Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern

 , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
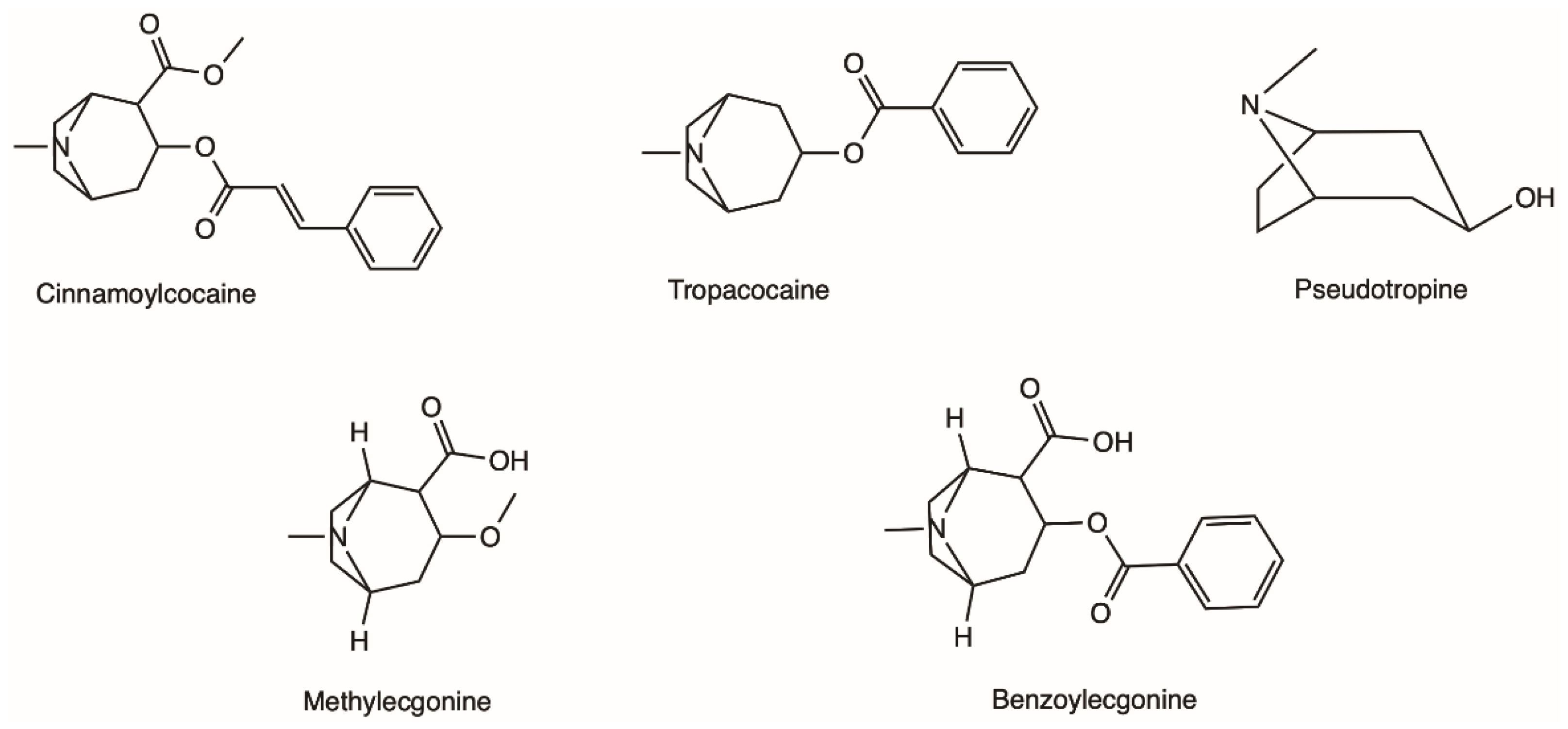
2.1. Natural Occurrence and Chemical Characterisation of Erythroxylum coca
2.2. Physicochemical Properties of Cocaine and Analytical Methods for Identification
2.3. Legal Status
2.4. Prevalence, Patterns of (Ab) Use and Public Health Concerns
2.5. Pharmacokinetics
2.5.1. Absorption
2.5.2. Distribution
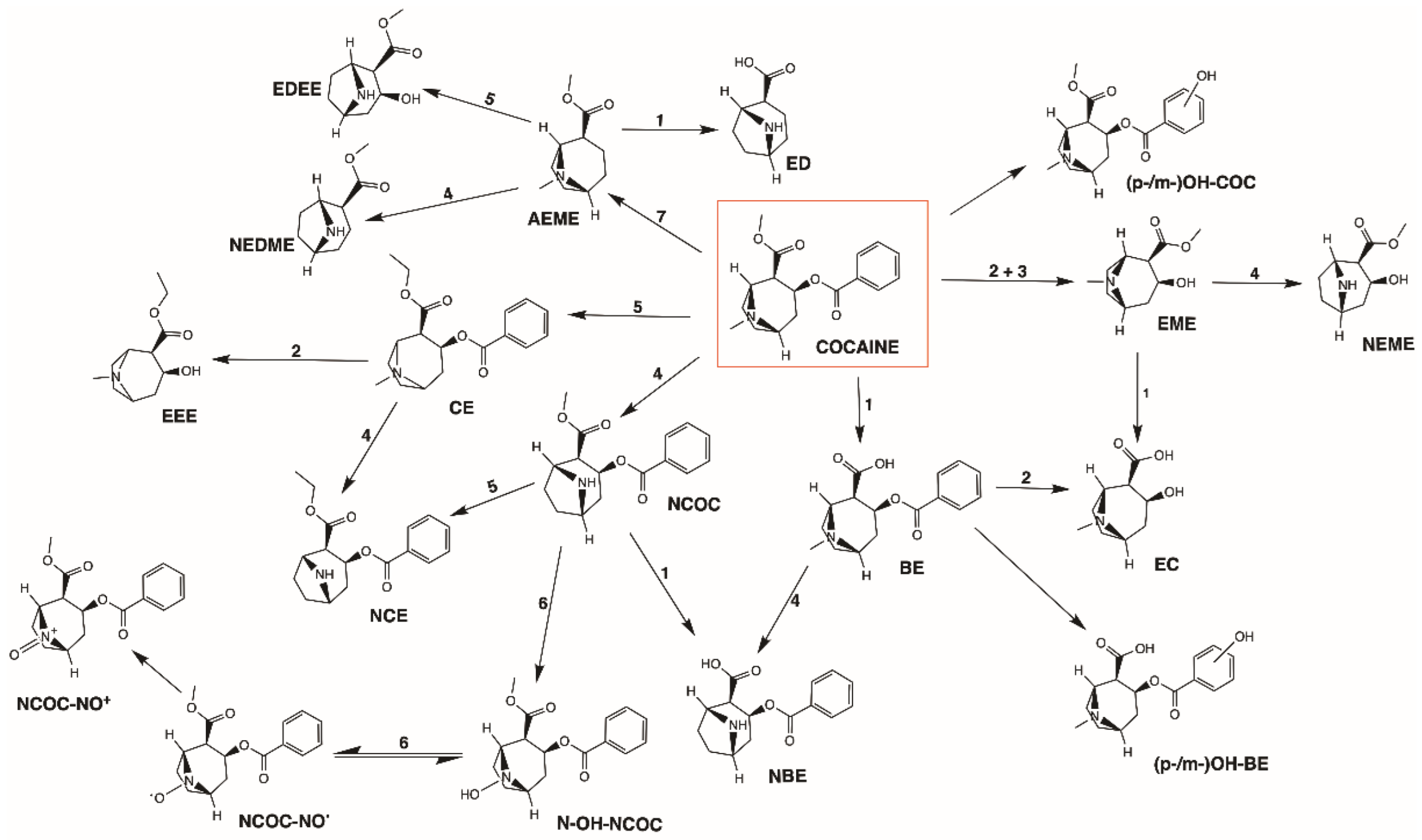
2.5.3. Metabolism
2.5.4. Excretion
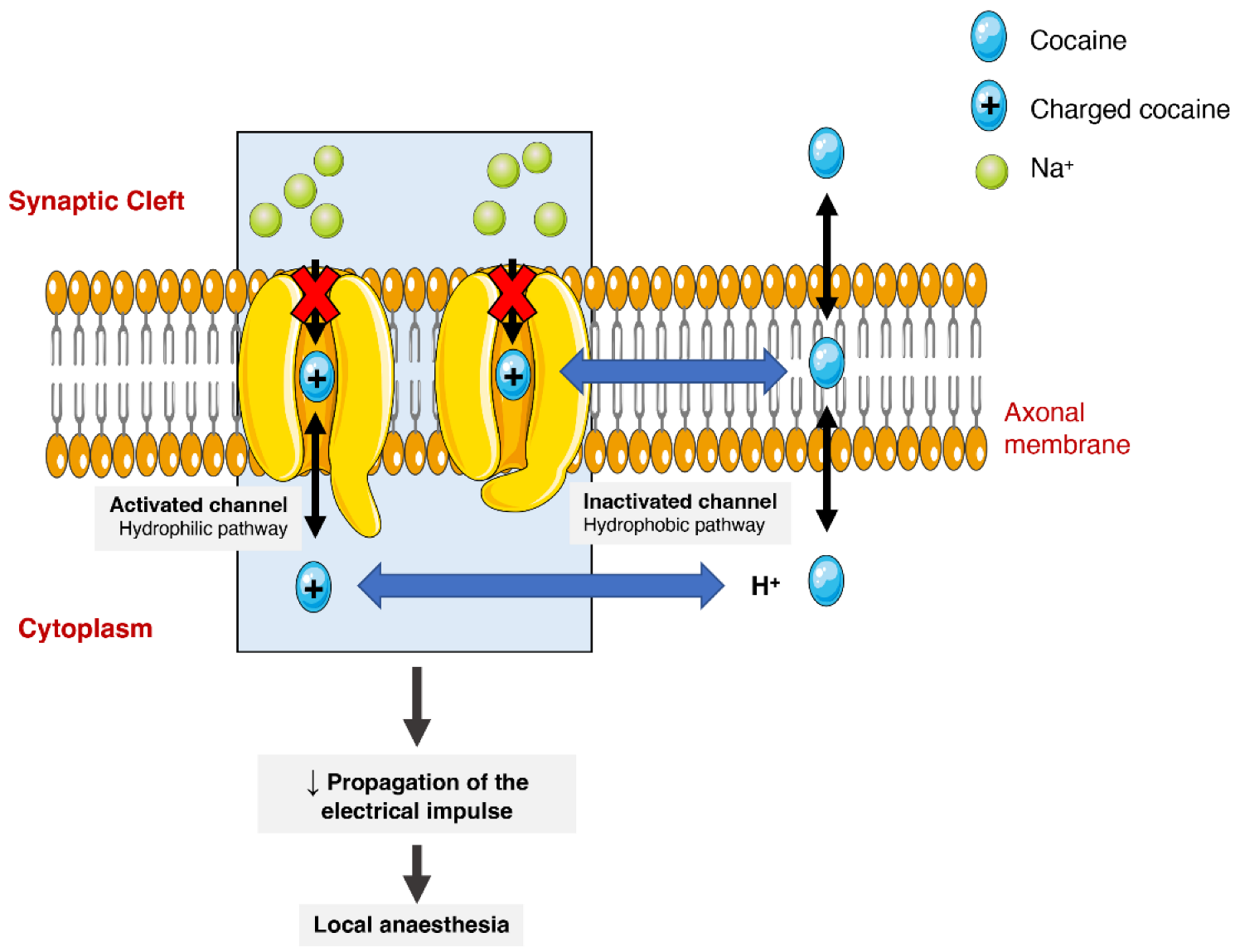
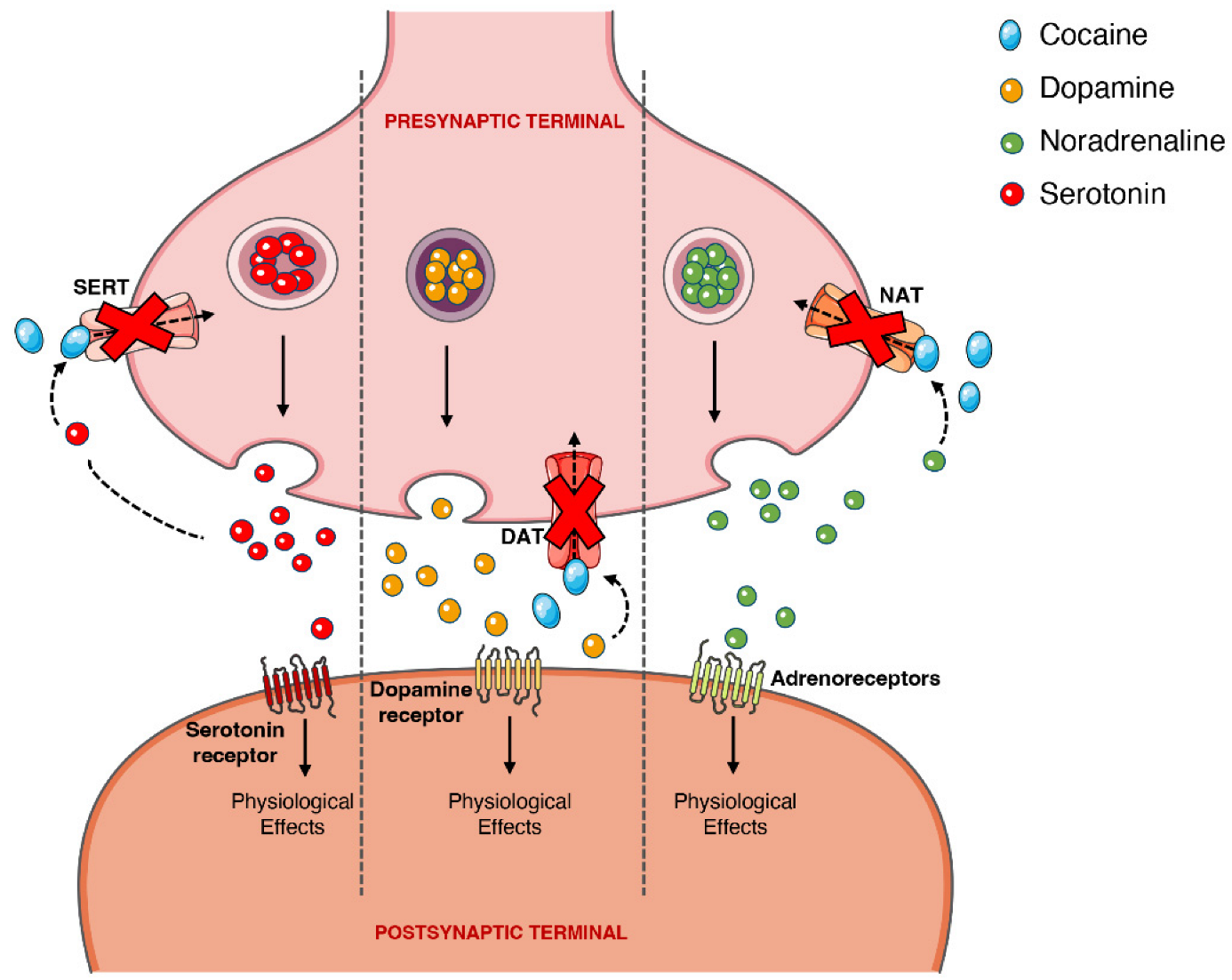
2.6. Pharmacodynamics
2.7. Effects and Toxicity of Cocaine
2.7.1. Subjective and Physiological Effects
2.7.2. Hyperthermia
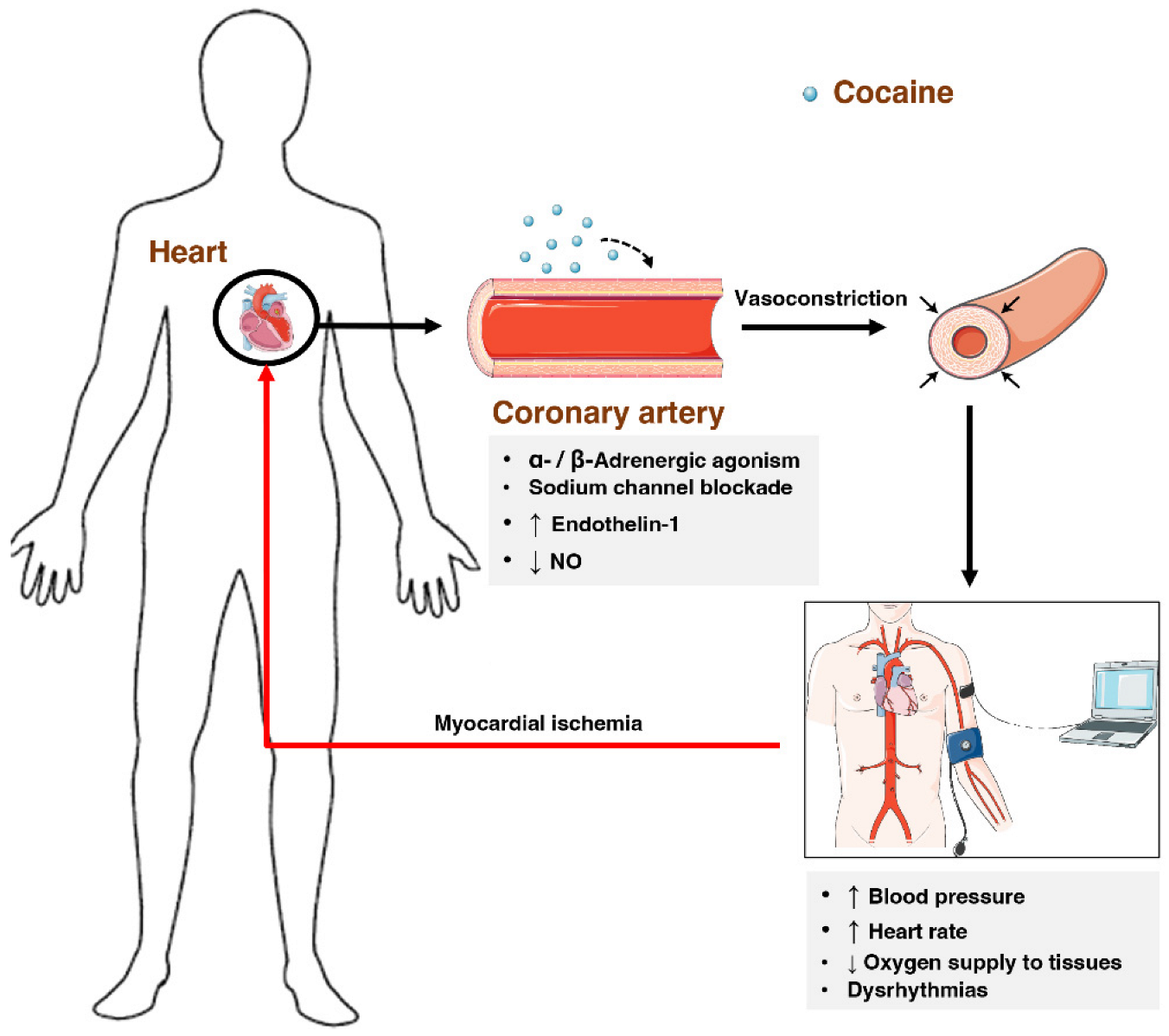
2.7.3. Cardiovascular System
2.7.4. Respiratory System
2.7.5. Renal System
2.7.6. Brain
2.7.7. Liver
2.8. Abuse Potential, Dependence, and Tolerance
2.9. Polydrug Use
2.9.1. Alcohol
2.9.2. Heroin/Opioids
2.10. Management of Acute Intoxications and Cocaine Use Disorder
2.10.1. Treatment of Acute Intoxication
2.10.2. Treatment of Cocaine Addiction/Dependence
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AEME | anhydroecgonine methyl ester |
| ATP | adenosine triphosphate |
| BBB | blood brain barrier |
| BE | benzoylecgonine |
| CE | cocaethylene |
| CNS | central nervous system |
| CUD | cocaine use disorder |
| CYP450 | cytochrome P450 |
| DAT | dopamine transporter |
| DSM-5 | Diagnostics and Statistics Manual of Mental Disorders 5th edition |
| EC | ecgonine |
| ED | ecgonidine |
| EDEE | ecgonidine ethyl ester |
| EEE | ecgonine ethyl ester |
| EMCDDA | European Monitoring Centre for Drug and Drug Addiction |
| EME | ecgonine methyl ester |
| FADM | flavin adenine dinucleotide-containing monooxygenase |
| GC-MS | gas chromatography coupled with mass spectrometry |
| GPx | glutathione peroxidase |
| GSH | reduced glutathione |
| hCE1 | human carboxylesterase type 1 |
| hCE2 | human carboxylesterase type 2 |
| HPLC | high performance liquid chromatography |
| LC-MS/MS | liquid chromatography coupled with tandem mass spectrometry |
| NAT | noradrenaline transporter |
| NBE | norbenzoylecgonine |
| NCE | norcocaethylene |
| NCOC | norcocaine |
| NCOC-NO• | orcocaine nitroxide |
| NCOC-NO+ | norcocaine nitrosonium |
| NEDME | norecgonidine methyl ester |
| NEME | norecgonine methyl ester |
| NMDA | N-methyl-D-aspartate |
| N-OH-NCOC | N-hydroxy-norcocaine |
| (p-/m-)OH-BE | (para-/meta-)hydroxybenzoylecgonine |
| (p-/m-)OH-COC | (para-/meta-)hydroxycocaine |
| PChE | pseudocholinesterase |
| ROS | reactive oxygen species |
| SERT | serotonin transporter |
| SOD | superoxide dismutase |
| UPLC-MS/MS | ultra-performance liquid chromatography coupled with tandem mass spectrometry |
References
- Drake, L.R.; Scott, P.J.H. DARK Classics in Chemical Neuroscience: Cocaine. ACS Chem. Neurosci. 2018, 9, 2358–2372. [Google Scholar] [CrossRef]
- Phillips, K.; Luk, A.; Soor, G.S.; Abraham, J.R.; Leong, S.; Butany, J. Cocaine cardiotoxicity: A review of the pathophysiology, pathology, and treatment options. Am. J. Cardiovasc. Drugs. 2009, 9, 177–196. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Carvalho, F.; Duarte, J.A.; Proenca, J.B.; Santos, A.; Magalhaes, T. Clinical and forensic signs related to cocaine abuse. Curr. Drug. Abuse. Rev. 2012, 5, 64–83. [Google Scholar] [CrossRef]
