Simultaneous Determination of 15 Mycotoxins in Aquaculture Feed by Liquid Chromatography–Tandem Mass Spectrometry




Abstract
:1. Introduction
2. Results and Discussion
2.1. HPLC-MS/MS Determination
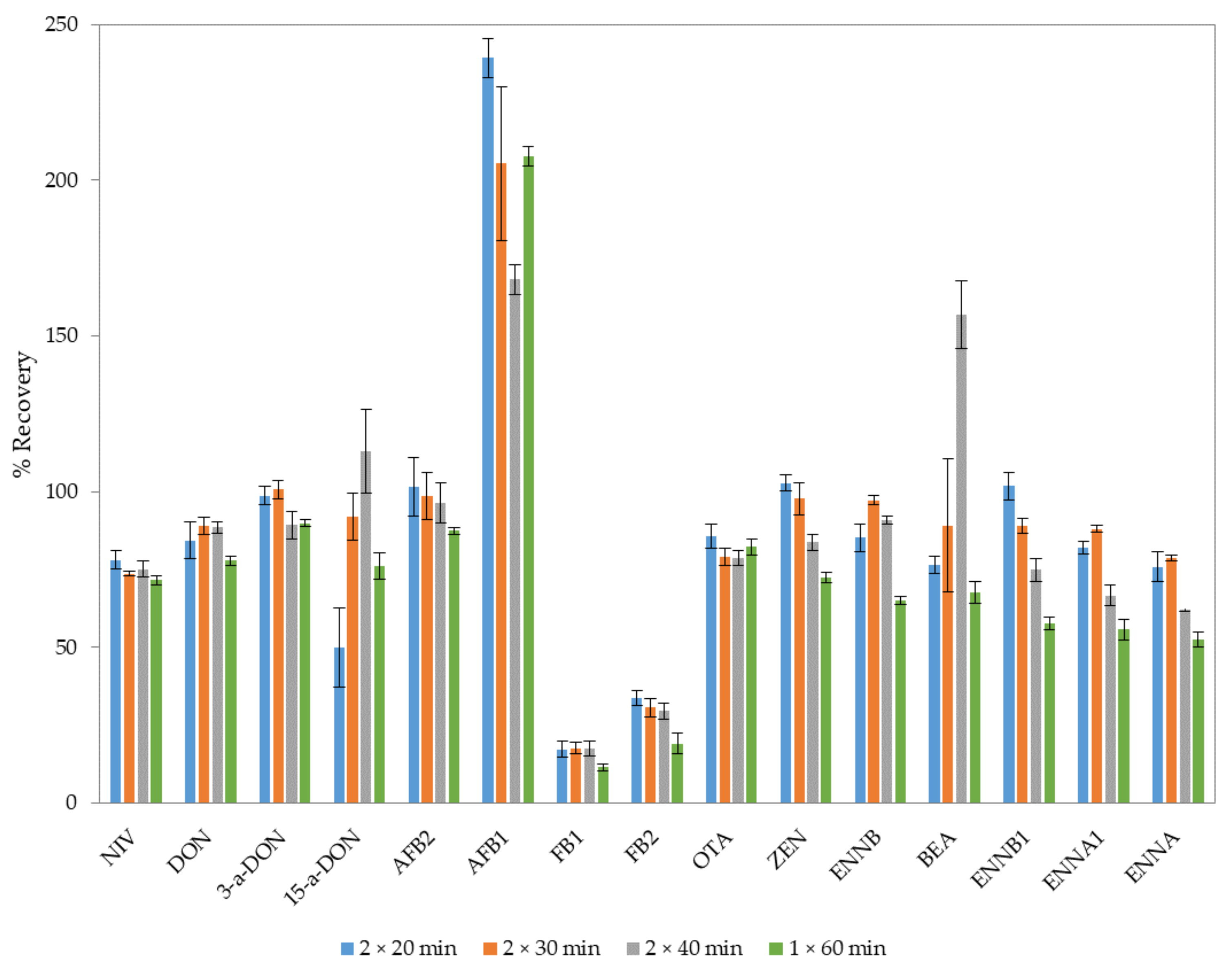
2.2. Optimization of the Extraction Procedure
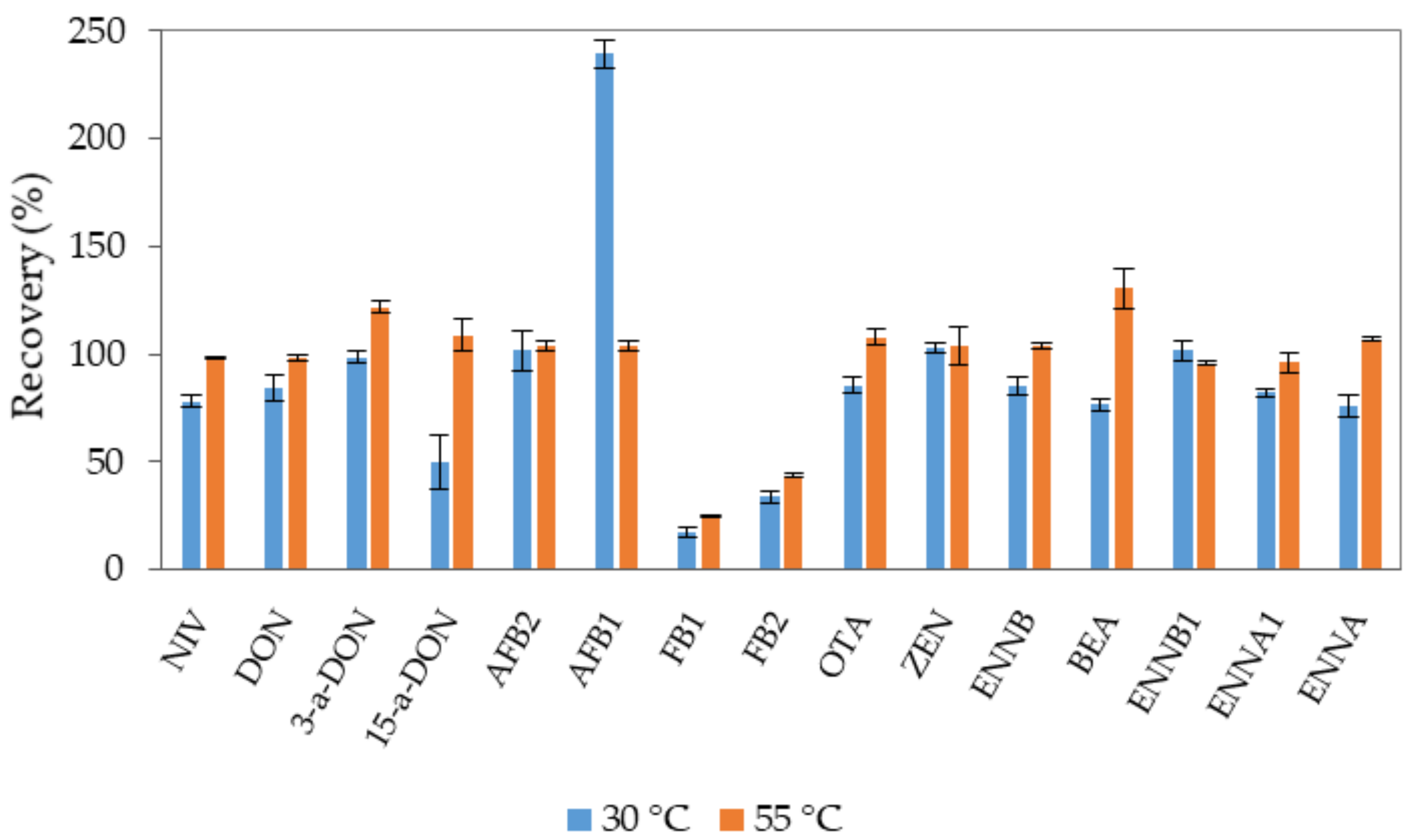
2.3. Method Validation
2.4. Real Samples
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Samples
4.3. Sample Preparation
4.4. LC-MS/MS Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. The Future of Food and Agriculture: Trends and Challenges; FAO: Rome, Italy, 2017; ISBN 978-92-5-109551-5. [Google Scholar]
- FAO. The State of World Fisheries and Aquaculture 2020. Sustainability in Action; FAO: Rome, Italy, 2020. [Google Scholar]
- Nácher-Mestre, J.; Beltrán, E.; Strachan, F.; Dick, J.R.; Pérez-Sánchez, J.; Berntssen, M.H.G.; Tocher, D.R. No transfer of the non-regulated mycotoxins, beauvericin and enniatins, from feeds to farmed fish reared on plant-based diets. Food Chem. 2020, 323, 126773. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.S.; Cunha, S.C.; Fernandes, J.O. Prevalent mycotoxins in animal feed: Occurrence and analytical methods. Toxins 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.; Vasconcelos, V. Occurrence of mycotoxins in fish feed and its effects: A review. Toxins 2020, 12, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, R.A.; Schatzmayr, D.; Albalat, A.; Mackenzie, S. Mycotoxins in aquaculture: Feed and food. Rev. Aquac. 2020, 12, 145–175. [Google Scholar] [CrossRef]
- Directive 2002/32/EC of the European Parliament and of the Council of 7 May 2002 on Undesirable Substances in Animal Feed—Council Statement. Available online: http://data.europa.eu/eli/dir/2002/32/oj (accessed on 28 February 2022).
- Commission Recommendation of 17 August 2006 on the Presence of Deoxynivalenol, Zearalenone, Ochratoxin A, T-2 and HT-2 and Fumonisins in Products Intended for Animal Feeding. Available online: http://data.europa.eu/eli/reco/2006/576/oj (accessed on 28 February 2022).
