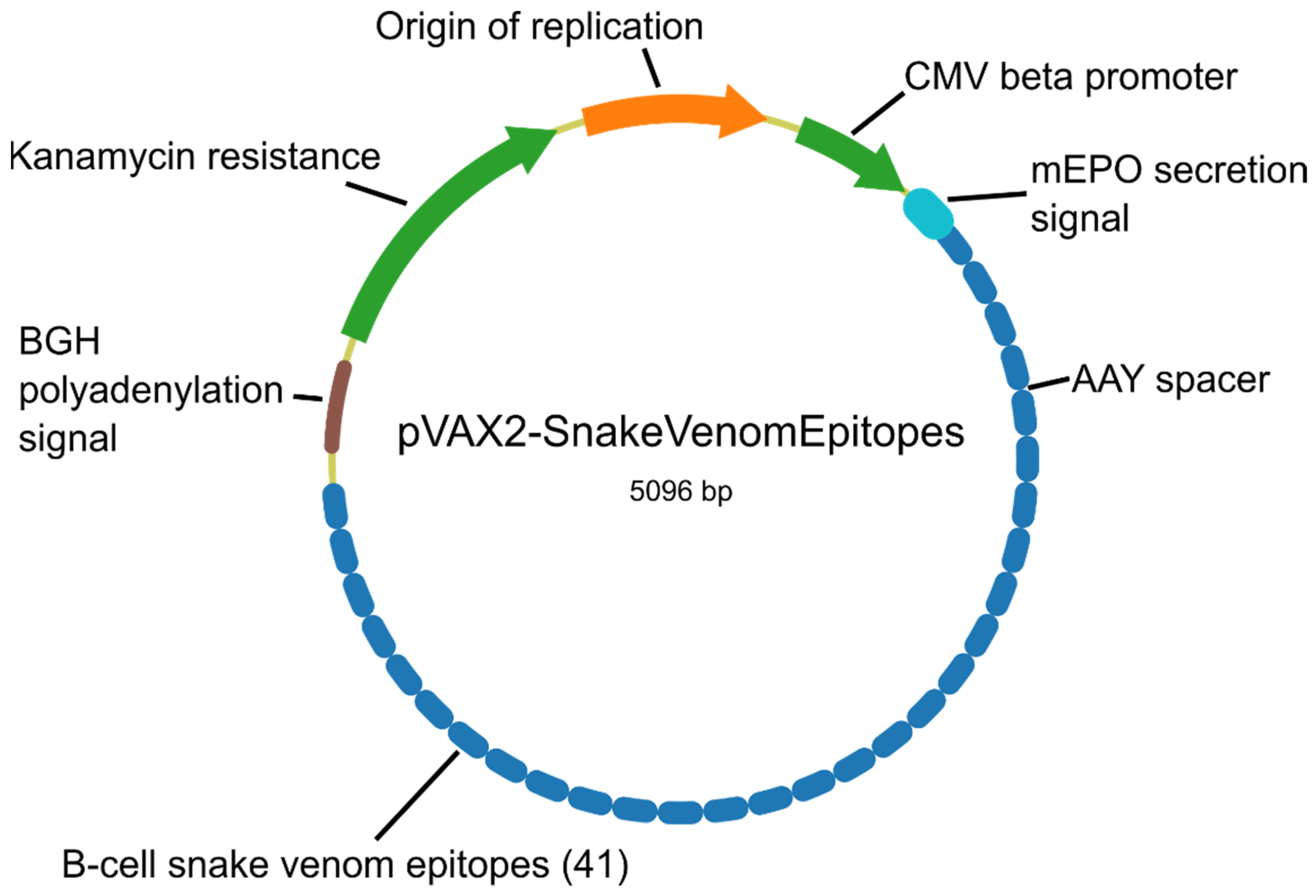
4.7. Genetic Construct and Codon Optimization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chippaux, J.-P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 38. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.A.; Armitage, J.O.; Sanchez, E.E. Snake Envenomation. N. Engl. J. Med. 2022, 386, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Prim. 2017, 3, nrdp201763. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Cerni, F.A.; Janke, R.; Bermúdez-Méndez, E.; Ledsgaard, L.; Barbosa, J.E.; Laustsen, A.H. History of Envenoming Therapy and Current Perspectives. Front. Immunol. 2019, 10, 1598. [Google Scholar] [CrossRef]
- Chippaux, J.-P.; Habib, A.G. Antivenom shortage is not circumstantial but structural. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 747–748. [Google Scholar] [CrossRef]
- Habib, A.G.; Brown, N.I. The snakebite problem and antivenom crisis from a health-economic perspective. Toxicon 2018, 150, 115–123. [Google Scholar] [CrossRef]
- Tasoulis, T.; Isbister, G.K. A Review and Database of Snake Venom Proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef] [Green Version]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef]
- Senji Laxme, R.R.; Khochare, S.; De Souza, H.F.; Ahuja, B.; Suranse, V.; Martin, G.; Whitaker, R.; Sunagar, K. Beyond the ‘big four’: Venom profiling of the medically important yet neglected Indian snakes reveals disturbing antivenom deficiencies. PLoS Negl. Trop. Dis. 2019, 13, e0007899. [Google Scholar] [CrossRef]
- Harrison, R. Development of venom toxin-specific antibodies by DNA immunisation: Rationale and strategies to improve therapy of viper envenoming. Vaccine 2004, 22, 1648–1655. [Google Scholar] [CrossRef]