- Wang, J.; Deng, X.; Wu, Y.; Huang, Y.; Hou, S.; Zhang, Y.; Qiu, T.; Tong, J.; Chen, X. Sub-lethal toxicity and elimination of the cocaine metabolite, benzoylecgonine: A narrative review. Ann. Palliat. Med. 2021, 10, 6936–6947. [Google Scholar] [CrossRef] [PubMed]
- UNODC. World Drug Report 2021; United Nations: Vienna, Austria, 2021. [Google Scholar]
- EMCDDA. European Drug Report 2021: Trends and Development; 1977-9860; European Monitoring Centre for Drugs and Drug Addiction: Luxembourg, 2021. [Google Scholar]
- Ali, S.F.; Hoglund, J.R.; Gibbs, M.A.; Littmann, L. Unusual electrocardiographic manifestations of lethal cocaine toxicity. Clin. Toxicol. 2021, 60, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Arenas, D.J.; Beltran, S.; Zhou, S.; Goldberg, L.R. Cocaine, cardiomyopathy, and heart failure: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 19795. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Roby, A.; Jaconelli, T. Pneumomediastinum, subcutaneous emphysema and pneumorrhachis following cocaine insufflation: A case report. Acute. Med. 2020, 19, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, O.; Garcia de de Jesus, K.; Then, E.O.; Rehmani, R. Bilateral Basal Ganglia Infarction After Intranasal Use of Cocaine: A Case Report. Cureus 2019, 11, e4405. [Google Scholar] [CrossRef]
- Cosenza, M.; Panza, L.; Califano, A.P.; Defendini, C.; D’Andria, M.; Romiti, R.; Vainieri, A.F.M.; Morelli, S. Carotid Thrombosis in a Crack Cocaine Smoker Woman. Case Rep. Vasc. Med. 2020, 2020, 4894825. [Google Scholar] [CrossRef]
- Deivasigamani, S.; Irrinki, S.; Shah, J.; Sakaray, Y. Rare cause of acute abdomen-cocaine-induced small intestinal perforation with coexisting lower gastrointestinal bleed: An unusual presentation. BMJ Case Rep. 2021, 14, e239981. [Google Scholar] [CrossRef]
- Gill, D.; Sheikh, N.; Ruiz, V.G.; Liu, K. Case report: Cocaine-induced takotsubo cardiomyopathy. Hellenic. J. Cardiol. 2018, 59, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Manninger, M.; Perl, S.; Brussee, H.; G, G.T. Sniff of coke breaks the heart: Cocaine-induced coronary vasospasm aggravated by therapeutic hypothermia and vasopressors after aborted sudden cardiac death: A case report. Eur. Heart J. Case Rep. 2018, 2, yty041. [Google Scholar] [CrossRef] [PubMed]
- Mullaguri, N.; Battineni, A.; Narayan, A.; Guddeti, R. Cocaine Induced Bilateral Posterior Inferior Cerebellar Artery and Hippocampal Infarction. Cureus 2018, 10, e2576. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Seller, A.; Hernandez-Pons, A.; Pascual, E.V.; Comin Perez, A.; Dolz Gaiton, R.; Albert-Fort, M. Severe Cocaine-Induced Midline Destructive Lesions (CIMDL) Leading to Orbital Apex Syndrome and Peripheral Ulcerative Keratitis. Ocul. Immunol. Inflamm. 2021, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Padilha, W.S.C.; Annes, M.; Massant, C.G.; Kaup, A.O.; Affonso, B.B.; Batista, M.C. Cocaine-Induced Renal Artery Dissection as a Cause of Secondary Hypertension: A Rare Presentation. Am. J. Case Rep. 2020, 21, e921565. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Konala, V.M.; Adapa, S.; Naramala, S.; Bose, S. Cocaine and Alcohol Co-Ingestion-Induced Severe Rhabdomyolysis With Acute Kidney Injury Culminating in Hemodialysis-Dependent End-Stage Renal Disease: A Case Report and Literature Review. Cureus 2020, 12, e8595. [Google Scholar] [CrossRef]