- Tebele, S.M.; Gbashi, S.; Adebo, O.; Changwa, R.; Naidu, K.; Njobeh, P.B. Quantification of multi-mycotoxin in cereals (maize, maize porridge, sorghum and wheat) from Limpopo province of South Africa. Food Addit. Contam. Part A 2020, 37, 1922–1938. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Glauner, T.; Berthiller, F.; Krska, R.; Schuhmacher, R.; Sulyok, M. Development and validation of a (semi-)quantitative UHPLC-MS/MS method for the determination of 191 mycotoxins and other fungal metabolites in almonds, hazelnuts, peanuts and pistachios. Anal. Bioanal. Chem. 2013, 405, 5087–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.B.; Song, N.E.; Nam, T.G.; Lee, S.; Seo, D.; Yoo, M. Occurrence of emerging mycotoxins in cereals and cereal-based products from the Korean market using LC-MS/MS. Food Addit. Contam. Part A 2019, 36, 289–295. [Google Scholar] [CrossRef]
- Rausch, A.; Brockmeyer, R.; Schwerdtle, T. Development and Validation of a QuEChERS-Based Liquid Chromatography Tandem Mass Spectrometry Multi-Method for the Determination of 38 Native and Modi fi ed Mycotoxins in Cereals. J. Agric. Food Chem. 2020, 68, 4657–4669. [Google Scholar] [CrossRef]
- Anater, A.; Manyes, L.; Meca, G.; Ferrer, E.; Luciano, F.B.; Pimpão, C.T.; Font, G. Mycotoxins and their consequences in aquaculture: A review. Aquaculture 2016, 451, 1–10. [Google Scholar] [CrossRef]
- Scarpino, V.; Reyneri, A.; Blandino, M. Development and comparison of two multiresidue methods for the determination of 17 Aspergillus and Fusarium mycotoxins in cereals using HPLC-ESI-TQ-MS/MS. Front. Microbiol. 2019, 10, 361. [Google Scholar] [CrossRef]
- Juan, C.; Oueslati, S.; Mañes, J.; Berrada, H. Multimycotoxin determination in Tunisian farm animal feed. J. Food Sci. 2019, 84, 3885–3893. [Google Scholar] [CrossRef] [PubMed]
- Dagnac, T.; Latorre, A.; Fernández Lorenzo, B.; Llompart, M. Validation and application of a liquid chromatography-tandem mass spectrometry based method for the assessment of the co-occurrence of mycotoxins in maize silages from dairy farms in NW Spain. Food Addit. Contam. Part A 2016, 33, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Albero, B.; Sánchez-Brunete, C.; Miguel, E.; Tadeo, J.L. Application of matrix solid-phase dispersion followed by GC–MS/MS to the analysis of emerging contaminants in vegetables. Food Chem. 2017, 217, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Albero, B.; Tadeo, J.L.; Pérez, R.A. Determination of emerging contaminants in cereals by gas chromatography-tandem mass spectrometry. Front. Chem. 2020, 8, 571668. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.L.; Fernandes, J.O.; Cunha, S.C. Mycotoxins in cereals and related foodstuffs: A review on occurrence and recent methods of analysis. Trends Food Sci. Technol. 2014, 36, 96–136. [Google Scholar] [CrossRef]
- De Boevre, M.; Di Mavungu, J.D.; Maene, P.; Audenaert, K. Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolites in different cereals and cereal-derived food. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Natural occurrence of emerging Fusarium mycotoxins in feed and fish from aquaculture. J. Agric. Food Chem. 2014, 62, 12462–12470. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Panel Scientific Opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. Efsa J. 2014, 12, 3802. [Google Scholar]
- Lhotská, I.; Gajdošová, B.; Solich, P.; Šatínský, D. Molecularly imprinted vs. reversed-phase extraction for the determination of zearalenone: A method development and critical comparison of sample clean-up efficiency achieved in an on-line coupled SPE chromatography system. Anal. Bioanal. Chem. 2018, 410, 3265–3273. [Google Scholar] [CrossRef]
- López-Blanco, R.; Nortes-Méndez, R.; Robles-Molina, J.; Molina-Díaz, A. Evaluation of different cleanup sorbents for multiresidue pesticide analysis in fatty vegetable matrices by liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1456, 89–104. [Google Scholar] [CrossRef]
- Slámová, T.; Sadowska-Rociek, A.; Fraňková, A.; Surma, M.; Banout, J. Application of QuEChERS-EMR-Lipid-DLLME method for the determination of polycyclic aromatic hydrocarbons in smoked food of animal origin. J. Food Compos. Anal. 2020, 87, 103420. [Google Scholar] [CrossRef]
- Lucas, D.; Zhao, L. Multiclass Mycotoxin Analysis in Cheese Using Agilent Captiva EMR—Lipid Cleanup and LC/MS/MS; Agilent Technologies Application Note; Agilent Technologies: Santa Clara, CA, USA, 2017; Volume 5991-8694, pp. 1–8. [Google Scholar]
