Emetic Response to T-2 Toxin Correspond to Secretion of Glucagon-like Peptide-17–36 Amide and Glucose-Dependent Insulinotropic Polypeptide

 ,
, 

Abstract
:1. Introduction
2. Results
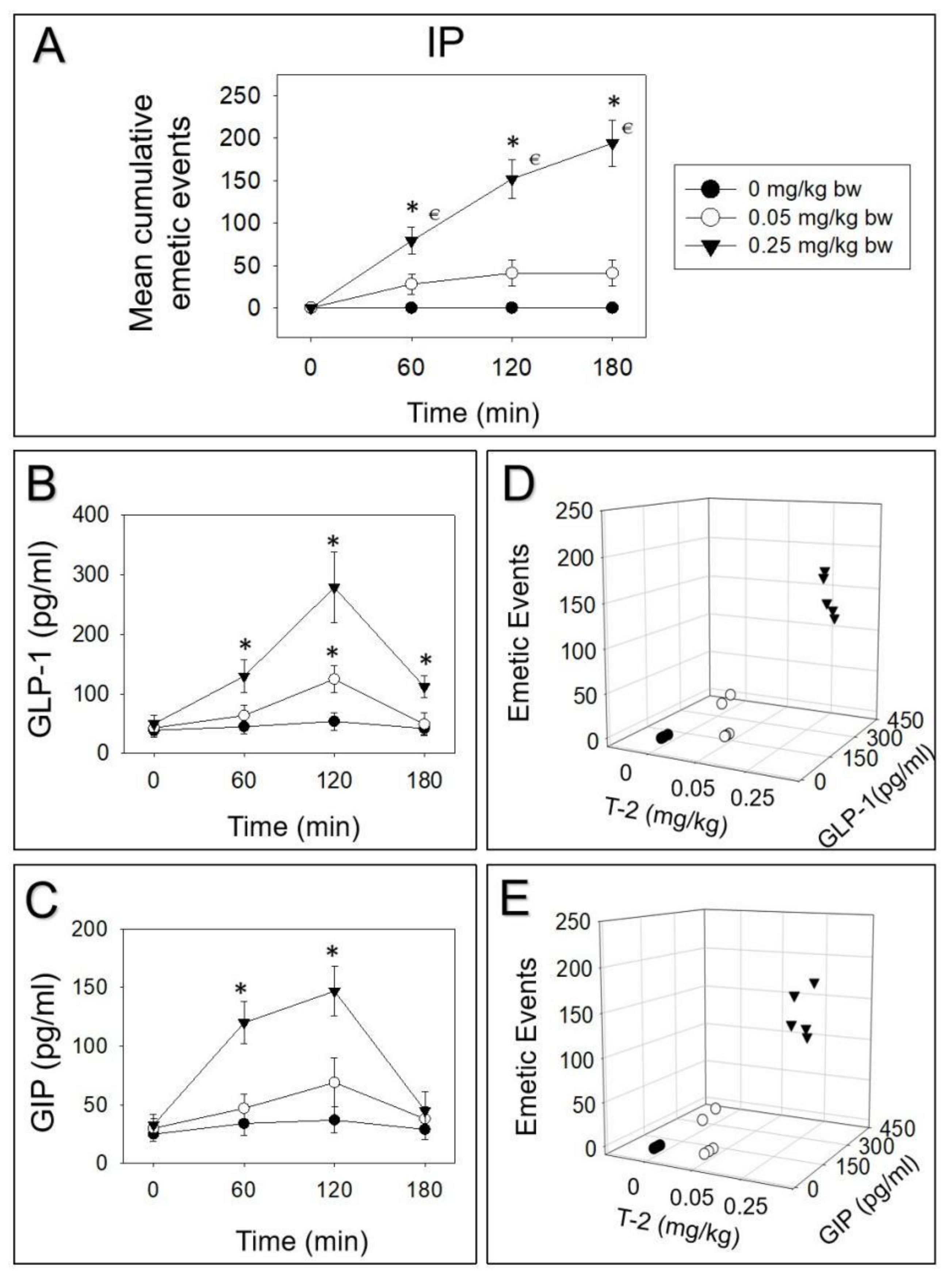
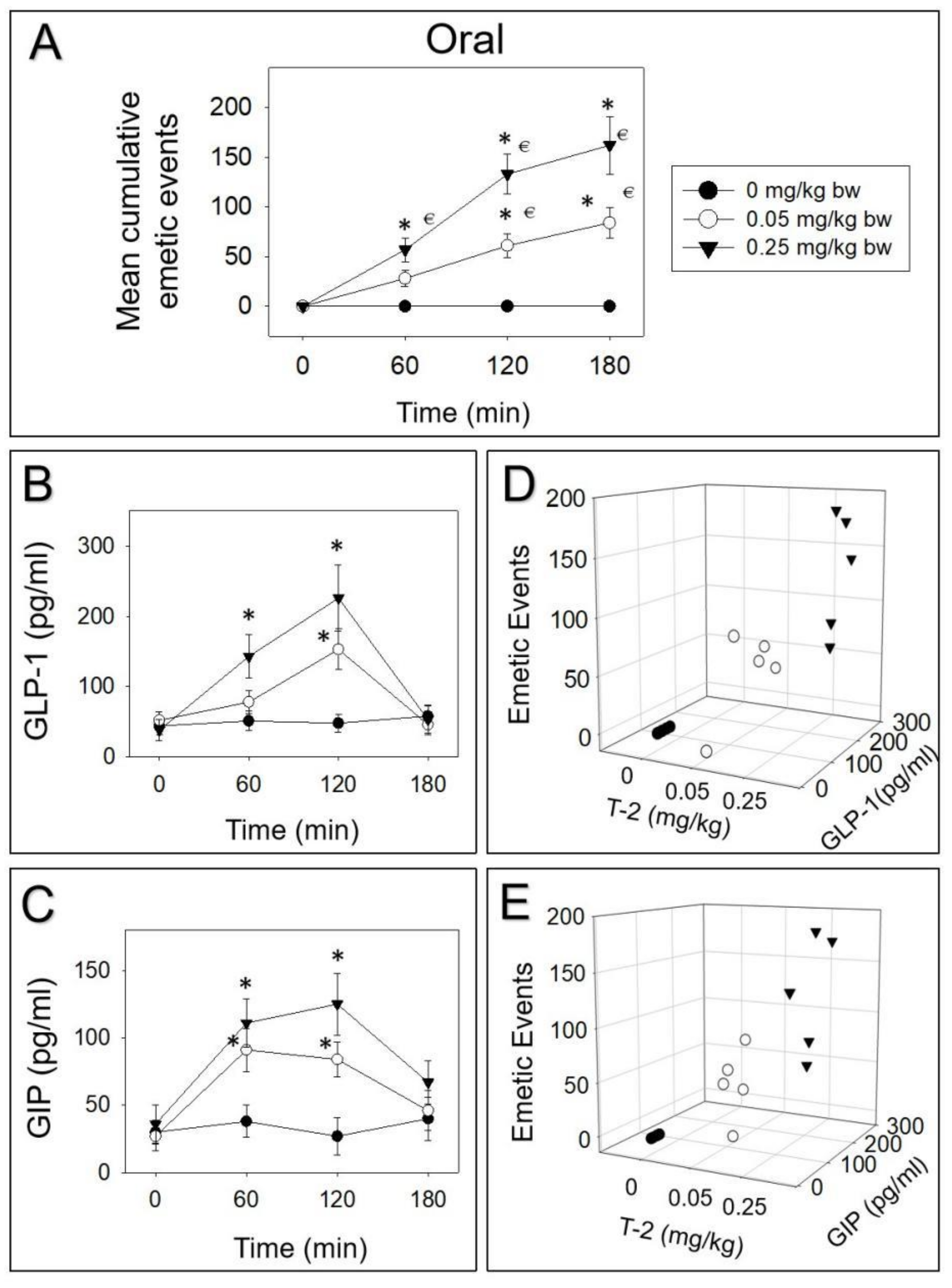
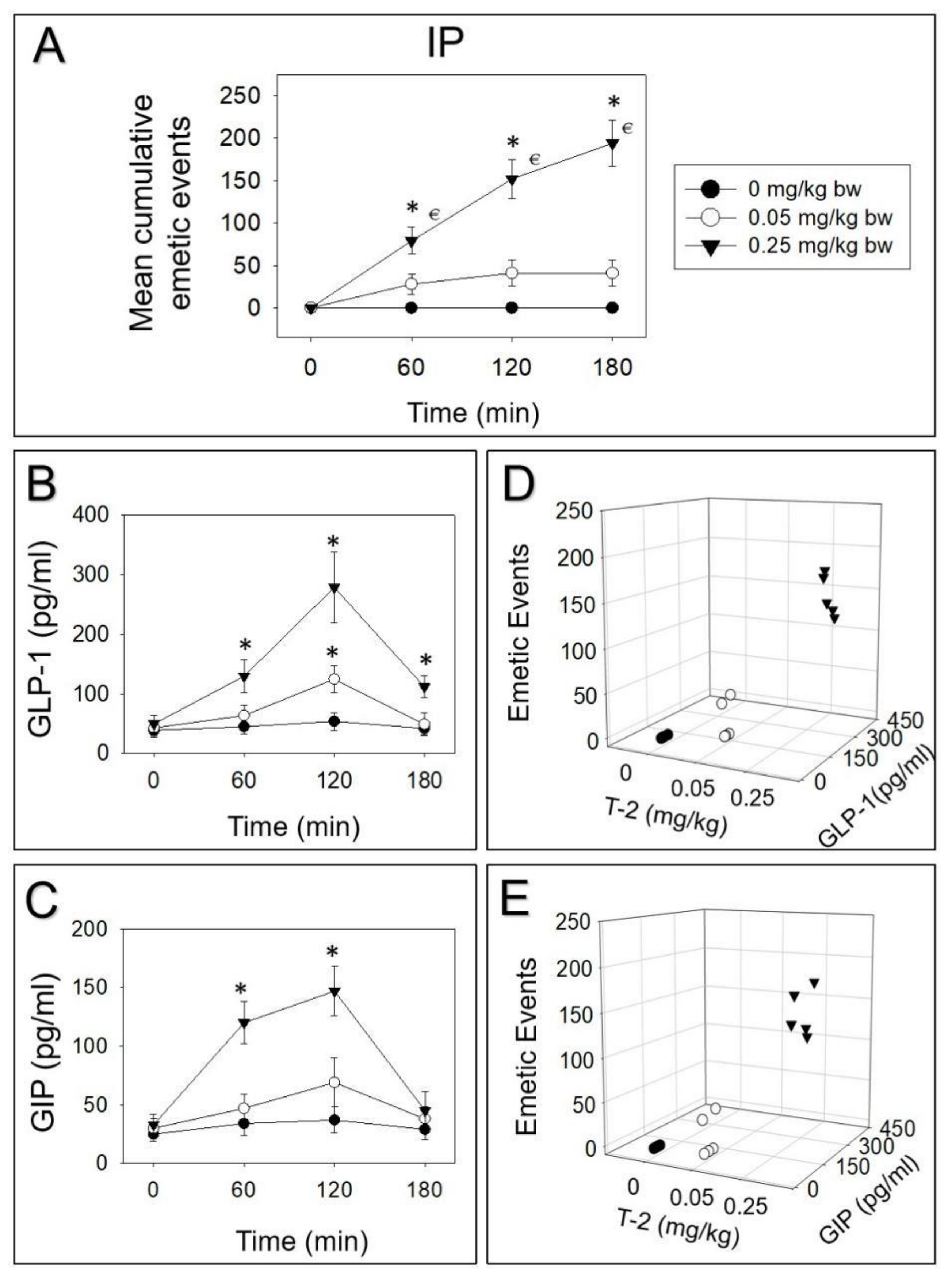
2.1. Emetic Potencies of T-2 Toxin Following Oral and IP Dosing
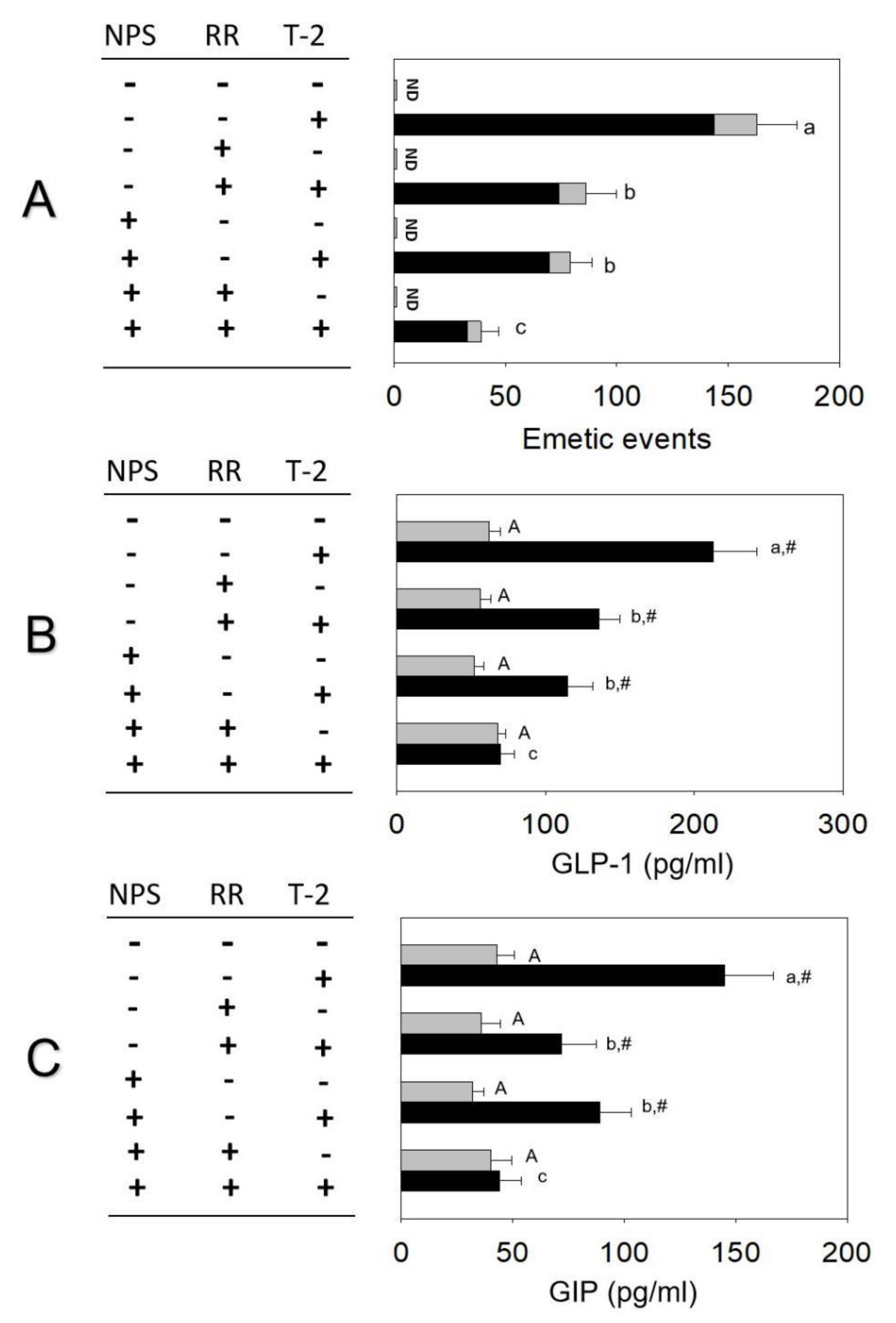
2.2. T-2 Toxin-Induced Emetic Effect Corresponds to Elevation of GLP-1 and GIP
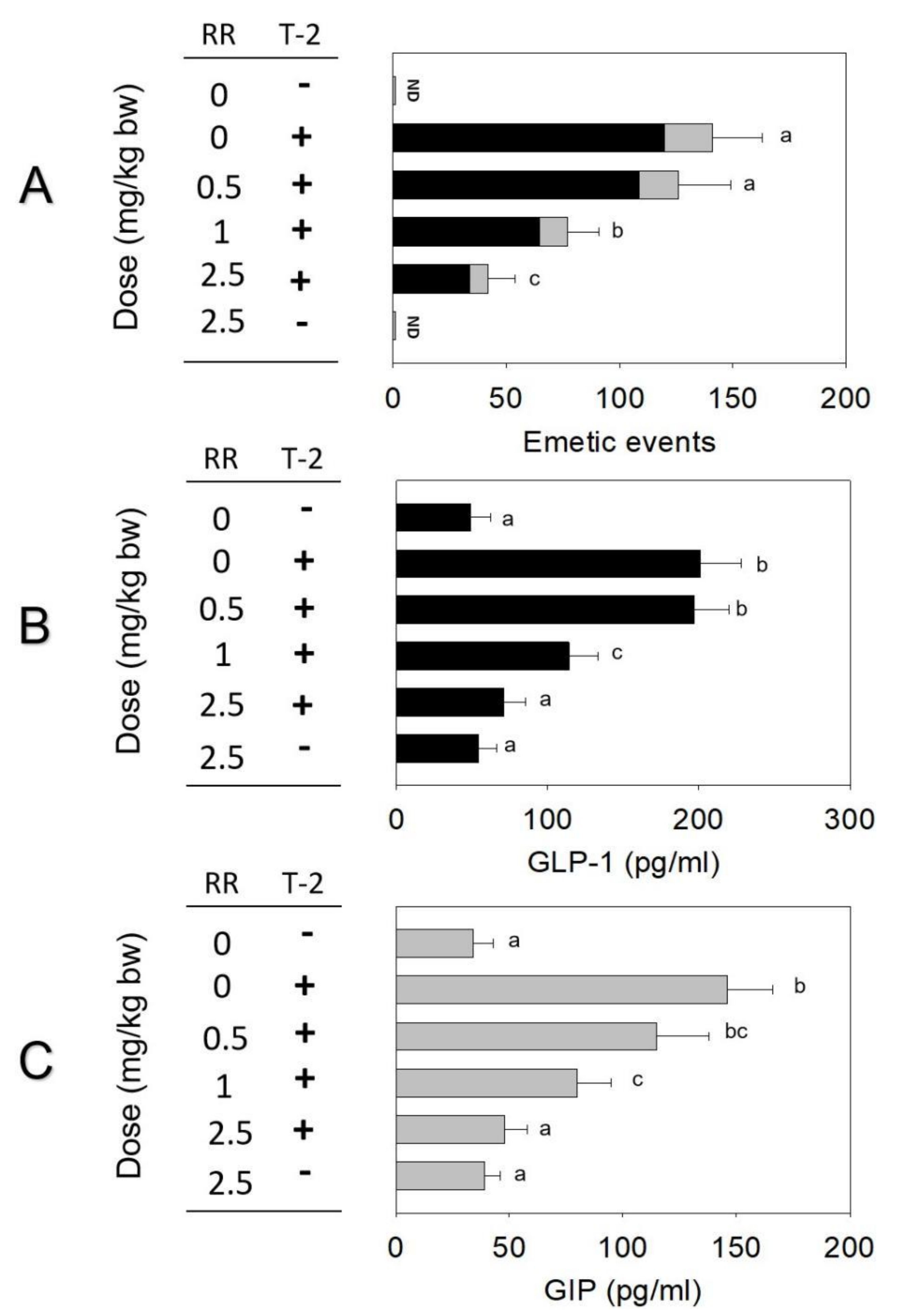
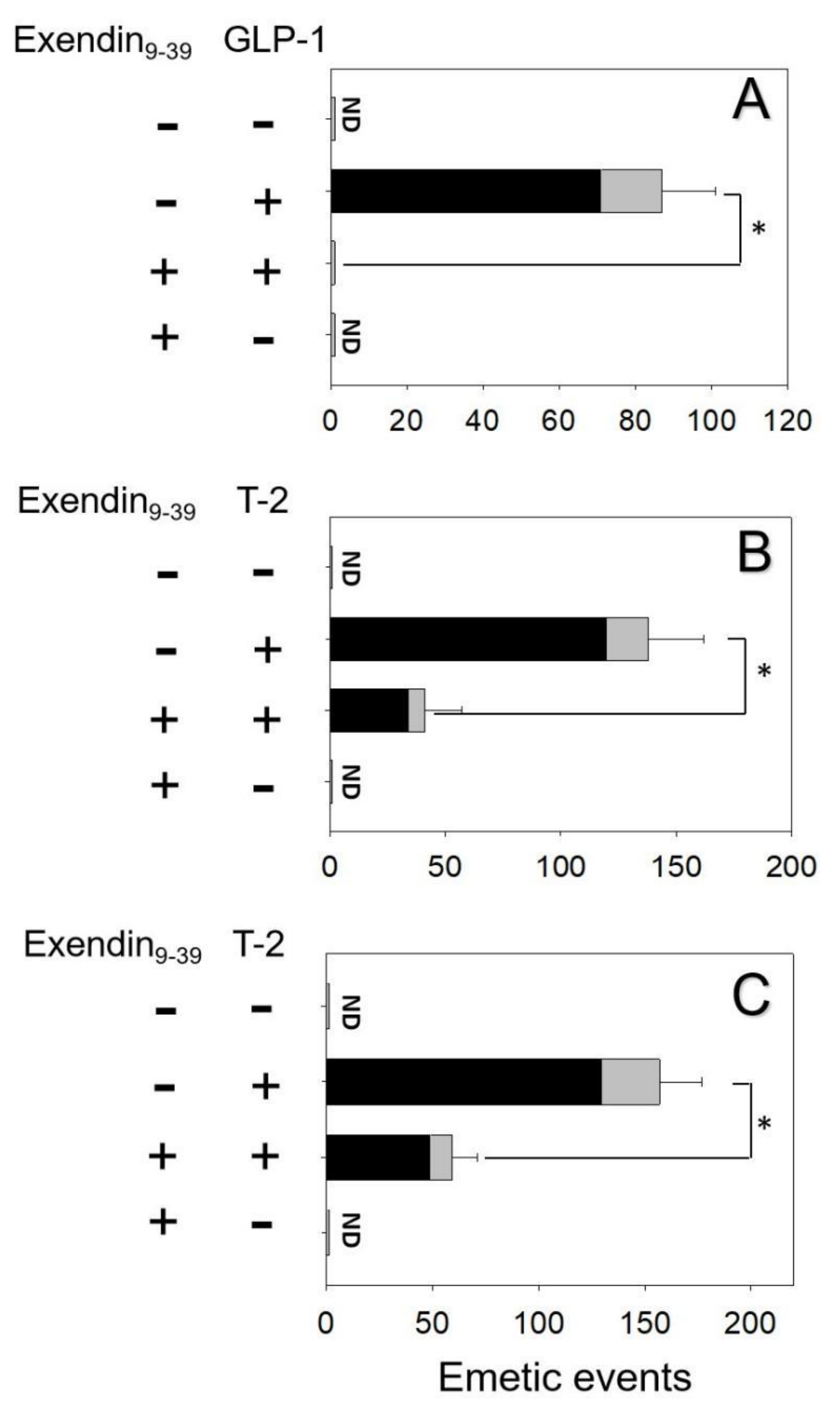
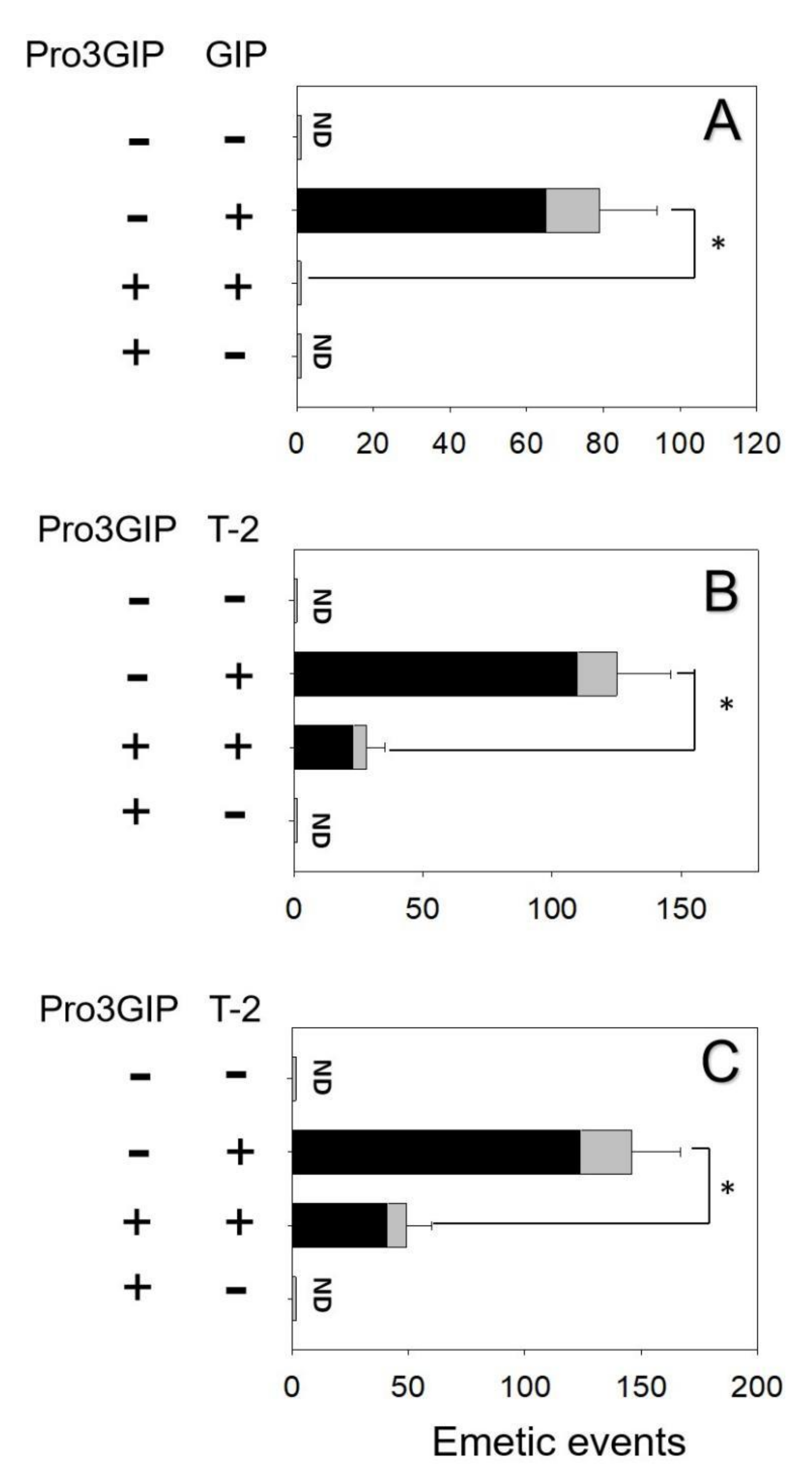
2.3. Effects of Brain–Gut Peptide Receptor Inhibitor on T-2 Toxin-Induced Emesis
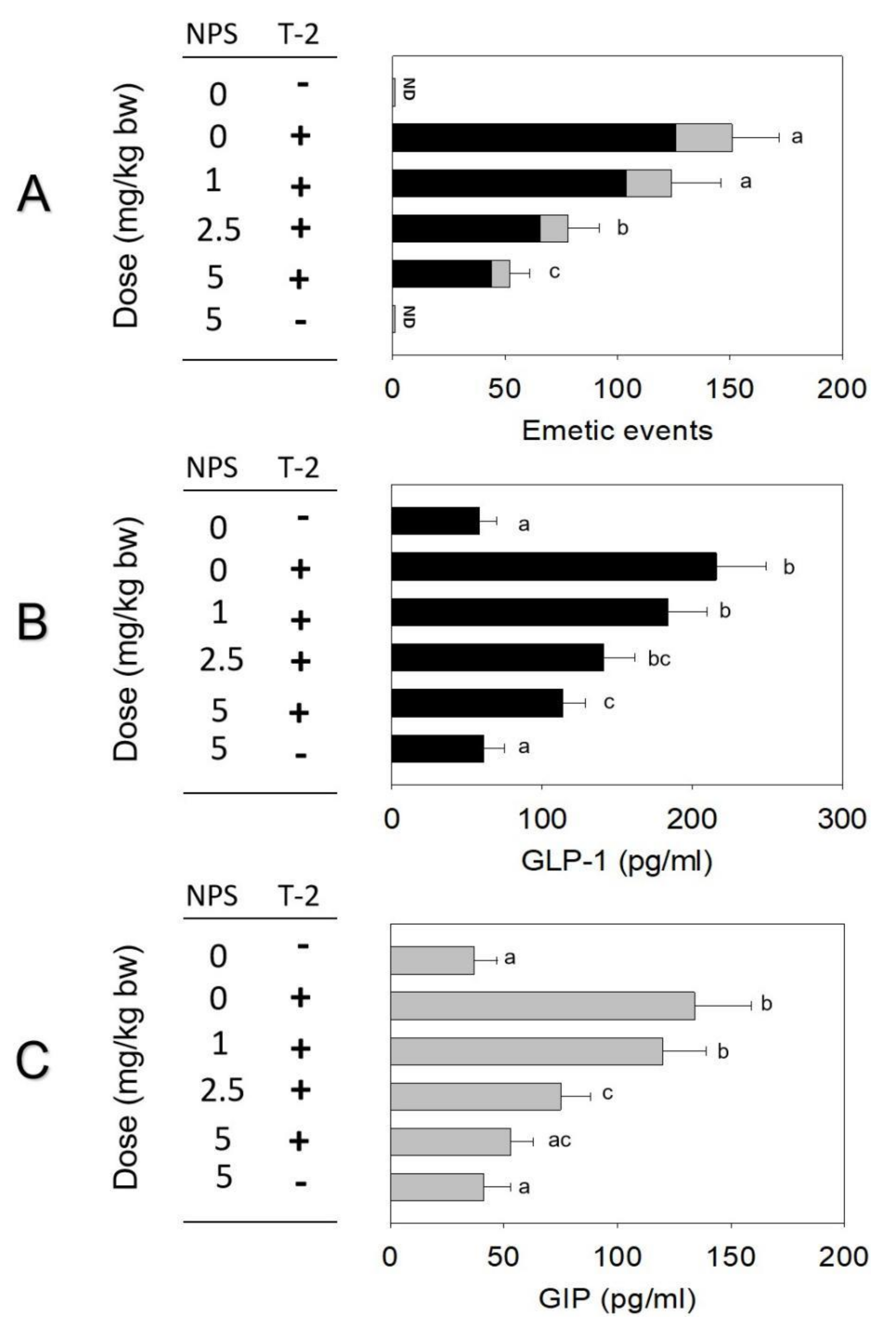
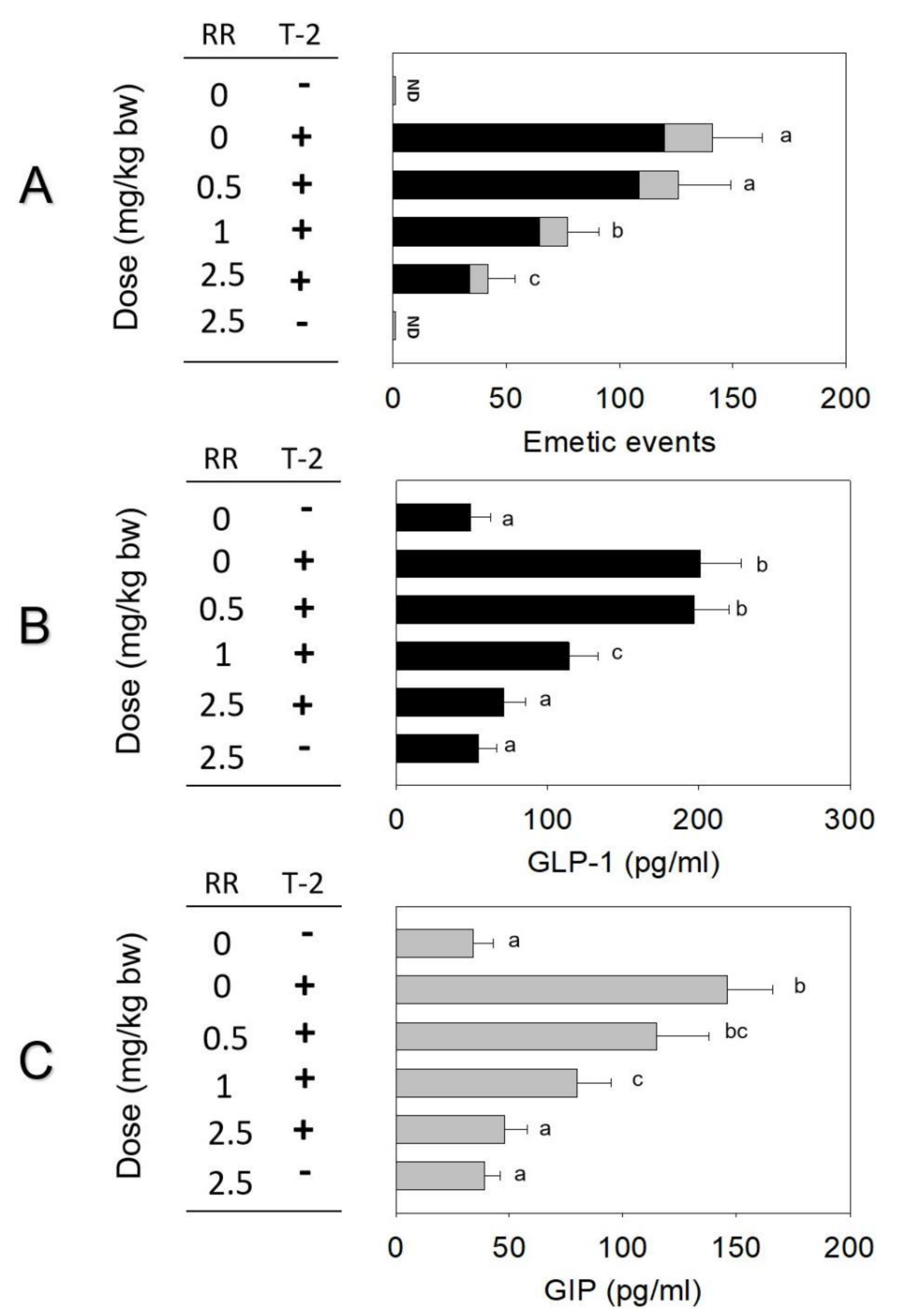
2.4. Roles of CaSR and TRP Channel on T-2 Toxin-Induced Emesis and Brain–Gut Peptides
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animal and Reagent
5.2. Experimental Design
5.3. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hjelkrem, A.-G.R.; Aamot, H.U.; Brodal, G.; Strand, E.C.; Torp, T.; Edwards, S.G.; Dill-Macky, R.; Hofgaard, I.S. HT-2 and T-2 toxins in Norwegian oat grains related to weather conditions at different growth stages. Eur. J. Plant Pathol. 2018, 151, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Kiš, M.; Vulić, A.; Kudumija, N.; Šarkanj, B.; Jaki Tkalec, V.; Aladić, K.; Škrivanko, M.; Furmeg, S.; Pleadin, J. A two-year occurrence of fusarium T-2 and HT-2 toxin in Croatian cereals relative of the regional weather. Toxins 2021, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, L.; Xu, Z.; Liu, X.; Chen, L.; Dai, J.; Karrow, N.A.; Sun, L. Occurrence of Aflatoxin B1, deoxynivalenol and zearalenone in feeds in China during 2018–2020. J. Anim. Sci. Biotechnol. 2021, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.; Liu, S.L.; Wu, W.D.; Zhang, H.B. Comparison of anorectic potencies of type A trichothecenes T-2 toxin, HT-2 toxin, diacetoxyscirpenol, and neosolaniol. Toxins 2018, 10, 179. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.D.; Zhou, H.R.; Bursian, S.J.; Link, J.E.; Pestka, J.J. Emetic responses to T-2 toxin, HT-2 toxin and emetine correspond to plasma elevations of peptide YY3-36 and 5-hydroxytryptamine. Arch. Toxicol. 2016, 90, 997–1007. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, H.; Wang, Q.Q.; Zhang, Y.J.; Wu, W.D.; Zhang, H.B. Role of peptide YY3-36 and glucose-dependent insulinotropic polypeptide in anorexia induction by trichothecences T-2 toxin, HT-2 toxin, diacetoxyscirpenol, and neosolaniol. Toxicol. Sci. 2017, 159, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Zhang, L.; Chu, X.