- Sharma, R.; Kapoor, N.; Chaudhari, K.S.; Scofield, R.H. Reversible Fulminant Hepatitis Secondary to Cocaine in the Setting of beta-Blocker Use. J. Investig. Med. High. Impact. Case Rep. 2020, 8, 2324709620924203. [Google Scholar] [CrossRef]
- Vermeulen, L.; Dirix, M.; Dendooven, A. Cocaine Consumption and Antineutrophil Cytoplasmic Antibody-associated Glomerulonephritis: A Case Report. Am. J. Forensic. Med. Pathol. 2021, 42, 198–200. [Google Scholar] [CrossRef]
- Biondich, A.S.; Joslin, J.D. Coca: The History and Medical Significance of an Ancient Andean Tradition. Emerg. Med. Int. 2016, 2016, 4048764. [Google Scholar] [CrossRef]
- Stolberg, V.B. The use of coca: Prehistory, history, and ethnography. J. Ethn. Subst. Abuse 2011, 10, 126–146. [Google Scholar] [CrossRef]
- Plowman, T. Botanical perspectives on coca. J. Psychedelic. Drugs. 1979, 11, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.A.; DesLauriers, C.; Burda, A.; Johnson-Arbor, K. Cocaine: History, social implications, and toxicity: A review. Semin. Diagn. Pathol. 2009, 26, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Plowman, T.; Rivier, L. Cocaine and Cinnamoylcocaine Content of Erythroxylum Species. Ann. Bot 1983, 51, 641–659. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 446220, Cocaine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cocaine (accessed on 13 September 2021).
- Siegrist, M.; Wiegand, T.J. Cocaine. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 999–1002. [Google Scholar]
- Chronister, C.W.; Walrath, J.C.; Goldberger, B.A. Rapid detection of benzoylecgonine in vitreous humor by enzyme immunoassay. J. Anal. Toxicol. 2001, 25, 621–624. [Google Scholar] [CrossRef]
- Liakoni, E.; Yates, C.; Dines, A.M.; Dargan, P.I.; Heyerdahl, F.; Hovda, K.E.; Wood, D.M.; Eyer, F.; Liechti, M.E.; Euro, D.E.N.P.R.G. Acute recreational drug toxicity: Comparison of self-reports and results of immunoassay and additional analytical methods in a multicenter European case series. Medicine 2018, 97, e9784. [Google Scholar] [CrossRef]
- Niedbala, R.S.; Kardos, K.; Fries, T.; Cannon, A.; Davis, A. Immunoassay for detection of cocaine/metabolites in oral fluids. J. Anal. Toxicol. 2001, 25, 62–68. [Google Scholar] [CrossRef]
- Cone, E.J.; Sampson-Cone, A.H.; Darwin, W.D.; Huestis, M.A.; Oyler, J.M. Urine testing for cocaine abuse: Metabolic and excretion patterns following different routes of administration and methods for detection of false-negative results. J. Anal. Toxicol. 2003, 27, 386–401. [Google Scholar] [CrossRef]
- Cone, E.J.; Tsadik, A.; Oyler, J.; Darwin, W.D. Cocaine metabolism and urinary excretion after different routes of administration. Ther. Drug. Monit. 1998, 20, 556–560. [Google Scholar] [CrossRef]
- Huestis, M.A.; Darwin, W.D.; Shimomura, E.; Lalani, S.A.; Trinidad, D.V.; Jenkins, A.J.; Cone, E.J.; Jacobs, A.J.; Smith, M.L.; Paul, B.D. Cocaine and metabolites urinary excretion after controlled smoked administration. J. Anal. Toxicol. 2007, 31, 462–468. [Google Scholar] [CrossRef]