- Lucas, D. Mycotoxin Analysis in Infant Formula Using Captiva EMR—Lipid Cleanup and LC/MS/MS; Agilent Technologies Application Note; Agilent Technologies: Santa Clara, CA, USA, 2019; Volume 5994-0365, pp. 1–6. [Google Scholar]
- Arce-López, B.; Lizarraga, E.; Flores-Flores, M.; Irigoyen, Á.; González-Peñas, E. Development and validation of a methodology based on Captiva EMR-lipid clean-up and LC-MS/MS analysis for the simultaneous determination of mycotoxins in human plasma. Talanta 2020, 206, 120193. [Google Scholar] [CrossRef]
- Nakhjavan, B.; Ahmed, N.S.; Khosravifard, M. Development of an improved method of sample extraction and quantitation of multi-mycotoxin in feed by LC-MS/MS. Toxins 2020, 12, 462. [Google Scholar] [CrossRef]
- Stroka, J.; Petz, M.; Joerissen, U.; Anklam, E. Investigation of various extractants for the analysis of aflatoxin B1 in different food and feed matrices. Food Addit. Contam. 1999, 16, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, C.; Zhu, Q.; Du, H.; Guan, S.; Wang, F.; Zhang, W.; Fan, W.; Chen, Z.; Yang, G.; et al. Reduction of matrix effects through a simplified QuEChERS method and using small injection volumes in a LC-MS/MS system for the determination of 28 pesticides in fruits and vegetables. Anal. Methods 2016, 8, 5061–5069. [Google Scholar] [CrossRef]
- Mwihia, E.W.; Lyche, J.L.; Mbuthia, P.G.; Ivanova, L.; Uhlig, S.; Gathumbi, J.K.; Maina, J.G.; Eshitera, E.E.; Eriksen, G.S. Co-Occurrence and levels of mycotoxins in fish feeds in Kenya. Toxins 2020, 12, 627. [Google Scholar] [CrossRef]
- Konak, I.; Yatmaz, H.A.; Nilüfer; Erkaymaz, T.; Certel, M. Multiresidue method for the simultaneous analysis of antibiotics and mycotoxins in feeds by ultra-high performance liquid chromatography coupled to tandem mass spectrometry. Acta Aliment. 2021, 50, 74–82. [Google Scholar] [CrossRef]
- Bernal, E. Limit of detection and limit of quantification determination in gas chromatography. Adv. Gas Chromatogr. 2014, 3, 57–63. [Google Scholar]
- Jedziniak, P.; Pietruszka, K.; Burek, O.; Wiśniewska-Dmytrow, H. Mycotoxin determination in animal feed by UPLC-MS/MS Development of a UPLC-MS/MS method for determination of mycotoxins in animal Feed. Euroreference 2016, 1, 63–69. [Google Scholar]