- Potet, J.; Smith, J.; McIver, L. Reviewing evidence of the clinical effectiveness of commercially available antivenoms in sub-Saharan Africa identifies the need for a multi-centre, multi-antivenom clinical trial. PLoS Neglected Trop. Dis. 2019, 13, e0007551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermúdez-Méndez, E.; Fuglsang-Madsen, A.; Føns, S.; Lomonte, B.; Gutiérrez, J.M.; Laustsen, A.H. Innovative Immunization Strategies for Antivenom Development. Toxins 2018, 10, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arce-Estrada, V.; Azofeifa-Cordero, G.; Estrada, R.; Alape-Girón, A.; Flores-Díaz, M. Neutralization of venom-induced hemorrhage by equine antibodies raised by immunization with a plasmid encoding a novel P-II metalloproteinase from the lancehead pitviper Bothrops asper. Vaccine 2009, 27, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa-Cordero, G.; Arce-Estrada, V.; Flores-Díaz, M.; Alape-Girón, A. Immunization with cDNA of a novel P-III type metalloproteinase from the rattlesnake Crotalus durissus durissus elicits antibodies which neutralize 69% of the hemorrhage induced by the whole venom. Toxicon 2008, 52, 302–308. [Google Scholar] [CrossRef]
- Hasson, S.S.A.A. Generation of antibodies against disintegrin and cysteine-rich domains by DNA immunization: An approach to neutralize snake venom-induced haemorrhage. Asian Pac. J. Trop. Biomed. 2017, 7, 198–207. [Google Scholar] [CrossRef]
- Ramos, H.R.; Junqueira-De-Azevedo, I.; Novo, J.B.; Castro, K.; Guerra-Duarte, C.; de Ávila, R.A.M.; Olórtegui, C.D.C.; Ho, P.L. A Heterologous Multiepitope DNA Prime/Recombinant Protein Boost Immunisation Strategy for the Development of an Antiserum against Micrurus corallinus (Coral Snake) Venom. PLoS Negl. Trop. Dis. 2016, 10, e0004484. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, S.C.; Laing, G.D.; Theakston, R.D.G.; Papaspyridis, C.; A Harrison, R. Bioinformatics and Multiepitope DNA Immunization to Design Rational Snake Antivenom. PLoS Med. 2006, 3, e184. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Lafuente, E.M.; Reche, P.A. Computer-Aided Design of an Epitope-Based Vaccine against Epstein-Barr Virus. J. Immunol. Res. 2017, 2017, 9363750-15. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.4.1. 2015. Available online: https://pymol.org (accessed on 6 June 2022).