-H.; Ma, R.; Wang, Y.-W.; Liu, Q.; Zhang, N.-Y.; Karrow, N.A.; Sun, L.-H. Effects of deoxynivalenol on the porcine growth performance and intestinal microbiota and potential remediation by a modified HSCAS binder. Food Chem. Toxicol. 2020, 141, 111373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, R.; Zhu, M.-X.; Zhang, N.-Y.; Liu, X.-L.; Wang, Y.-W.; Qin, T.; Zheng, L.-Y.; Liu, Q.; Zhang, W.-P.; et al. Effect of deoxynivalenol on the porcine acquired immune response and potential remediation by a novel modified HSCAS adsorbent. Food Chem. Toxicol. 2020, 138, 111187. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.D.; Sheng, K.; Xu, X.; Zhang, H.; Zhou, G. Potential roles for glucagon-like peptide-17–36 amide and cholecystokinin in anorectic response to the trichothecene mycotoxin T-2 toxin. Ecotoxicol. Environ. Saf. 2018, 153, 181–187. [Google Scholar] [CrossRef]
- Vidal, A.; Bouzaghnane, N.; De Saeger, S.; De Boevre, M. Human mycotoxin biomonitoring: Conclusive remarks on direct or indirect assessment of urinary deoxynivalenol. Toxins 2020, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Knutsen, H.K.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B. Appropriateness to set a group health based guidance value for T2 and HT 2 toxin and its modified forms. EFSA J. 2017, 15, e04655. [Google Scholar] [PubMed]
- Koch, K.L.; Andrews, P.; Stern, R.M. Nausea: Mechanisms and Management; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Andrews, P.; Sanger, G.J. Nausea and the quest for the perfect anti-emetic. Eur. J. Pharmacol. 2014, 722, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.; Horn, C. Signals for nausea and emesis: Implications for models of upper gastrointestinal diseases. Auton. Neurosci. 2006, 125, 100–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornby, P.J. Central neurocircuitry associated with emesis. Am. J. Med. 2001, 111, 106–112. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.L.; Zhang, H.; Li, Y.Y.; Wu, W.D.; Zhang, H.B. Gut satiety hormones cholecystokinin and glucagon-like Peptide-1(7–36) amide mediate anorexia induction by trichothecenes T-2 toxin, HT-2 toxin, diacetoxyscirpenol and neosolaniol. Toxicol. Appl. Pharmacol. 