- Myers, A.L.; Williams, H.E.; Kraner, J.C.; Callery, P.S. Identification of anhydroecgonine ethyl ester in the urine of a drug overdose victim. J. Forensic. Sci. 2005, 50, 1481–1485. [Google Scholar] [CrossRef]
- Smith, M.L.; Shimomura, E.; Paul, B.D.; Cone, E.J.; Darwin, W.D.; Huestis, M.A. Urinary excretion of ecgonine and five other cocaine metabolites following controlled oral, intravenous, intranasal, and smoked administration of cocaine. J. Anal. Toxicol. 2010, 34, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Dias, M.; Vieira, D.N.; Queiroz, J.A.; Lopez-Rivadulla, M. Development and validation of an analytical method for the simultaneous determination of cocaine and its main metabolite, benzoylecgonine, in human hair by gas chromatography/mass spectrometry. Rapid. Commun. Mass. Spectrom. 2008, 22, 3320–3326. [Google Scholar] [CrossRef] [PubMed]
- Kintz, P.; Sengler, C.; Cirimele, V.; Mangin, P. Evidence of crack use by anhydroecgonine methylester identification. Hum. Exp. Toxicol. 1997, 16, 123–127. [Google Scholar] [CrossRef]
- Menzies, E.L.; Archer, J.R.H.; Dargan, P.I.; Parkin, M.C.; Yamamoto, T.; Wood, D.M.; Braithwaite, R.A.; Elliott, S.P.; Kicman, A.T. Detection of cocaine and its metabolites in whole blood and plasma following a single dose, controlled administration of intranasal cocaine. Drug. Test. Anal. 2019, 11, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.L.; Tebbett, I.R.; Bertholf, R.L. Comparison of HPLC and GC-MS for measurement cocaine and metabolites in human urine. J. Anal. Toxicol. 1996, 20, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.; Cabanillas, L.M.; Olivera, N.M.; Quiroga, P.N. Optimization and validation of simultaneous analyses of ecgonine, cocaine, and seven metabolites in human urine by gas chromatography-mass spectrometry using a one-step solid-phase extraction. Drug. Test. Anal. 2019, 11, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Melanson, S.E.F.; Petrides, A.K.; Khaliq, T.; Griggs, D.A.; Flood, J.G. Comparison of Oral Fluid and Urine for Detection of Cocaine Abuse Using Liquid Chromatography with Tandem Mass Spectrometry. J. Appl. Lab. Med. 2020, 5, 935–942. [Google Scholar] [CrossRef]
- de Lima Feltraco Lizot, L.; da Silva, A.C.C.; Bastiani, M.F.; Hahn, R.Z.; Bulcao, R.; Perassolo, M.S.; Antunes, M.V.; Linden, R. Simultaneous determination of cocaine, ecgonine methyl ester, benzoylecgonine, cocaethylene and norcocaine in dried blood spots by ultra-performance liquid chromatography coupled to tandem mass spectrometry. Forensic. Sci. Int. 2019, 298, 408–416. [Google Scholar] [CrossRef]
- Room, R.; Reuter, P. How well do international drug conventions protect public health? Lancet 2012, 379, 84–91. [Google Scholar] [CrossRef]
- United States Drug Enforcement Administration. Drug Scheduling. Available online: https://www.dea.gov/drug-information/drug-scheduling (accessed on 17 September 2021).
- Talking Drugs. Drug Decriminalization Across the World. Available online: https://www.talkingdrugs.org/drug-decriminalisation (accessed on 23 September 2021).
- Misuse of Drugs Act. Misuse of Drugs Act 1971. Available online: https://www.legislation.gov.uk/ukpga/1971/38/section/5 (accessed on 22 September 2021).
- SICAD. SICAD > Política Portuguesa. Available online: http://www.sicad.pt/PT/PoliticaPortuguesa/SitePages/Home%20Page.aspx (accessed on 21 September 2021).