- Lalloo, D.G.; Theakston, R.D.G. Snake Antivenoms. J. Toxicol. Clin. Toxicol. 2003, 41, 277–290. [Google Scholar] [CrossRef] [PubMed]
- McCleary, R.; Sridharan, S.; Dunstan, N.L.; Mirtschin, P.J.; Kini, R.M. Proteomic comparisons of venoms of long-term captive and recently wild-caught Eastern brown snakes (Pseudonaja textilis) indicate venom does not change due to captivity. J. Proteom. 2016, 144, 51–62. [Google Scholar] [CrossRef]
- Rex, C.J.; Mackessy, S.P. Venom composition of adult Western Diamondback Rattlesnakes (Crotalus atrox) maintained under controlled diet and environmental conditions shows only minor changes. Toxicon 2019, 164, 51–60. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Johansen, K.H.; Engmark, M.; Andersen, M.R. Recombinant snakebite antivenoms: A cost-competitive solution to a neglected tropical disease? PLoS Negl. Trop. Dis. 2017, 11, e0005361. [Google Scholar] [CrossRef] [Green Version]
- Chippaux, J.-P.; Massougbodji, A.; Habib, A.G. The WHO strategy for prevention and control of snakebite envenoming: A sub-Saharan Africa plan. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, 83. [Google Scholar] [CrossRef]
- Ferraz, C.R.; Arrahman, A.; Xie, C.; Casewell, N.R.; Lewis, R.J.; Kool, J.; Cardoso, F.C. Multifunctional Toxins in Snake Venoms and Therapeutic Implications: From Pain to Hemorrhage and Necrosis. Front. Ecol. Evol. 2019, 7, 218. [Google Scholar] [CrossRef] [Green Version]
- Antúnez, J.; Fernández, J.; Lomonte, B.; Angulo, Y.; Sanz, L.; Pérez, A.; Calvete, J.J.; Gutiérrez, J.M. Antivenomics of Atropoides mexicanus and Atropoides picadoi snake venoms: Relationship to the neutralization of toxic and enzymatic activities. J. Venom Res. 2010, 1, 8–17. [Google Scholar]
- Fernández, J.; Alape-Girón, A.; Angulo, Y.; Sanz, L.; Gutiérrez, J.M.; Calvete, J.J.; Lomonte, B. Venomic and Antivenomic Analyses of the Central American Coral Snake, Micrurus nigrocinctus (Elapidae). J. Proteome Res. 2011, 10, 1816–1827. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Lomonte, B.; Lohse, B.; Fernández, J.; Gutiérrez, J.M. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: Identification of key toxin targets for antivenom development. J. Proteom. 2015, 119, 126–142. [Google Scholar] [CrossRef]
- Lomonte, B.; Escolano, J.; Fernández, J.; Sanz, L.; Angulo, Y.; Gutiérrez, J.M.; Calvete, J.J. Snake Venomics and Antivenomics of the Arboreal Neotropical Pitvipers Bothriechis lateralis and Bothriechis schlegelii. J. Proteome Res. 2008, 7, 2445–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alomran, N.; Alsolaiss, J.; Albulescu, L.-O.; Crittenden, E.; Harrison, R.A.; Ainsworth, S.; Casewell, N.R. Pathology-specific experimental antivenoms for haemotoxic snakebite: The impact of immunogen diversity on the in vitro cross-reactivity and in vivo neutralisation of geographically diverse snake venoms. PLoS Negl. Trop. Dis. 2021, 15, e0009659. [Google Scholar] [CrossRef] [PubMed]
- Damm, M.; Hempel, B.-F.; Süssmuth, R. Old World Vipers—A Review about Snake Venom Proteomics of Viperinae and Their Variations. Toxins 2021, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- De Castro, K.L.P.; Lopes-De-Souza, L.; De Oliveira, D.; de Ávila, R.A.M.; Paiva, A.L.B.; De Freitas, C.F.; Ho, P.L.; Olórtegui, C.D.C.; Guerra-Duarte, C. A Combined Strategy to Improve the Development of a Coral Antivenom Against Micrurus spp. Front. Immunol. 2019, 10, 2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolimbek, B.Z.; Atassi, M.Z. Protection against α-bungarotoxin poisoning by immunization with synthetic toxin peptides. Mol. Immunol. 1996, 33, 681–689. [Google Scholar] [CrossRef]