2017, 335, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.M.; Guo, D.W.; Gao, X.G.; Wang, J.C.; Nepovimova, E.; Wu, W.D.; Kuca, K. Deoxynivalenol (vomitoxin)-induced anorexia is Induced by the release of intestinal hormones in mice. Toxins 2021, 13, 512. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.; Lu, X.; Yue, J.; Gu, W.; Gu, C.; Zhang, H.; Wu, W.D. Role of neurotransmitters 5-hydroxytryptamine and substance P in anorexia induction following oral exposure to the trichothecene T-2 toxin. Food Chem. Toxicol. 2019, 123, 1–8. [Google Scholar] [CrossRef]
- Wu, W.D.; Zhou, H.-R.; Bursian, S.J.; Link, J.E.; Pestka, J.J. Calcium-sensing receptor and transient receptor ankyrin-1 mediate emesis induction by deoxynivalenol (Vomitoxin). Toxicol. Sci. 2017, 155, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- López-Ferreras, L.; Richard, J.; Noble, E.; Eerola, K.; Anderberg, R.; Olandersson, K.; Taing, L.; Kanoski, S.; Hayes, M.; Skibicka, K. Lateral hypothalamic GLP-1 receptors are critical for the control of food reinforcement, ingestive behavior and body weight. Mol. Psychiatry 2018, 23, 1157–1168. [Google Scholar] [CrossRef] [Green Version]
- Kanoski, S.E.; Rupprecht, L.E.; Fortin, S.M.; De Jonghe, B.C.; Hayes, M.R. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology 2012, 62, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Chan, S.W.; Tu, L.; Ngan, M.P.; Rudd, J.A. GLP-1 receptors are involved in the GLP-1 (7–36) amide-induced modulation of glucose homoeostasis, emesis and feeding in Suncus murinus (house musk shrew). Eur. J. Pharmacol. 2020, 888, 173528. [Google Scholar] [CrossRef] [PubMed]
- Borner, T.; Geisler, C.E.; Fortin, S.M.; Cosgrove, R.; Alsina-Fernandez, J.; Dogra, M.; Doebley, S.; Sanchez-Navarro, M.J.; Leon, R.M.; Gaisinsky, J. GIP receptor agonism attenuates GLP-1 receptor agonist–induced nausea and emesis in preclinical models. Diabetes 2021, 70, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.M.; Polak, J.M.; Capella, C.; Solcia, E.; Pearse, A. Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) im man. Histochemistry 1978, 56, 37–44. [Google Scholar] [CrossRef]
- Usdin, T.; Mezey, E.; Button, D.; Brownstein, M.; Bonner, T. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993, 133, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Irwin, N.; Gault, V.A.; Green, B.D.; Greer, B.; Mccluskey, J.T.; Harriott, P.; Oharte, F.P.M.; Flatt, P.R. Effects of short-term chemical ablation of the GIP receptor on insulin secretion, islet morphology and glucose homeostasis in mice. Biol. Chem. 2004, 385, 845–852. [Google Scholar] [CrossRef]
