Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade


Abstract
:1. Venom: What’s in Thy Name?
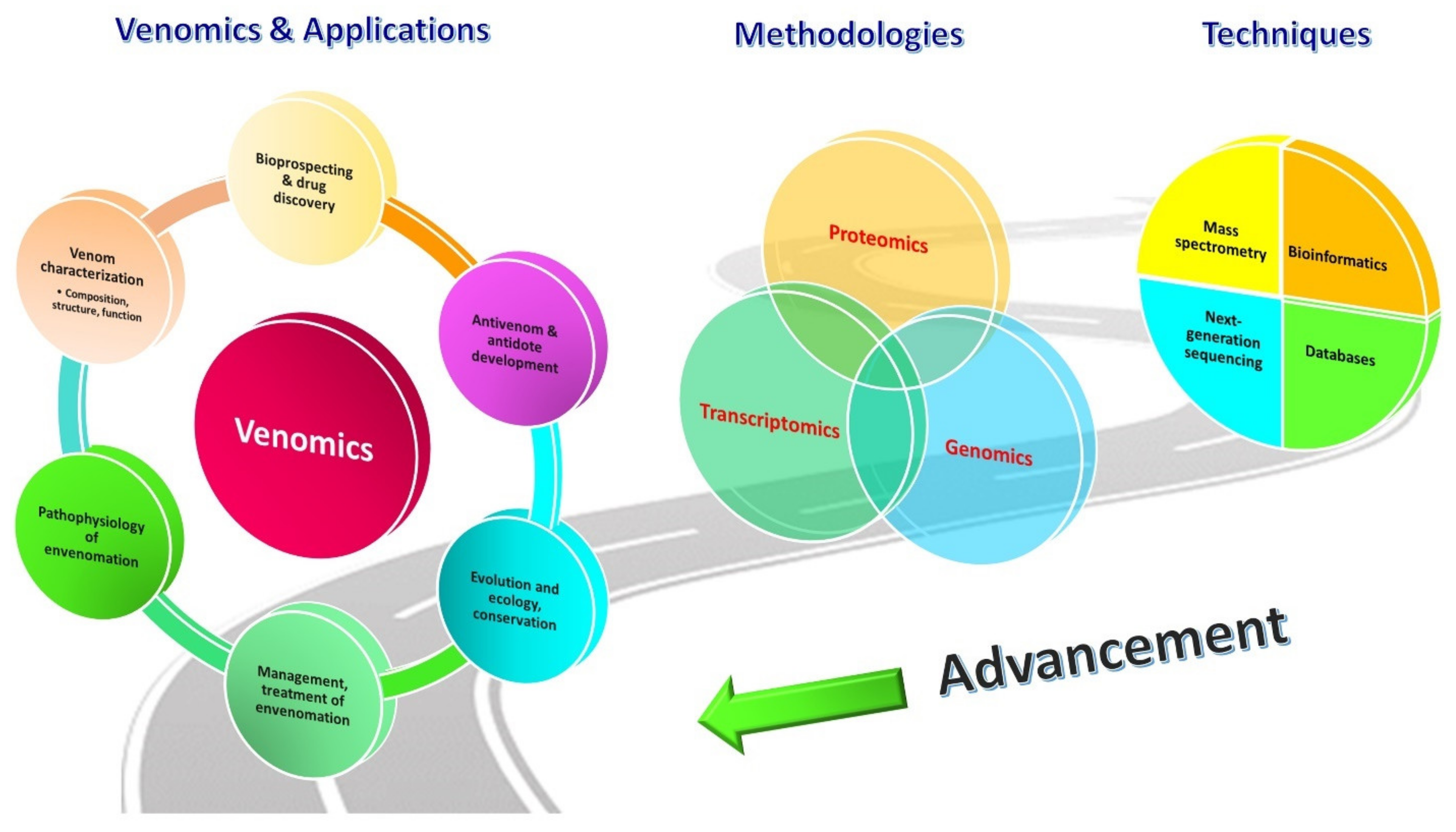
2. Venomics: The Unravelling Tool
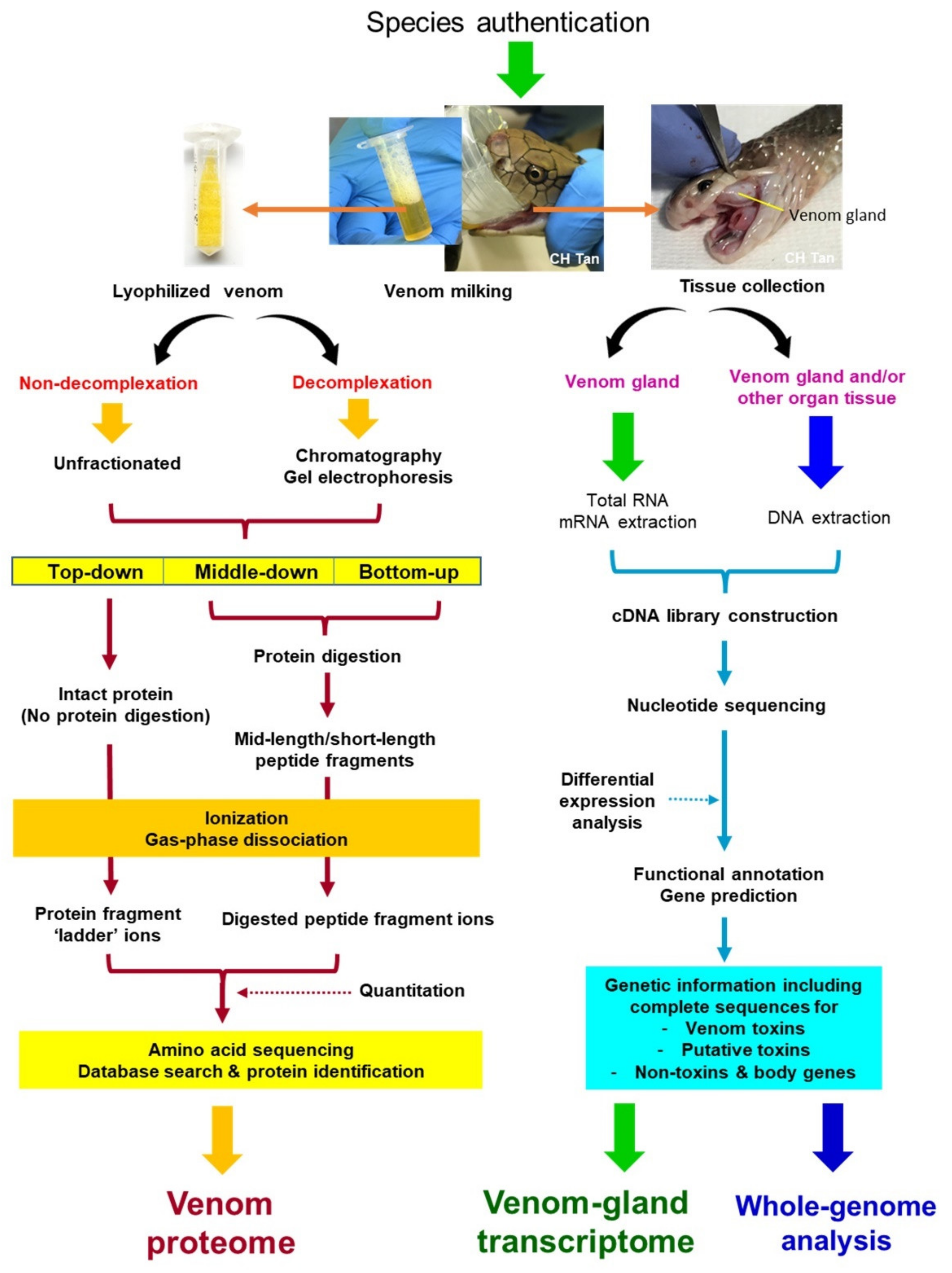