- Westhof, E.; Altschuh, D.; Moras, D.; Bloomer, A.C.; Mondragón, A.; Klug, A.; Van Regenmortel, M.H.V. Correlation between segmental mobility and the location of antigenic determinants in proteins. Nature 1984, 311, 123–126. [Google Scholar] [CrossRef]
- Ros-Lucas, A.; Correa-Fiz, F.; Bosch-Camós, L.; Rodriguez, F.; Alonso-Padilla, J. Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble. Pathogens 2020, 9, 1078. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Volkmuth, W.; Duca, J.; Corti, L.; Pallaoro, M.; Pezzicoli, A.; Karle, A.; Rigat, F.; Rappuoli, R.; Narasimhan, V.; et al. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci. Transl. Med. 2015, 7, 294ra105. [Google Scholar] [CrossRef] [Green Version]
- Petrova, G.; Ferrante, A.; Gorski, J. Cross-Reactivity of T Cells and Its Role in the Immune System. Crit. Rev. Immunol. 2012, 32, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, A.; Greenbaum, J.; Arlehamn, C.S.L.; Sette, A.; Nielsen, M.; Peters, B. The Interplay of Sequence Conservation and T Cell Immune Recognition. In Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, Newport Beach, CA, USA, 20–23 September 2014; Association for Computing Machinery: New York, NY, USA, 2014; pp. 739–743. [Google Scholar]
- Ansar, S.; Vetrivel, U. PepVis: An integrated peptide virtual screening pipeline for ensemble and flexible docking protocols. Chem. Biol. Drug Des. 2019, 94, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, W.; Warrell, D.; Whitaker, R.; Menon, G.; Rodrigues, R.; Fu, S.H.; Begum, R.; Sati, P.; Piyasena, K.; Bhatia, M.; et al. Trends in snakebite deaths in India from 2000 to 2019 in a nationally representative mortality study. eLife 2020, 9, e54076. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Venomous Snakes Distribution and Species Risk Categories. Available online: https://apps.who.int/bloodproducts/snakeantivenoms/database/default.htm (accessed on 6 October 2021).
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information (NCBI) Entrez Programming Utilities Help. 2010. Available online: https://www.ncbi.nlm.nih.gov/books/NBK25501/ (accessed on 3 March 2022).
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Michel-Todó, L.; Reche, P.; Bigey, P.; Pinazo, M.J.; Gascón, J.; Alonso-Padilla, J. In silico Design of an Epitope-Based Vaccine Ensemble for Chagas Disease. Front. Immunol. 2019, 10, 2698. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- John, B. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003, 31, 3982–3992. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.J.; Thornton, J.M. NACCESS; Department of Biochemistry and Molecular Biology University College: London, UK, 1993. [Google Scholar]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanami, S.; Azadegan-Dehkordi, F.; Rafieian-Kopaei, M.; Salehi, M.; Ghasemi-Dehnoo, M.; Mahooti, M.; Alizadeh, M.; Bagheri, N. Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Sci. Rep. 2021, 11, 12397. [Google Scholar] [CrossRef]