- Bernal-Algaba, E.; Pulgarín-Alfaro, M.; Fernández-Cruz, M.L. Cytotoxicity of mycotoxins frequently present in aquafeeds to the fish cell line RTGill-W1. Toxins 2021, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- Fegan, D.F.; Spring, P. Recognizing the reality of the aquaculture mycotoxin problem: Searching for a common and effective solution. In Nutritional Biotechnology in the Feed and Food Industries, Proceedings of Alltech’s 23rd Annual Symposium. The New Energy Crisis: Food, Feed or Fuel, Lexington, KY, USA, 20–23 May 2007; Alltech: Lexington, KY, USA, 2007; pp. 343–354. [Google Scholar]
- Woźny, M.; Obremski, K.; Jakimiuk, E.; Gusiatin, M.; Brzuzan, P. Zearalenone contamination in rainbow trout farms in north-eastern Poland. Aquaculture 2013, 416–417, 209–211. [Google Scholar] [CrossRef]
- Nácher-Mestre, J.; Serrano, R.; Beltrán, E.; Pérez-Sánchez, J.; Silva, J.; Karalazos, V.; Hernández, F.; Berntssen, M.H.G. Occurrence and potential transfer of mycotoxins in gilthead sea bream and Atlantic salmon by use of novel alternative feed ingredients. Chemosphere 2015, 128, 314–320. [Google Scholar] [CrossRef] [Green Version]
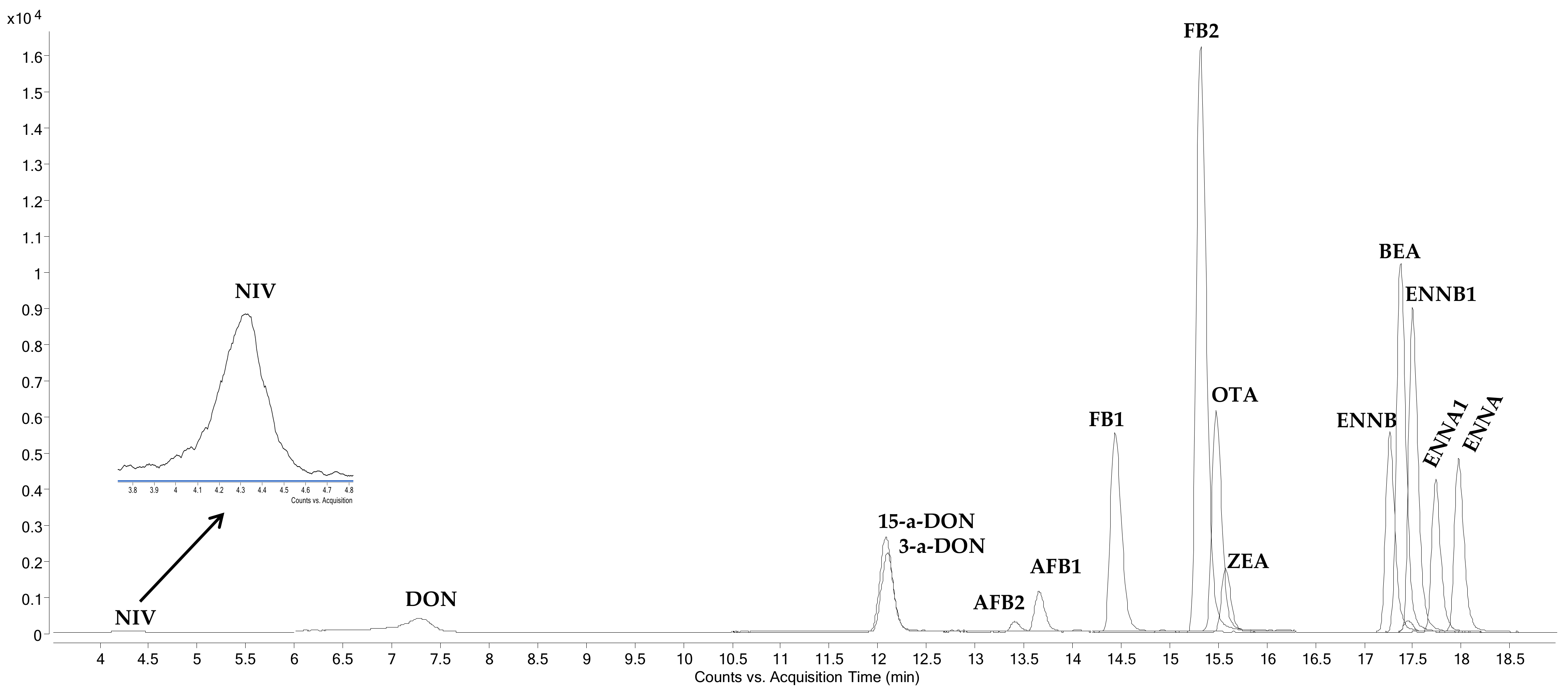


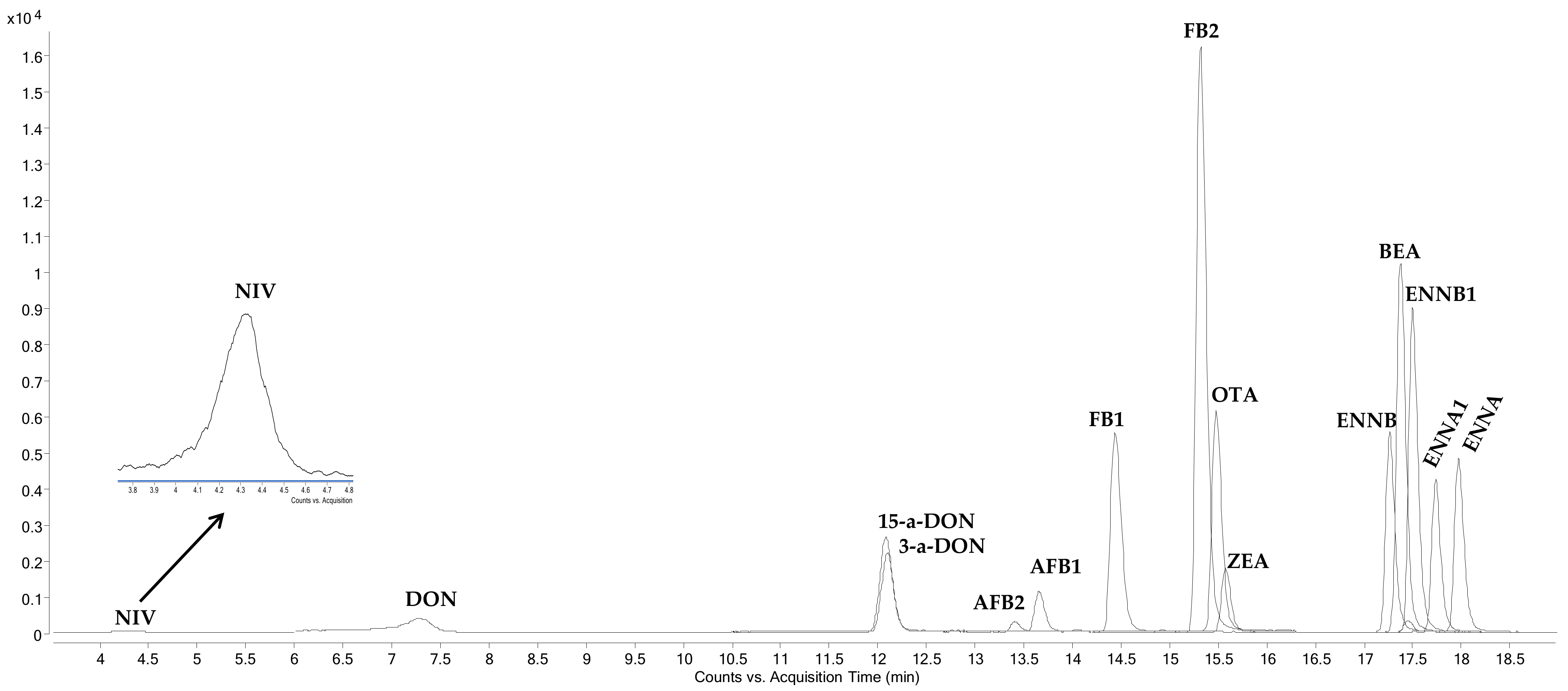{kind=link}
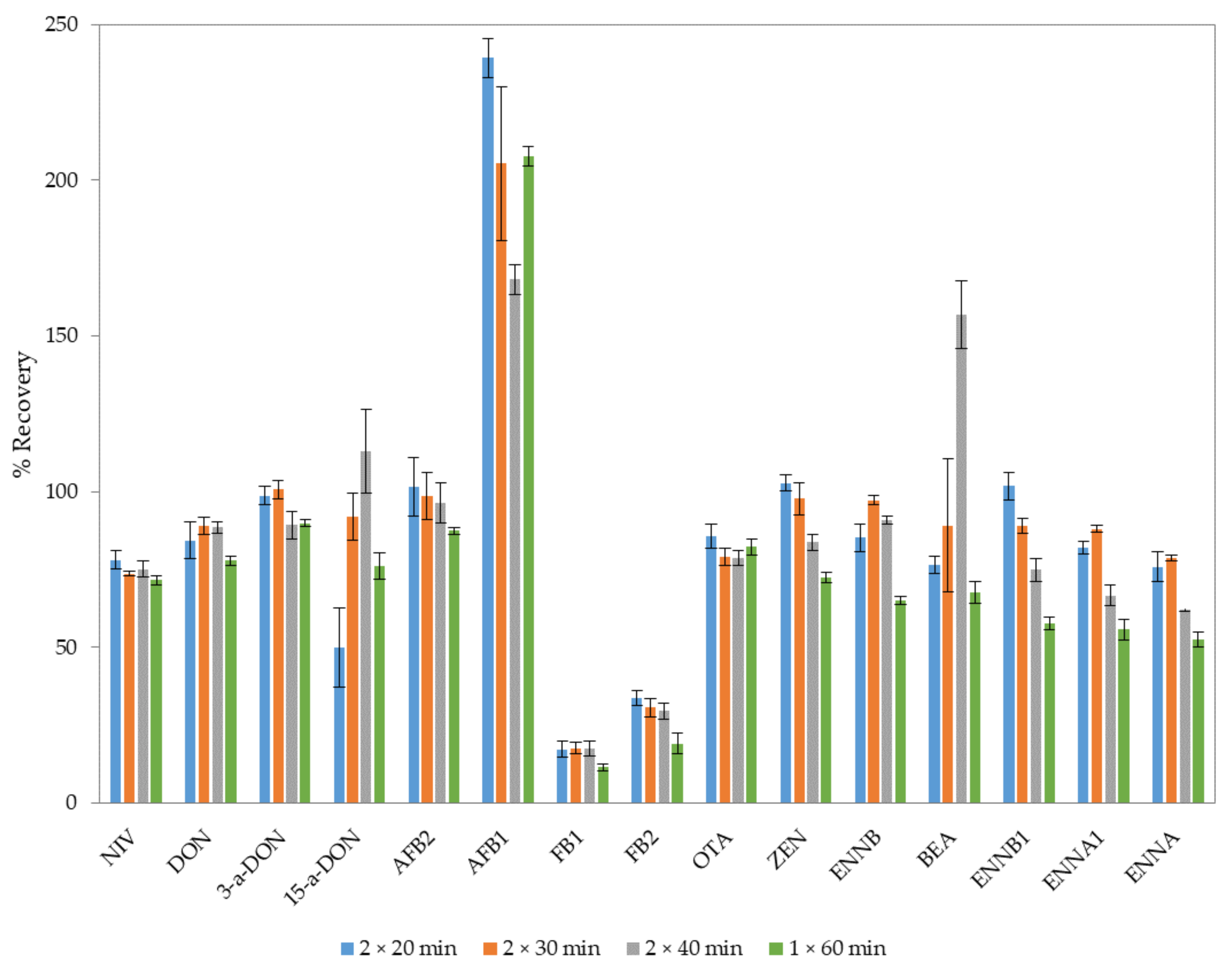{kind=link}
{kind=link}
| Compounds | After Extraction | Before Extraction | ||||
|---|---|---|---|---|---|---|
| dSPE-PSA | LLE-Hexane | Hexane Defatting | ||||
| Mean | SD | Mean | SD | Mean | SD | |
| NIV | 97.6 | 7.0 | 82.8 | 8.3 | 80.2 | 3.3 |
| DON | 104.0 | 4.9 | 85.8 | 7.1 | 90.2 | 2.6 |
| 3-a-DON | 97.5 | 2.4 | 85.7 | 2.9 | 110.9 | 14.0 |
| 15-a-DON | 48.1 | 3.1 | 43.3 | 2.1 | 89.2 | 1.9 |
| AFB2 | 75.9 | 5.3 | 74.2 | 2.5 | 97.4 | 5.5 |
| AFB1 | 72.7 | 4.1 | 63.2 | 0.9 | 96.4 | 2.6 |
| FB1 | 14.9 | 5.4 | 12.8 | 0.9 | 44.7 | 2.6 |
| FB2 | 42.0 | 3.2 | 4.3 | 1.1 | 43.7 | 3.5 |
| OTA | 12.9 | 0.5 | 14.4 | 2.0 | 60.4 | 4.2 |
| ZEN | 1.2 | 0.3 | 6.0 | 0.8 | 42.5 | 0.3 |
| ENNB | 0.8 | 0.3 | 73.6 | 10.5 | ||
| BEA | 1.2 | 1.1 | 2.3 | 0.1 | ||
| ENNB1 | 0.8 | 0.3 | 2.3 | 1.7 | 39.3 | 2.4 |
| ENNA1 | 2.0 | 0.9 | 1.2 | 0.3 | 33.7 | 3.3 |
| ENNA | 1.6 | 0.6 | 14.6 | 0.3 | 24.7 | 2.7 |
| Linearity Range μg/kg | Spiking Level μg/kg | Recovery (%) | Spiking Level μg/kg | Recovery (%) | Spiking Level μg/kg | Recovery (%) | LOD | LOQ | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | μg/kg | μg/kg | |||||
| NIV | 200–21,000 | 150 | 750 | 76 | 9 | 1500 | 88 | 1 | 54 | 180 | ||