- Eastwood, N.; Fox, E.; Rosmarin, A. A Quiet Revolution: Drug Decriminalization Across The Globe; Release: London, UK, 2016; p. 51. [Google Scholar]
- Prinzleve, M.; Haasen, C.; Zurhold, H.; Matali, J.L.; Bruguera, E.; Gerevich, J.; Bacskai, E.; Ryder, N.; Butler, S.; Manning, V.; et al. Cocaine use in Europe-a multi-centre study: Patterns of use in different groups. Eur Addict. Res. 2004, 10, 147–155. [Google Scholar] [CrossRef]
- Gossop, M.; Manning, V.; Ridge, G. Concurrent use and order of use of cocaine and alcohol: Behavioural differences between users of crack cocaine and cocaine powder. Addiction 2006, 101, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- SAMHSA. Drug Abuse Warning Network, 2011: National Estimates of Drug-Related Emergency Department Visits; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2013. [Google Scholar]
- EMCDDA. Drug-related Deaths and Mortality in Europe: Update from the EMCDDA Expert Network; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar]
- Cunha-Oliveira, T.; Rego, A.C.; Carvalho, F.; Oliveira, C.R. Chapter 17-Medical Toxicology of Drugs of Abuse. In Principles of Addiction, Miller, P.M., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 159–175. [Google Scholar]
- Pomara, C.; Cassano, T.; D’Errico, S.; Bello, S.; Romano, A.D.; Riezzo, I.; Serviddio, G. Data available on the extent of cocaine use and dependence: Biochemistry, pharmacologic effects and global burden of disease of cocaine abusers. Curr. Med. Chem. 2012, 19, 5647–5657. [Google Scholar] [CrossRef] [PubMed]
- Cone, E.J. Pharmacokinetics and Pharmacodynamics of Cocaine. J. Anal. Toxicol. 1995, 19, 459–478. [Google Scholar] [CrossRef] [PubMed]
- Homstedt, B.; Lindgren, J.E.; Rivier, L.; Plowman, T. Cocaine in blood of coca chewers. J. Ethnopharmacol. 1979, 1, 69–78. [Google Scholar] [CrossRef]
- Clapp, L.; Martin, B.; Beresford, T.P. Sublingual cocaine: Novel recurrence of an ancient practice. Clin. Neuropharmacol. 2004, 27, 93–94. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Llosa, T.; Montoya, I.; Cone, E.J. Identification and quantitation of alkaloids in coca tea. Forensic. Sci. Int. 1996, 77, 179–189. [Google Scholar] [CrossRef]
- Edwards, D.J.; Bowles, S.K. Protein binding of cocaine in human serum. Pharm. Res. 1988, 5, 440–442. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Cone, E.J. Pharmacokinetics: Drug absorption, distribution, and elimination. In Drug Abuse Handbook; Karch, S.B., Ed.; CRC Press: New York, NY, USA, 1998; pp. 184–187. [Google Scholar]
- Kolbrich, E.A.; Barnes, A.J.; Gorelick, D.A.; Boyd, S.J.; Cone, E.J.; Huestis, M.A. Major and minor metabolites of cocaine in human plasma following controlled subcutaneous cocaine administration. J. Anal. Toxicol. 2006, 30, 501–510. [Google Scholar] [CrossRef]
- Valente, M.J.; Carvalho, F.; Bastos, M.; de Pinho, P.G.; Carvalho, M. Contribution of oxidative metabolism to cocaine-induced liver and kidney damage. Curr. Med. Chem. 2012, 19, 5601–5606. [Google Scholar] [CrossRef]
- Wang, Q.; Simpao, A.; Sun, L.; Falk, J.L.; Lau, C.E. Contribution of the active metabolite, norcocaine, to cocaine’s effects after intravenous and oral administration in rats: Pharmacodynamics. Psychopharmacology 2001, 153, 341–352. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Foltz, R.L. Cocaine metabolism in man: Identification of four previously unreported cocaine metabolites in human urine. J. Anal. Toxicol. 1990, 14, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Laizure, S.C.; Mandrell, T.; Gades, N.M.; Parker, R.B. Cocaethylene metabolism and interaction with cocaine and ethanol: Role of carboxylesterases. Drug. Metab. Dispos. 2003, 31, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.S.; Everhart, E.T.; Mendelson, J.; Jones, R.T. The pharmacology of cocaethylene in humans following cocaine and ethanol administration. Drug. Alcohol. Depend. 2003, 72, 169–182. [Google Scholar] [CrossRef]
- Herbst, E.D.; Harris, D.S.; Everhart, E.T.; Mendelson, J.; Jacob, P.; Jones, R.T. Cocaethylene formation following ethanol and cocaine administration by different routes. Exp. Clin. Psychopharmacol. 2011, 19, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rush, C.R.; Roll, J.M.; Higgins, S.T. Controlled laboratory studies on the effects of cocaine in combination with other commonly abused drugs in humans. In Cocaine Abuse: Behavior, Pharmacology and Clinical Applications; HIggins, S.T., Katz, J.L., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; p. 248. [Google Scholar]
- Musshoff, F. Chromatographic methods for the determination of markers of chronic and acute alcohol consumption. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 781, 457–480. [Google Scholar] [CrossRef]
- Politi, L.; Zucchella, A.; Morini, L.; Stramesi, C.; Polettini, A. Markers of chronic alcohol use in hair: Comparison of ethyl glucuronide and cocaethylene in cocaine users. Forensic. Sci. Int. 2007, 172, 23–27. [Google Scholar] [CrossRef]
- Gomes, E.F.; Lipaus, I.F.S.; Martins, C.W.; Araujo, A.M.; Mendonca, J.B.; Pelicao, F.S.; Lebarch, E.C.; de Melo Rodrigues, L.C.; Nakamura-Palacios, E.M. Anhydroecgonine Methyl Ester (AEME), a Product of Cocaine Pyrolysis, Impairs Spatial Working Memory and Induces Striatal Oxidative Stress in Rats. Neurotox. Res. 2018, 34, 834–847. [Google Scholar] [CrossRef]
- Scheidweiler, K.B.; Plessinger, M.A.; Shojaie, J.; Wood, R.W.; Kwong, T.C. Pharmacokinetics and pharmacodynamics of methylecgonidine, a crack cocaine pyrolyzate. J. Pharmacol. Exp. Ther. 2003, 307, 1179–1187. [Google Scholar] [CrossRef]
- Garcia, R.C.; Dati, L.M.; Fukuda, S.; Torres, L.H.; Moura, S.; de Carvalho, N.D.; Carrettiero, D.C.; Camarini, R.; Levada-Pires, A.C.; Yonamine, M.; et al. Neurotoxicity of anhydroecgonine methyl ester, a crack cocaine pyrolysis product. Toxicol. Sci. 2012, 128, 223–234. [Google Scholar] [CrossRef]
- Jeffcoat, A.R.; Perez-Reyes, M.; Hill, J.M.; Sadler, B.M.; Cook, C.E. Cocaine disposition in humans after intravenous injection, nasal insufflation (snorting), or smoking. Drug. Metab. Dispos. 1989, 17, 153–159. [Google Scholar]
- O’Leary, M.E.; Hancox, J.C. Role of voltage-gated sodium, potassium and calcium channels in the development of cocaine-associated cardiac arrhythmias. Br. J. Clin. Pharmacol. 2010, 69, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.; Sowerby, L.; Rotenberg, B.W. Is cocaine a safe topical agent for use during endoscopic sinus surgery? Laryngoscope 2016, 126, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Proebstl, L.; Kamp, F.; Manz, K.; Krause, D.; Adorjan, K.; Pogarell, O.; Koller, G.; Soyka, M.; Falkai, P.; Kambeitz, J. Effects of stimulant drug use on the dopaminergic system: A systematic review and meta-analysis of in vivo neuroimaging studies. Eur. Psychiatry 2019, 59, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, S.B.; Halbout, B. Chapter 29-Mesolimbic Dopamine Signaling in Cocaine Addiction. In The Neuroscience of Cocaine; Preedy, V.R., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 287–295. [Google Scholar]