3. Strategies in Venomics: No ‘One-Size-Fits-All’
3.1. Proteomics of Snake Venom
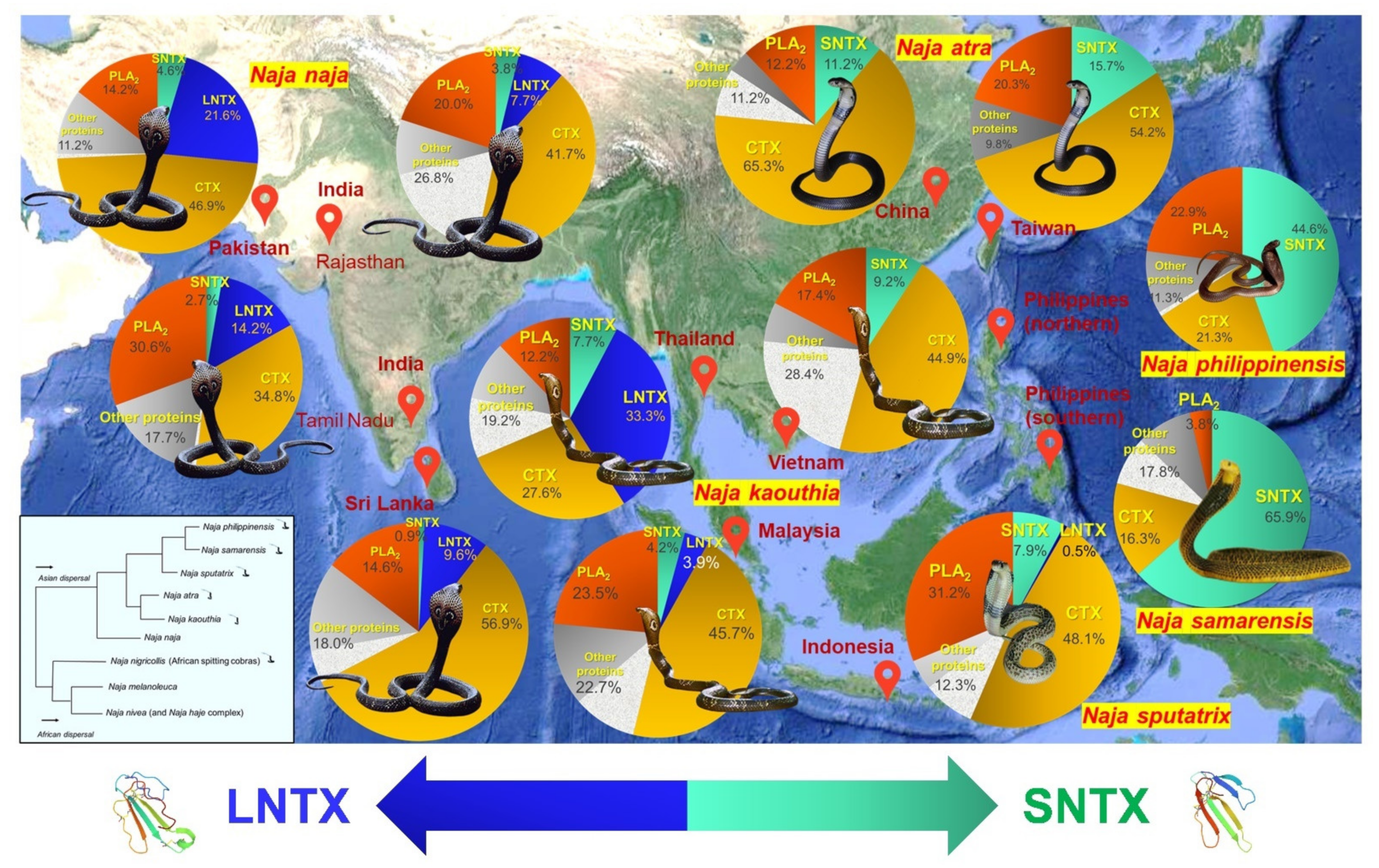