| DON | 70–1400 | 100 | 104 | 3 | 500 | 91 | 4 | 1000 | 102 | 2 | 11 | 32 |
| 3-a-DON | 18–3500 | 25 | 127 | 8 | 125 | 91 | 6 | 250 | 122 | 5 | 3 | 9 |
| 15-a-DON | 18–3500 | 25 | 90 | 7 | 125 | 91 | 7 | 250 | 109 | 8 | 3 | 9 |
| AFB2 | 0.7–140 | 1 | 113 | 8 | 5 | 87 | 6 | 10 | 94 | 3 | 0.2 | 0.6 |
| AFB1 | 0.7–140 | 1 | 102 | 10 | 5 | 92 | 6 | 10 | 95 | 4 | 0.1 | 0.4 |
| FB1 | 70–14,000 | 100 | 10 | 2 | 500 | 22. | 7 | 1000 | 25 | 1 | 6 | 18 |
| FB2 | 70–14,000 | 100 | 21 | 6 | 500 | 35 | 7 | 1000 | 44 | 1 | 9 | 27 |
| OTA | 7–1400 | 10 | 100 | 16 | 50 | 79 | 6 | 100 | 108 | 3 | 2 | 6 |
| ZEN | 105–21,000 | 150 | 119 | 18 | 750 | 51 | 5 | 1500 | 104 | 9 | 20 | 59 |
| ENNB | 0.7–140 | 1 | 104 | 5 | 5 | 75 | 6 | 10 | 104 | 2 | 0.08 | 0.25 |
| BEA | 0.7–140 | 1 | 118 | 9 | 5 | 96 | 6 | 10 | 130 | 9 | 0.05 | 0.16 |
| ENNB1 | 7–1400 | 10 | 64 | 6 | 50 | 70 | 7 | 100 | 96 | 1 | 0.1 | 0.3 |
| ENNA1 | 0.7–140 | 1 | 66 | 5 | 5 | 72 | 9 | 10 | 96 | 5 | 0.2 | 0.5 |
| ENNA | 0.7–140 | 1 | 58 | 6 | 5 | 66 | 9 | 10 | 107 | 1 | 0.2 | 0.7 |
| Mycotoxin | Matrix | ||||||
|---|---|---|---|---|---|---|---|
| Cereals a | Maize Silage b | Animal Feed c | Cereal and Derivatives d | Fish Feed e | Fish Feed f | Fish Feed g | |
| NIV | 100 | 134 | 180 | ||||
| DON | 60–214 | 57 | 100 | 135 | 32 | ||
| 3-a-DON | 0.15–0.4 | 2.6 h | 50 | 120 | 9 | ||
| 15-a-DON | 59.6–110.8 | 2.6 h | 409 | 9 | |||
| AFB1 | 0.04–12.3 | 0.08–0.5 | 1 | 49 | 1.2 | ||
| AFB2 | 0.04–9.3 | 1 | 60 | 0.4 | |||
| FB1 | 0.03–22.8 | 2.9 | 50 | 209 | 18 | ||
| FB2 | 1.2–20.8 | 6.4 | 50 | 230 | 27 | ||
| OTA | 0.3–1.9 | 0.48 | 5 | 91 | 6 | ||
| ZEN | 0.05–11.4 | 5.6 | 10 | 127 | 59 | ||
| ENNB | 0.08 | 0.2 | 129 | 0.1 | 0.25 | ||
| BEA | 6.1 | 53 | 0.1 | 0.16 | |||
| ENNB1 | 0.08 | 7.2 | 43 | 0.1 | 0.3 | ||
| ENNA1 | 4.1 | 45 | 0.25 | 0.5 | |||
| ENNA | 1.3 | 87 | 0.5 | ||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| 15-a-DON | 55 ± 2 | 21 ± 3 | <LOQ | ||||||
| FB1 | <LOQ | <LOQ | <LOQ | 136 ± 16 | <LOQ | ||||
| FB2 | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | 105 ± 1 | <LOQ | <LOQ | <LOQ |
| ZEN | <LOQ | 121 ± 14 | |||||||
| ENNB | 1.1 ± 0.1 | 1.6 ± 0.1 | 21 ± 1 | 0.94 ± 0.02 | 6.1 ± 1.0 | 6.5 ± 0.6 | 0.6 ± 0.1 | 2.6 ± 0.2 | 3.1 ± 0.1 |