- Sheng, K.; Zhang, H.; Yue, J.M.; Gu, W.; Gu, C.; Zhang, H.B.; Wu, W.D. Anorectic response to the trichothecene T-2 toxin correspond to plasma elevations of the satiety hormone glucose-dependent insulinotropic polypeptide and peptide YY3-36. Toxicology 2018, 402, 28–36. [Google Scholar] [CrossRef]
- Zhao, X.; Xian, Y.; Wang, C.; Ding, L.; Meng, X.; Zhu, W.; Hang, S. Calcium-sensing receptor-mediated L-tryptophan-induced secretion of cholecystokinin and glucose-dependent insulinotropic peptide in swine duodenum. J. Vet. Sci. 2018, 19, 179–187. [Google Scholar] [CrossRef]
- Van Liefferinge, E.; Van Noten, N.; Degroote, J.; Vrolix, G.; Van Poucke, M.; Peelman, L.; Van Ginneken, C.; Roura, E.; Michiels, J. Expression of transient receptor potential ankyrin 1 and transient receptor potential vanilloid 1 in the gut of the peri-weaning pig is strongly dependent on age and intestinal site. Animals 2020, 10, 2417. [Google Scholar] [CrossRef]
- Acar, I.; Cetinkaya, A.; Lay, I.; Ileri-Gurel, E. The role of calcium sensing receptors in GLP-1 and PYY secretion after acute intraduodenal administration of L-Tryptophan in rats. Nutr. Neurosci. 2020, 23, 481–489. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, H.-R.; Pestka, J.J. Potential roles for calcium-sensing receptor (CaSR) and transient receptor potential ankyrin-1 (TRPA1) in murine anorectic response to deoxynivalenol (vomitoxin). Arch. Toxicol. 2017, 91, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, S.; Marrs, T.; Parker, H.; Scawin, J.; Swanston, D. Acute toxicity of T2 toxin in rats, mice, guinea pigs, and pigeons. Toxicology 1987, 43, 31–49. [Google Scholar] [CrossRef]
- Mace, O.J.; Schindler, M.; Patel, S. The regulation of K-and L-cell activity by GLUT2 and the calcium-sensing receptor CasR in rat small intestine. J. Physiol. 2012, 590, 2917–2936. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Hesketh, P.J.; Somerfield, M.R.; Feyer, P.; Clark-Snow, R.; Koeller, J.M.; Morrow, G.R.; Chinnery, L.W.; Chesney, M.J.; Gralla, R.J. American Society of Clinical Oncology guideline for antiemetics in oncology: Update 2006. J. Clin. Oncol. 2006, 24, 2932–2947. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, H.; Mousavi, M. Review on T-2 toxin. Jundishapur J. Nat. Pharm. Prod. 2010, 5, 26–38. [Google Scholar]
- Beasley, V.R.; Swanson, S.P.; Corley, R.A.; Buck, W.B.; Koritz, G.D.; Burmeister, H.R. Pharmacokinetics of the trichothecene mycotoxin, T-2 toxin, in swine and cattle. Toxicon 1986, 24, 13–23. [Google Scholar] [CrossRef]