- Chen, R.; Tilley, M.R.; Wei, H.; Zhou, F.; Zhou, F.M.; Ching, S.; Quan, N.; Stephens, R.L.; Hill, E.R.; Nottoli, T.; et al. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc. Natl. Acad. Sci. USA 2006, 103, 9333–9338. [Google Scholar] [CrossRef] [PubMed]
- Filip, M.; Frankowska, M.; Zaniewska, M.; Golda, A.; Przegalinski, E. The serotonergic system and its role in cocaine addiction. Pharmacol. Rep. 2005, 57, 685–700. [Google Scholar]
- Shanti, C.M.; Lucas, C.E. Cocaine and the critical care challenge. Crit Care Med. 2003, 31, 1851–1859. [Google Scholar] [CrossRef]
- Richards, J.R.; Hollander, J.E.; Ramoska, E.A.; Fareed, F.N.; Sand, I.C.; Izquierdo Gomez, M.M.; Lange, R.A. beta-Blockers, Cocaine, and the Unopposed alpha-Stimulation Phenomenon. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 239–249. [Google Scholar] [CrossRef]
- Riezzo, I.; Fiore, C.; De Carlo, D.; Pascale, N.; Neri, M.; Turillazzi, E.; Fineschi, V. Side effects of cocaine abuse: Multiorgan toxicity and pathological consequences. Curr. Med. Chem. 2012, 19, 5624–5646. [Google Scholar] [CrossRef]
- Ortinski, P.I. Cocaine-induced changes in NMDA receptor signaling. Mol. Neurobiol. 2014, 50, 494–506. [Google Scholar] [CrossRef]
- Lever, J.R.; Fergason-Cantrell, E.A.; Watkinson, L.D.; Carmack, T.L.; Lord, S.A.; Xu, R.; Miller, D.K.; Lever, S.Z. Cocaine occupancy of sigma1 receptors and dopamine transporters in mice. Synapse 2016, 70, 98–111. [Google Scholar] [CrossRef]
- Heal, D.J.; Gosden, J.; Smith, S.L. Dopamine reuptake transporter (DAT) “inverse agonism”—A novel hypothesis to explain the enigmatic pharmacology of cocaine. Neuropharmacology 2014, 87, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, D. Stimulant abuse: Pharmacology, cocaine, methamphetamine, treatment, attempts at pharmacotherapy. Prim. Care 2011, 38, 41–58. [Google Scholar] [CrossRef] [PubMed]
- NIDA. What Are the Short-Term Effects of Cocaine Use? National Institute on Drug Abuse: North Bethesda, ML, USA, 2021. [Google Scholar]
- Zimmerman, J.L. Cocaine intoxication. Crit Care Clin. 2012, 28, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kosten, T.R.; Kleber, H.D. Rapid death during cocaine abuse: A variant of the neuroleptic malignant syndrome? Am. J. Drug Alcohol. Abuse 1988, 14, 335–346. [Google Scholar] [CrossRef]
- Docherty, J.R.; Alsufyani, H.A. Pharmacology of D.Drugs Used as Stimulants. J. Clin. Pharmacol. 2021, 61 (Suppl. 2), S53–S69. [Google Scholar] [CrossRef]
- Marco, C.A.; Gupta, K.; Lubov, J.; Jamison, A.; Murray, B.P. Hyperthermia associated with methamphetamine and cocaine use. Am. J. Emerg. Med. 2021, 42, 20–22. [Google Scholar] [CrossRef]
- Okada, T.; Shioda, K.; Makiguchi, A.; Suda, S. Risperidone and 5-HT2A Receptor Antagonists Attenuate and Reverse Cocaine-Induced Hyperthermia in Rats. Int. J. Neuropsychopharmacol. 2020, 23, 811–820. [Google Scholar] [CrossRef]
- Crandall, C.G.; Vongpatanasin, W.; Victor, R.G. Mechanism of cocaine-induced hyperthermia in humans. Ann. Intern. Med. 2002, 136, 785–791. [Google Scholar] [CrossRef]
- Elkattawy, S.; Alyacoub, R.; Al-Nassarei, A.; Younes, I.; Ayad, S.; Habib, M. Cocaine induced heart failure: Report and literature review. J. Community Hosp. Intern. Med. Perspect. 2021, 11, 547–550. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; Magnusson, P.; LeQuang, J.A.K.; Breve, F.; Varrassi, G. Cocaine and Cardiotoxicity: A Literature Review. Cureus 2021, 13, e14594. [Google Scholar] [CrossRef]
- Lange, R.A.; Hillis, L.D. Cardiovascular complications of cocaine use. N. Engl. J. Med. 2001, 345, 351–358. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.; Jneid, H.; Hollander, J.E.; de Lemos, J.A.; Cercek, B.; Hsue, P.; Gibler, W.B.; Ohman, E.M.; Drew, B.; Philippides, G.; et al. Management of cocaine-associated chest pain and myocardial infarction: A scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation 2008, 117, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Afonso, L.; Mohammad, T.; Thatai, D. Crack whips the heart: A review of the cardiovascular toxicity of cocaine. Am. J. Cardiol 2007, 100, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.J.; Roque Bravo, R.; Enea, M.; Carmo, H.; Carvalho, F.; Bastos, M.L.; Dinis-Oliveira, R.J.; Dias da Silva, D. Ethanol addictively enhances the in vitro cardiotoxicity of cocaine through oxidative damage, energetic deregulation, and apoptosis. Arch. Toxicol. 2018, 92, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Glauser, J.; Queen, J.R. An overview of non-cardiac cocaine toxicity. J. Emerg Med. 2007, 32, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Herculiani, P.P.; Pires-Neto, R.C.; Bueno, H.M.; Zorzetto, J.C.; Silva, L.C.; Santos, A.B.; Garcia, R.C.; Yonamine, M.; Detregiachi, C.R.; Saldiva, P.H.; et al. Effects of chronic exposure to crack cocaine on the respiratory tract of mice. Toxicol. Pathol. 2009, 37, 324–332. [Google Scholar] [CrossRef]
- Restrepo, C.S.; Rojas, C.A.; Martinez, S.; Riascos, R.; Marmol-Velez, A.; Carrillo, J.; Vargas, D. Cardiovascular complications of cocaine: Imaging findings. Emerg. Radiol. 2009, 16, 11–19. [Google Scholar] [CrossRef]
- Filho, J.; Ogawa, M.Y.; de Souza Andrade, T.H.; de Andrade Cordeiro Gadelha, S.; Fernandes, P.; Queiroz, A.L.; Daher, E.F. Spectrum of acute kidney injury associated with cocaine use: Report of three cases. BMC Nephrol. 2019, 20, 99. [Google Scholar] [CrossRef]
- Goel, N.; Pullman, J.M.; Coco, M. Cocaine and kidney injury: A kaleidoscope of pathology. Clin. Kidney J. 2014, 7, 513–517. [Google Scholar] [CrossRef]
- Valente, M.J.; Henrique, R.; Vilas-Boas, V.; Silva, R.; Bastos Mde, L.; Carvalho, F.; Guedes de Pinho, P.; Carvalho, M. Cocaine-induced kidney toxicity: An in vitro study using primary cultured human proximal tubular epithelial cells. Arch. Toxicol. 2012, 86, 249–261. [Google Scholar] [CrossRef]
- Mai, H.N.; Jeong, J.H.; Kim, D.J.; Chung, Y.H.; Shin, E.J.; Nguyen, L.T.; Nam, Y.; Lee, Y.J.; Cho, E.H.; Nah, S.Y.; et al. Genetic overexpressing of GPx-1 attenuates cocaine-induced renal toxicity via induction of anti-apoptotic factors. Clin. Exp. Pharmacol. Physiol. 2016, 43, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk-Pachel, D.; Iciek, M.; Bilska-Wilkosz, A.; Gorny, M.; Jastrzebska, J.; Kaminska, K.; Dudzik, P.; Filip, M.; Lorenc-Koci, E. Evaluation of Cysteine Metabolism in the Rat Liver and Kidney Following Intravenous Cocaine Administration and Abstinence. Antioxidants 2021, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk-Pachel, D.; Iciek, M.; Wydra, K.; Nowak, E.; Gorny, M.; Filip, M.; Wlodek, L.; Lorenc-Koci, E. Cysteine Metabolism and Oxidative Processes in the Rat Liver and Kidney after Acute and Repeated Cocaine Treatment. PLoS ONE 2016, 11, e0147238. [Google Scholar] [CrossRef] [PubMed]
- Farooque, U.; Okorie, N.; Kataria, S.; Shah, S.F.; Bollampally, V.C. Cocaine-Induced Headache: A Review of Pathogenesis, Presentation, Diagnosis, and Management. Cureus 2020, 12, e10128. [Google Scholar] [CrossRef] [PubMed]
- Mai, H.N.; Sharma, N.; Jeong, J.H.; Shin, E.J.; Pham, D.T.; Trinh, Q.D.; Lee, Y.J.; Jang, C.G.; Nah, S.Y.; Bing, G.; et al. P53 knockout mice are protected from cocaine-induced kindling behaviors via inhibiting mitochondrial oxidative burdens, mitochondrial dysfunction, and proapoptotic changes. Neurochem. Int. 2019, 124, 68–81. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Garrido, J.; Borges, F.; Macedo, T.; Oliveira, C.R. Neurotoxicity of heroin-cocaine combinations in rat cortical neurons. Toxicology 2010, 276, 11–17. [Google Scholar] [CrossRef]