| BEA | 6.6 ± 0.1 | 0.56 ± 0.04 | 0.5 ± 0.1 | 6.5 ± 0.3 | 9.4 ± 1.0 | 30 ± 2 | 0.9 ± 0.1 | 0.54 ± 0.03 | 16 ± 0.5 |
| ENNB1 | 0.7 ± 0.2 | 1.5 ± 0.7 | 7.9 ± 1.2 | 0.7 ± 0.3 | 3.4 ± 0.2 | 2.3 ± 0.9 | 1.9 ± 0.8 | 2.4 ± 0.2 | 1.8 ± 0.3 |
| ENNA1 | <LOQ | 1.9 ± 0.5 | 0.5 ± 0.1 | 0.47 ± 0.01 |
| Compound | MRM 1 | CE (eV) | MRM 2 | CE (eV) | Fragmentor (V) | Polarity |
|---|---|---|---|---|---|---|
| NIV | 357.1 > 45 | 42 | 357.1 > 281.1 | 22 | 150 | Negative |
| DON | 297.1 > 249.1 | 10 | 297.1 > 203.1 | 10 | 100 | Positive |
| 15-a-DON | 356.1 > 137.1 | 15 | 356.1 > 261.2 | 15 | 90 | Positive |
| 3-a-DON | 339.1 > 231.1 | 10 | 339.1 > 213.1 | 20 | 110 | Positive |
| AFB2 | 315.2 > 287 | 24 | 315.2 > 259 | 28 | 190 | Positive |
| AFB1 | 313 > 285.2 | 20 | 313.2 > 259 | 38 | 190 | Positive |
| FB1 | 722.5 > 334.4 | 44 | 722.5 > 352.3 | 36 | 210 | Positive |
| FB2 | 706.3 > 336.3 | 40 | 706.3 > 318.5 | 40 | 220 | Positive |
| OTA | 404 > 239 | 20 | 404.1 > 221 | 36 | 115 | Positive |
| ZEN | 317 > 131 | 28 | 317 > 175 | 20 | 195 | Negative |
| ENNB | 657 > 196 | 32 | 657 > 214 | 32 | 160 | Positive |
| BEA | 801.5 > 244 | 36 | 801.5 > 262 | 32 | 180 | Positive |
| ENNB1 | 672 > 196 | 32 | 671.4 > 214 | 60 | 170 | Positive |
| ENNA1 | 685 > 210 | 32 | 685 > 228 | 32 | 150 | Positive |
| ENNA | 699 > 210 | 32 | 699 > 228 | 32 | 170 | Positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albero, B.; Fernández-Cruz, M.L.; Pérez, R.A. Simultaneous Determination of 15 Mycotoxins in Aquaculture Feed by Liquid Chromatography–Tandem Mass Spectrometry. Toxins 2022, 14, 316. https://doi.org/10.3390/toxins14050316
Albero B, Fernández-Cruz ML, Pérez RA. Simultaneous Determination of 15 Mycotoxins in Aquaculture Feed by Liquid Chromatography–Tandem Mass Spectrometry. Toxins. 2022; 14(5):316. https://doi.org/10.3390/toxins14050316
Chicago/Turabian StyleAlbero, Beatriz, María Luisa Fernández-Cruz, and Rosa Ana Pérez. 2022. "Simultaneous Determination of 15 Mycotoxins in Aquaculture Feed by Liquid Chromatography–Tandem Mass Spectrometry" Toxins 14, no. 5: 316. https://doi.org/10.3390/toxins14050316
APA StyleAlbero, B., Fernández-Cruz, M. L., & Pérez, R. A. (2022). Simultaneous Determination of 15 Mycotoxins in Aquaculture Feed by Liquid Chromatography–Tandem Mass Spectrometry. Toxins, 14(5), 316. https://doi.org/10.3390/toxins14050316