- Bigey, P.; Gnidehou, S.; Doritchamou, J.; Quiviger, M.; Viwami, F.; Couturier, A.; Salanti, A.; Nielsen, M.A.; Scherman, D.; Deloron, P.; et al. The NTS-DBL2X Region of VAR2CSA Induces Cross-Reactive Antibodies that Inhibit Adhesion of Several Plasmodium falciparum Isolates to Chondroitin Sulfate A. J. Infect. Dis. 2011, 204, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Rajanathan, T.C.; Chandra, H.; Pericherla, H.P.; Kumar, S.; Choonia, H.S.; Bajpai, M.; Singh, A.K.; Sinha, A.; Saini, G.; et al. Immunogenic potential of DNA vaccine candidate, ZyCoV-D against SARS-CoV-2 in animal models. Vaccine 2021, 39, 4108–4116. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | Taxid | Family 1 | Cat. 2 |
|---|---|---|---|---|
| Bitis arietans | Puff adder | 8692 | V | 1 |
| Bitis gabonica | East African Gaboon viper | 8694 | V | 1 |
| Bitis nasicornis | Rhinoceros viper | 8695 | V | 1 |
| Bitis rhinoceros | West African Gaboon viper | 715877 | V | 1 |
| Cerastes cerastes | Horned viper | 8697 | V | 1 |
| Dendroaspis jamesoni | Jameson’s mamba | 8623 | E | 1 |
| Dendroaspis polylepis | Black mamba | 8624 | E | 1 |
| Dendroaspis viridis | Western green mamba | 8621 | E | 1 |
| Echis jogeri | Joger’s carpet viper | 696809 | V | 1 |
| Echis leucogaster | White-bellied carpet viper | 504457 | V | 1 |
| Echis ocellatus | West African carpet viper | 99586 | V | 1 |
| Naja haje | Egyptian cobra | 8639 | E | 1 |
| Naja katiensis | West African brown spitting cobra | 409859 | E | 1 |
| Naja melanoleuca | Forest cobra | 8643 | E | 1 |
| Naja nigricollis | Black-necked spitting cobra | 8654 | E | 1 |
| Naja senegalensis | Senegalese cobra | 862238 | E | 1 |
| Atheris broadleyi 3 | Broadley’s bush viper | NA | V | 2 |
| Atheris chlorechis | West African bush viper | 110216 | V | 2 |
| Atheris squamigera | Variable bush viper | 110225 | V | 2 |
| Atractaspis irregularis | Variable burrowing asp | 512568 | L | 2 |
| Dispholidus typus | Boomslang | 46295 | C | 2 |
| Naja annulata | Banded water cobra | 8609 | E | 2 |
| Naja nubiae | Nubian spitting cobra | 186441 | E | 2 |
| Pseudohaje goldii | Gold’s tree cobra | 1545503 | E | 2 |
| Pseudohaje nigra 3 | Black tree cobra | NA | E | 2 |
| Thelotornis kirtlandii | Forest vine or twig snake | 292880 | C | 2 |
| Tier | BepiPred2 | Hydrophobicity | RSA | Flexibility |
|---|---|---|---|---|
| Tier 1 | X | X | X | X |
| Tier 2 | X | X | X | |
| Tier 3 | X | X | X | |
| Tier 4 | X | X | ||
| Tier 5 | X | X | X | |
| Tier 6 | X | X | ||
| Tier 7 | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ros-Lucas, A.; Bigey, P.; Chippaux, J.-P.; Gascón, J.; Alonso-Padilla, J. Computer-Aided Analysis of West Sub-Saharan Africa Snakes Venom towards the Design of Epitope-Based Poly-Specific Antivenoms. Toxins 2022, 14, 418. https://doi.org/10.3390/toxins14060418
Ros-Lucas A, Bigey P, Chippaux J-P, Gascón J, Alonso-Padilla J. Computer-Aided Analysis of West Sub-Saharan Africa Snakes Venom towards the Design of Epitope-Based Poly-Specific Antivenoms. Toxins. 2022; 14(6):418. https://doi.org/10.3390/toxins14060418
Chicago/Turabian StyleRos-Lucas, Albert, Pascal Bigey, Jean-Philippe Chippaux, Joaquim Gascón, and Julio Alonso-Padilla. 2022. "Computer-Aided Analysis of West Sub-Saharan Africa Snakes Venom towards the Design of Epitope-Based Poly-Specific Antivenoms" Toxins 14, no. 6: 418. https://doi.org/10.3390/toxins14060418
APA StyleRos-Lucas, A., Bigey, P., Chippaux, J.-P., Gascón, J., & Alonso-Padilla, J. (2022). Computer-Aided Analysis of West Sub-Saharan Africa Snakes Venom towards the Design of Epitope-Based Poly-Specific Antivenoms. Toxins, 14(6), 418. https://doi.org/10.3390/toxins14060418