- Guha, P.; Harraz, M.M.; Snyder, S.H. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc. Natl. Acad. Sci. USA 2016, 113, 1417–1422. [Google Scholar] [CrossRef]
- Udo, M.S.B.; da Silva, M.A.A.; de Souza Prates, S.; Dal’Jovem, L.F.; de Oliveira Duro, S.; Faiao-Flores, F.; Garcia, R.C.T.; Maria-Engler, S.S.; Marcourakis, T. Anhydroecgonine methyl ester, a cocaine pyrolysis product, contributes to cocaine-induced rat primary hippocampal neuronal death in a synergistic and time-dependent manner. Arch. Toxicol. 2021, 95, 1779–1791. [Google Scholar] [CrossRef]
- Du, C.; Park, K.; Allen, C.P.; Hu, X.T.; Volkow, N.D.; Pan, Y. Ca(2+) channel blockade reduces cocaine’s vasoconstriction and neurotoxicity in the prefrontal cortex. Transl. Psychiatry 2021, 11, 459. [Google Scholar] [CrossRef]
- Lopez-Pedrajas, R.; Ramirez-Lamelas, D.T.; Muriach, B.; Sanchez-Villarejo, M.V.; Almansa, I.; Vidal-Gil, L.; Romero, F.J.; Barcia, J.M.; Muriach, M. Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front. Cell Neurosci. 2015, 9, 279. [Google Scholar] [CrossRef]
- Bittencourt, A.M.L.; Bampi, V.F.; Sommer, R.C.; Schaker, V.; Juruena, M.F.P.; Soder, R.B.; Franco, A.R.; Sanvicente-Vieira, B.; Grassi-Oliveira, R.; Ferreira, P. Cortical thickness and subcortical volume abnormalities in male crack-cocaine users. Psychiatry Res. Neuroimaging 2021, 310, 111232. [Google Scholar] [CrossRef] [PubMed]
- Schuch-Goi, S.B.; Goi, P.D.; Bermudez, M.; Fara, L.S.; Kessler, F.P.; Pechansky, F.; Gama, C.S.; Massuda, R.; von Diemen, L. Accumbens volumes are reduced among crack-cocaine users. Neurosci. Lett. 2017, 645, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Harbison, R.D. Cocaine-induced hepatotoxicity in mice. Toxicol. Appl. Pharmacol. 1978, 45, 739–754. [Google Scholar] [CrossRef]
- Mehanny, S.Z.; Abdel-Rahman, M.S. Cocaine hepatotoxicity in mice: Histologic and enzymatic studies. Toxicol. Pathol. 1991, 19, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Perino, L.E.; Warren, G.H.; Levine, J.S. Cocaine-induced hepatotoxicity in humans. Gastroenterology 1987, 93, 176–180. [Google Scholar] [CrossRef]
- Vitcheva, V. Cocaine toxicity and hepatic oxidative stress. Curr. Med. Chem. 2012, 19, 5677–5682. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J. Metabolomics of cocaine: Implications in toxicity. Toxicol. Mech. Methods 2015, 25, 494–500. [Google Scholar]
- Mai, H.N.; Jung, T.W.; Kim, D.J.; Sharma, G.; Sharma, N.; Shin, E.J.; Jang, C.G.; Nah, S.Y.; Lee, S.H.; Chung, Y.H.; et al. Protective potential of glutathione peroxidase-1 gene against cocaine-induced acute hepatotoxic consequences in mice. J. Appl. Toxicol. 2018, 38, 1502–1520. [Google Scholar] [CrossRef]
- Mai, H.N.; Sharma, G.; Sharma, N.; Shin, E.J.; Kim, D.J.; Pham, D.T.; Trinh, Q.D.; Jang, C.G.; Nah, S.Y.; Jeong, J.H.; et al. Genetic depletion of p53 attenuates cocaine-induced hepatotoxicity in mice. Biochimie 2019, 158, 53–61. [Google Scholar] [CrossRef]
- Boess, F.; Ndikum-Moffor, F.M.; Boelsterli, U.A.; Roberts, S.M. Effects of cocaine and its oxidative metabolites on mitochondrial respiration and generation of reactive oxygen species. Biochem. Pharmacol. 2000, 60, 615–623. [Google Scholar] [CrossRef]
- Krokos, A.; Deda, O.; Virgiliou, C.; Gika, H.; Raikos, N.; Aggelidou, E.; Kritis, A.; Theodoridis, G. Evaluation of Cocaine Effect on Endogenous Metabolites of HepG2 Cells Using Targeted Metabolomics. Molecules 2021, 26, 4610. [Google Scholar] [CrossRef] [PubMed]
- Zaitsu, K.; Miyawaki, I.; Bando, K.; Horie, H.; Shima, N.; Katagi, M.; Tatsuno, M.; Bamba, T.; Sato, T.; Ishii, A.; et al. Metabolic profiling of urine and blood plasma in rat models of drug addiction on the basis of morphine, methamphetamine, and cocaine-induced conditioned place preference. Anal. Bioanal. Chem. 2014, 406, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- NIDA. Cocaine DrugFacts. Available online: https://www.drugabuse.gov/publications/drugfacts/cocaine (accessed on 24 September 2021).
- American Psychiatric Association. Stimulant-Related Disorders. In Diagnostics and Statistical Manual of Mental health Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, 2013; p. 561. [Google Scholar]
- Barbosa-Mendez, S.; Perez-Sanchez, G.; Becerril-Villanueva, E.; Salazar-Juarez, A. Melatonin decreases cocaine-induced locomotor sensitization and cocaine-conditioned place preference in rats. J. Psychiatr. Res. 2021, 132, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Caffino, L.; Moro, F.; Mottarlini, F.; Targa, G.; Di Clemente, A.; Toia, M.; Orru, A.; Giannotti, G.; Fumagalli, F.; Cervo, L. Repeated exposure to cocaine during adolescence enhances the rewarding threshold for cocaine-conditioned place preference in adulthood. Addict. Biol. 2021, 26, e13012. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Ohnishi, Y.N.; Ohnishi, Y.H.; Kawahara, H.; Nishi, A. Distinct role of dopamine in the PFC and NAc during exposure to cocaine-associated cues. Int. J. Neuropsychopharmacol. 2021, 24, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Zinani, D.B.; Wetzel, H.N.; Norman, A.B. The compulsion zone explains the self-administration of cocaine, RTI-55 and bupropion in rats. Brain Res. 2021, 147707. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Childress, A.R.; Jayne, M.; Ma, Y.; Wong, C. Cocaine cues and dopamine in dorsal striatum: Mechanism of craving in cocaine addiction. J. Neurosci. 2006, 26, 6583–6588. [Google Scholar] [CrossRef]
- Volkow, N.D.; Tomasi, D.; Wang, G.J.; Logan, J.; Alexoff, D.L.; Jayne, M.; Fowler, J.S.; Wong, C.; Yin, P.; Du, C. Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol. Psychiatry 2014, 19, 1037–1043. [Google Scholar] [CrossRef]
- Andrianarivelo, A.; Saint-Jour, E.; Pousinha, P.; Fernandez, S.P.; Petitbon, A.; De Smedt-Peyrusse, V.; Heck, N.; Ortiz, V.; Allichon, M.C.; Kappes, V.; et al. Disrupting D1-NMDA or D2-NMDA receptor heteromerization prevents cocaine’s rewarding effects but preserves natural reward processing. Sci. Adv. 2021, 7, eabg5970. [Google Scholar] [CrossRef]
- Sanvicente-Vieira, B.; Kommers-Molina, J.; De Nardi, T.; Francke, I.; Grassi-Oliveira, R. Crack-cocaine dependence and aging: Effects on working memory. Braz J. Psychiatry 2016, 38, 58–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pudiak, C.M.; KuoLee, R.; Bozarth, M.A. Tolerance to cocaine in brain stimulation reward following continuous cocaine infusions. Pharmacol. Biochem. Behav. 2014, 122, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Small, A.C.; Kampman, K.M.; Plebani, J.; De Jesus Quinn, M.; Peoples, L.; Lynch, K.G. Tolerance and sensitization to the effects of cocaine use in humans: A retrospective study of long-term cocaine users in Philadelphia. Subst. Use Misuse 2009, 44, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Ferris, M.J.; Calipari, E.S.; Mateo, Y.; Melchior, J.R.; Roberts, D.C.; Jones, S.R. Cocaine self-administration produces pharmacodynamic tolerance: Differential effects on the potency.y of dopamine transporter blockers, releasers, and methylphenidate. Neuropsychopharmacology 2012, 37, 1708–1716. [Google Scholar] [CrossRef]