- Osselaere, A.; Devreese, M.; Goossens, J.; Vandenbroucke, V.; De Baere, S.; De Backer, P.; Croubels, S. Toxicokinetic study and absolute oral bioavailability of deoxynivalenol, T-2 toxin and zearalenone in broiler chickens. Food Chem. Toxicol. 2013, 51, 350–355. [Google Scholar] [CrossRef]
- Horn, C.C. Measuring the nausea-to-emesis continuum in non-human animals: Refocusing on gastrointestinal vagal signaling. Exp. Brain Res. 2014, 232, 2471–2481. [Google Scholar] [CrossRef]
- Babic, T.; Browning, K.N. The role of vagal neurocircuits in the regulation of nausea and vomiting. Eur. J. Pharmacol. 2014, 722, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.D.; Leslie, R.A. The area postrema and vomiting. Front. Neuroendocrinol. 1994, 15, 301–320. [Google Scholar] [CrossRef]
- Baker, P.D.; Morzorati, S.L.; Ellett, M.L. The pathophysiology of chemotherapy-induced nausea and vomiting. Gastroenterol. Nurs. 2005, 28, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.R.; Borner, T.; De Jonghe, B.C. The role of GIP in the regulation of GLP-1 satiety and nausea. Diabetes 2021, 70, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Price, C.J.; Hoyda, T.D.; Ferguson, A.V. The area postrema: A brain monitor and integrator of systemic autonomic state. Neuroscientist 2007, 14, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Pei, H.; Sandoval, D.A.; Seeley, R.J.; Chang, R.B.; Liberles, S.D.; Olson, D.P. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor–expressing glutamatergic neurons. Diabetes 2018, 67, 1538–1548. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Mesgarzadeh, S.; Ramesh, K.S.; Huey, E.L.; Liu, Y.; Gray, L.A.; Aitken, T.J.; Chen, Y.; Beutler, L.R.; Ahn, J.S. Genetic identification of vagal sensory neurons that control feeding. Cell 2019, 179, 1129–1143.e23. [Google Scholar] [CrossRef]
- Krieger, J.-P. Intestinal glucagon-like peptide-1 effects on food intake: Physiological relevance and emerging mechanisms. Peptides 2020, 131, 170342. [Google Scholar] [CrossRef]
- Trapp, S.; Brierley, D.I. Brain GLP-1 and the regulation of food intake: GLP-1 action in the brain and its implications for GLP-1 receptor agonists in obesity treatment. Br. J. Pharmacol. 2022, 179, 557–570. [Google Scholar] [CrossRef]