- Calipari, E.S.; Ferris, M.J.; Jones, S.R. Extended access of cocaine self-administration results in tolerance to the dopamine-elevating and locomotor-stimulating effects of cocaine. J. Neurochem. 2014, 128, 224–232. [Google Scholar] [CrossRef]
- NIDA. What are the Long-Term Effects of Cocaine Use? National Institute on Drug Abuse: North Bethesda, ML, USA, 2021. [Google Scholar]
- Gilmore, D.; Zorland, J.; Akin, J.; Johnson, J.A.; Emshoff, J.G.; Kuperminc, G.P. Mortality risk in a sample of emergency department patients who use cocaine with alcohol and/or cannabis. Subst. Abus. 2018, 39, 266–270. [Google Scholar] [CrossRef]
- Leri, F.; Bruneau, J.; Stewart, J. Understanding polydrug use: Review of heroin and cocaine co-use. Addiction 2003, 98, 7–22. [Google Scholar] [CrossRef]
- Motta-Ochoa, R.; Bertrand, K.; Arruda, N.; Jutras-Aswad, D.; Roy, E. I love having benzos after my coke shot: The use of psychotropic medication among cocaine users in downtown Montreal. Int. J. Drug. Policy 2017, 49, 15–23. [Google Scholar] [CrossRef]
- Tassoni, G.; Cippitelli, M.; Mietti, G.; Cerioni, A.; Buratti, E.; Bury, E.; Cingolani, M. Hair Analysis to Evaluate Polydrug Use. Healthcare 2021, 9, 972. [Google Scholar] [CrossRef]
- Obembe, S.B. Common Psychoactive Drugs. In Practical Skills and Clinical Management of Alcoholism & Drug Addiction; Obembe, S.B., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 11–32. [Google Scholar]
- Dean, R.A.; Harper, E.T.; Dumaual, N.; Stoeckel, D.A.; Bosron, W.F. Effects of ethanol on cocaine metabolism: Formation of cocaethylene and norcocaethylene. Toxicol. Appl. Pharmacol. 1992, 117, 1–8. [Google Scholar] [CrossRef]
- Uszenski, R.T.; Gillis, R.A.; Schaer, G.L.; Analouei, A.R.; Kuhn, F.E. Additive myocardial depressant effects of cocaine and ethanol. Am. Heart J. 1992, 124, 1276–1283. [Google Scholar] [CrossRef]
- Pereira, R.B.; Andrade, P.B.; Valentão, P. A Comprehensive View of the Neurotoxicity Mechanisms of Cocaine and Ethanol. Neurotox. Res. 2015, 28, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Zucoloto, A.D.; Eller, S.; de Oliveira, T.F.; Wagner, G.A.; Fruchtengarten, L.V.G.; de Oliveira, C.D.R.; Yonamine, M. Relationship between cocaine and cocaethylene blood concentration with the severity of clinical manifestations. Am. J. Emerg. Med. 2021, 50, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.A.; Perekopskiy, D.; Kiyatkin, E.A. Cocaine added to heroin fails to affect heroin-induced brain hypoxia. Brain Res. 2020, 1746, 147008. [Google Scholar] [CrossRef] [PubMed]
- Karch, S.B. Cocaine cardiovascular toxicity. South. Med. J. 2005, 98, 794–799. [Google Scholar] [CrossRef]
- Hearn, W.L.; Rose, S.; Wagner, J.; Ciarleglio, A.; Mash, D.C. Cocaethylene is more potent than cocaine in mediating lethality. Pharmacol. Biochem. Behav. 1991, 39, 531–533. [Google Scholar] [CrossRef]
- Mendelson, J.H.; Mello, N.K. Management of Cocaine Abuse and Dependence. N. Engl. J. Med. 1996, 334, 965–972. [Google Scholar] [CrossRef]
- McIntyre, K.M. Vasopressin in asystolic cardiac arrest. N. Engl. J. Med. 2004, 350, 179–181. [Google Scholar] [CrossRef]
- Wenzel, V.; Krismer, A.C.; Arntz, H.R.; Sitter, H.; Stadlbauer, K.H.; Lindner, K.H.; European Resuscitation Council Vasopressor during Cardiopulmonary Resuscitation Study Group. A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. N. Engl. J. Med. 2004, 350, 105–113. [Google Scholar] [CrossRef]
- Maraj, S.; Figueredo, V.M.; Lynn Morris, D. Cocaine and the heart. Clin. Cardiol. 2010, 33, 264–269. [Google Scholar] [CrossRef]
- Hollander, J.E.; Henry, T.D. Evaluation and management of the patient who has cocaine-associated chest pain. Cardiol. Clin. 2006, 24, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Witchel, H.J.; Hancox, J.C.; Nutt, D.J. Psychotropic drugs, cardiac arrhythmia, and sudden death. J. Clin. Psychopharmacol 2003, 23, 58–77. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, T.G.; DiMaio, V.J.M. Excited Delirium Syndrome: Causes of Death and Prevention; CRC Press: Boca Raton, 2005. [Google Scholar]
- Kampman, K.M. The treatment of cocaine use disorder. Sci. Adv. 2019, 5, eaax1532. [Google Scholar] [CrossRef] [PubMed]
- Nuijten, M.; Blanken, P.; van de Wetering, B.; Nuijten, B.; van den Brink, W.; Henrdriks, V. Sustained-release dexamfetamine in the treatment of chronic cocaine-dependent patients on heroin-assisted treatment: A randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 2226–2234. [Google Scholar] [CrossRef]
- Kampman, K.M.; Pettinati, H.M.; Lynch, K.G.; Spratt, K.; Wierzbicki, M.R.; O’Brien, C.P. A double-blind, placebo-controlled trial of topiramate for the treatment of comorbid cocaine and alcohol dependence. Drug. Alcohol. Depend. 2013, 133, 94–99. [Google Scholar] [CrossRef]
- Reith, M.E.; Blough, B.E.; Hong, W.C.; Jones, K.T.; Schmitt, K.C.; Baumann, M.H.; Partilla, J.S.; Rothman, R.B.; Katz, J.L. Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug. Alcohol. Depend. 2015, 147, 1–19. [Google Scholar] [CrossRef]
- Bentzley, B.S.; Han, S.S.; Neuner, S.; Humphreys, K.; Kampman, K.M.; Halpern, C.H. Comparison of Treatments for Cocaine Use Disorder Among Adults: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e218049. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocaine Form | Cocaine Hydrochloride | Cocaine Free Base |
|---|---|---|
| Other names it is known by | ‘coke’, ‘snow’, ‘blow’ | ‘crack’ |
| CAS | 53-21-4 | 50-36-2 |
| Molecular formula | C17H22ClNO4 | C17H21NO4 |
| Molecular weight | 339.8 g mol−1 | 303.35 g mol−1 |
| Boiling point | - | 187 °C |
| Melting point | 195 °C | 98 °C |
| Solubility in water | 2 g/mL | 1.7 × 10−3 g/mL |
| pKa; pKb (at 15 °C) | - | 8.61; 5.59 |
| Log P | - | 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roque Bravo, R.; Faria, A.C.; Brito-da-Costa, A.M.; Carmo, H.; Mladěnka, P.; Dias da Silva, D.; Remião, F.; on behalf of The OEMONOM Researchers. Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern. Toxins 2022, 14, 278. https://doi.org/10.3390/toxins14040278
Roque Bravo R, Faria AC, Brito-da-Costa AM, Carmo H, Mladěnka P, Dias da Silva D, Remião F, on behalf of The OEMONOM Researchers. Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern. Toxins. 2022; 14(4):278. https://doi.org/10.3390/toxins14040278
Chicago/Turabian StyleRoque Bravo, Rita, Ana Carolina Faria, Andreia Machado Brito-da-Costa, Helena Carmo, Přemysl Mladěnka, Diana Dias da Silva, Fernando Remião, and on behalf of The OEMONOM Researchers. 2022. "Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern" Toxins 14, no. 4: 278. https://doi.org/10.3390/toxins14040278
APA StyleRoque Bravo, R., Faria, A. C., Brito-da-Costa, A. M., Carmo, H., Mladěnka, P., Dias da Silva, D., Remião, F., & on behalf of The OEMONOM Researchers. (2022). Cocaine: An Updated Overview on Chemistry, Detection, Biokinetics, and Pharmacotoxicological Aspects including Abuse Pattern. Toxins, 14(4), 278. https://doi.org/10.3390/toxins14040278