- Williams, E.K.; Chang, R.B.; Strochlic, D.E.; Umans, B.D.; Lowell, B.B.; Liberles, S.D. Sensory neurons that detect stretch and nutrients in the digestive system. Cell 2016, 166, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Berthoud, H.-R.; Albaugh, V.L.; Neuhuber, W.L. Gut-brain communication and obesity: Understanding functions of the vagus nerve. J. Clin. Investig. 2021, 131, e143770. [Google Scholar] [CrossRef]
- Borner, T.; Tinsley, I.C.; Doyle, R.P.; Hayes, M.R.; De Jonghe, B.C. Glucagon-like peptide-1 in diabetes care: Can glycaemic control be achieved without nausea and vomiting? Br. J. Pharmacol. 2022, 179, 542–556. [Google Scholar] [CrossRef]
- Trujillo, J. Safety and tolerability of once-weekly GLP-1 receptor agonists in type 2 diabetes. J. Clin. Pharm. Ther. 2020, 45, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Bergenstal, R.M.; Wysham, C.; Macconell, L.; Malloy, J.; Walsh, B.; Yan, P.; Wilhelm, K.; Malone, J.; Porter, L.E. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): A randomised trial. Lancet 2010, 376, 431–439. [Google Scholar] [CrossRef]
- Ludwig, M.Q.; Todorov, P.V.; Egerod, K.L.; Olson, D.P.; Pers, T.H. Single-cell mapping of GLP-1 and GIP receptor expression in the dorsal vagal complex. Diabetes 2021, 70, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.E.; Biggs, E.K.; Darwish, T.; Tadross, J.; Sukthankar, T.; Girish, M.; Polex-Wolf, J.; Lam, B.Y.; Zvetkova, I.; Pan, W.; et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 2019, 30, 987–996.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Kaye, J.A.; Cai, Z.; Wang, Y.; Prescott, S.L.; Liberles, S.D. Area postrema cell types that mediate nausea-associated behaviors. Neuron 2021, 109, 461–472.e5. [Google Scholar] [CrossRef]
- Takeda, N.; Hasegawa, S.; Morita, M.; Matsunaga, T. Pica in rats is analogous to emesis: An animal model in emesis research. Pharmacol. Biochem. Behav. 1993, 45, 817–821. [Google Scholar] [CrossRef]
- Mcclean, P.L.; Gault, V.A.; Irwin, N.; Mccluskey, J.T.; Flatt, P.R. Daily administration of the GIP-R antagonist (Pro(3))GIP in streptozotocin-induced diabetes suggests that insulin-dependent mechanisms are critical to anti-obesity-diabetes actions of (Pro(3))GIP. Diabetes Obes. Metab. 2008, 10, 336–342. [Google Scholar] [CrossRef]
- Pala, L.; Ciani, S.; Dicembrini, I.; Bardini, G.; Cresci, B.; Pezzatini, A.; Giannini, S.; Mannucci, E.; Rotella, C.M. Relationship between GLP-1 levels and dipeptidyl peptidase-4 activity in different glucose tolerance conditions. Diabetic. Med. 2010, 27, 691–695. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Whittington, H.J.; Wynne, A.M.; Begum, S.S.; Theodorou, L.; Riksen, N.; Mocanu, M.M.; Yellon, D.M. Dipeptidyl peptidase-4 inhibitors and GLP-1 reduce myocardial infarct size in a glucose-dependent manner. Cardiovasc. Diabetol. 2013, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- Deacon, C.F. Metabolism of GIP and the contribution of GIP to the glucose-lowering properties of DPP-4 inhibitors. Peptides 2020, 125, 170196. [Google Scholar] [CrossRef]
- D’souza-Li, L. The calcium-sensing receptor and related diseases. Arq. Bras. Endocrinol. Metabol. 2006, 50, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roura, E.; Depoortere, I.; Navarro, M. Chemosensing of nutrients and non-nutrients in the human and porcine gastrointestinal tract. Animal 2019, 13, 2714–2726. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Bai, W.D.; Zeng, X.F.; Cui, C. γ-Glu ((n = 1,2))-Phe/-Met/-Val stimulates gastrointestinal hormone (CCK and GLP-1) secretion by activating the calcium-sensing receptor. Food Funct. 2019, 10, 4071–4080. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Ding, L.R.; Zhu, W.Y.; Hang, S.Q. Soybean protein hydrolysate stimulated cholecystokinin secretion and inhibited feed intake through calcium-sensing receptors and intracellular calcium signalling in pigs. Food Funct. 2021, 12, 9286–9299. [Google Scholar] [CrossRef]
- Boesmans, W.; Owsianik, G.; Tack, J.; Voets, T.; Vanden Berghe, P. TRP channels in neurogastroenterology: Opportunities for therapeutic intervention. Br. J. Pharmacol. 2011, 162, 18–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, E.C.; Diakogiannaki, E.; Gentry, C.; Psichas, A.; Habib, A.M.; Bevan, S.; Fischer, M.J.M.; Reimann, F.; Gribble, F.M. Stimulation of GLP-1 secretion downstream of the ligand-gated ion channel TRPA1. Diabetes 2014, 64, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yan, Z.; Zhong, J.; Chen, J.; Ni, Y.; Li, L.; Ma, L.; Zhao, Z.; Liu, D.; Zhu, Z. Transient receptor potential vanilloid 1 activation enhances gut glucagon-like peptide-1 secretion and improves glucose homeostasis. Diabetes 2012, 61, 2155–2165. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-R.; Pestka, J.J. Deoxynivalenol (vomitoxin)-induced cholecystokinin and glucagon-like peptide-1 release in the STC-1 enteroendocrine cell model is mediated by calcium-sensing receptor and transient receptor potential ankyrin-1 channel. Toxicol. Sci. 2015, 145, 407–417. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, L.; Gong, G.; Zhang, L.; Shi, L.; Dai, J.; Han, Y.; Wu, Y.; Khalil, M.M.; Sun, L. Invited review: Remediation strategies for mycotoxin control in feed. J. Anim. Sci. Biotechnol. 2022, 13, 19. [Google Scholar] [CrossRef]
- Jia, H.; Wu, W.; Lu, X.; Zhang, J.; He, C.H.; Zhang, H.B. Role of glucagon-like peptide-1 and gastric inhibitory peptide in anorexia induction following oral exposure to the trichothecene mycotoxin deoxynivalenol (Vomitoxin). Toxicol. Sci. 2017, 159, 16–24. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure Route | Dose (mg/kg bw) | Incidence (Responding/Tested) | Latency (min) A,B | Duration (min) A,B | Emetic Episodes C | ||
|---|---|---|---|---|---|---|---|
| Retch | Vomit | Total | |||||
| Oral | 0 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| 0.002 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| 0.01 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| 0.05 * | 4/5 | 28 ± 1 a | 63 ± 8 a | 72 ± 8 | 12 ± 3 | 84 ± 11 | |
| 0.25 * | 5/5 | 19 ± 5 a | 124 ± 13 b | 132 ± 21 | 30 ± 8 | 162 ± 29 | |
| IP | 0 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| 0.002 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| 0.01 | 0/5 | - | - | 0 ± 0 | 0 ± 0 | 0 ± 0 | |
| 0.05 | 2/5 | 32 ± 4 a | 48 ± 7 a | 34 ± 6 | 7 ± 2 | 41 ± 8 | |
| 0.25 * | 5/5 | 25 ± 6 a | 161 ± 11 b | 151 ± 20 | 43 ± 7 | 194 ± 27 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Li, T.; Wu, Q.; Qin, Z.; Wei, B.; Wu, R.; Guo, X.; Xiao, H.; Wu, W. Emetic Response to T-2 Toxin Correspond to Secretion of Glucagon-like Peptide-17–36 Amide and Glucose-Dependent Insulinotropic Polypeptide. Toxins 2022, 14, 389. https://doi.org/10.3390/toxins14060389
Zhang J, Li T, Wu Q, Qin Z, Wei B, Wu R, Guo X, Xiao H, Wu W. Emetic Response to T-2 Toxin Correspond to Secretion of Glucagon-like Peptide-17–36 Amide and Glucose-Dependent Insulinotropic Polypeptide. Toxins. 2022; 14(6):389. https://doi.org/10.3390/toxins14060389
Chicago/Turabian StyleZhang, Jie, Tushuai Li, Qinghua Wu, Zihui Qin, Ben Wei, Ran Wu, Xinyi Guo, Huiping Xiao, and Wenda Wu. 2022. "Emetic Response to T-2 Toxin Correspond to Secretion of Glucagon-like Peptide-17–36 Amide and Glucose-Dependent Insulinotropic Polypeptide" Toxins 14, no. 6: 389. https://doi.org/10.3390/toxins14060389
APA StyleZhang, J., Li, T., Wu, Q., Qin, Z., Wei, B., Wu, R., Guo, X., Xiao, H., & Wu, W. (2022). Emetic Response to T-2 Toxin Correspond to Secretion of Glucagon-like Peptide-17–36 Amide and Glucose-Dependent Insulinotropic Polypeptide. Toxins, 14(6), 389. https://doi.org/10.3390/toxins14060389