3.1.1. Evolutionary Significance and Medical Importance: A Case of Cobra (Naja spp.)
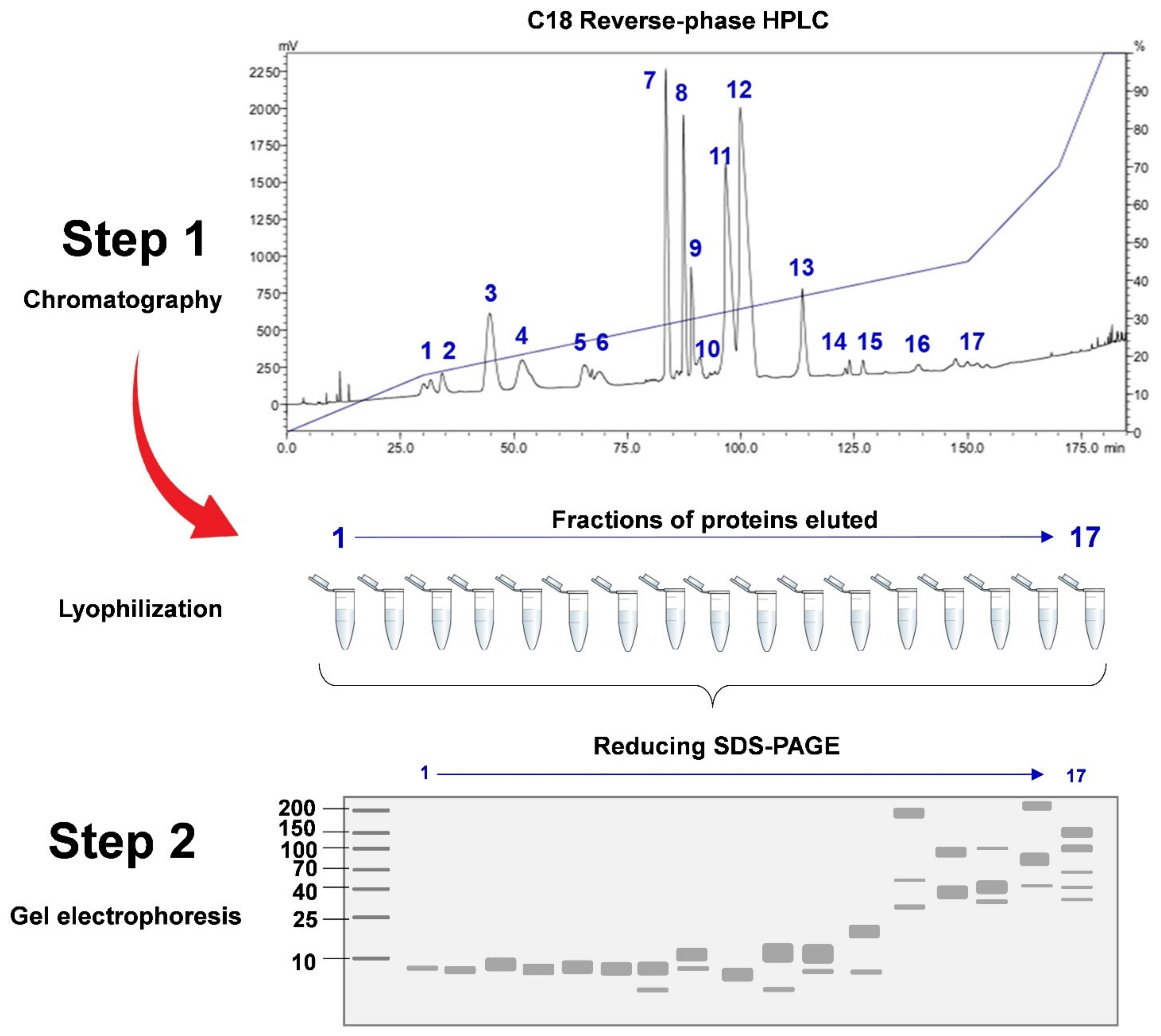
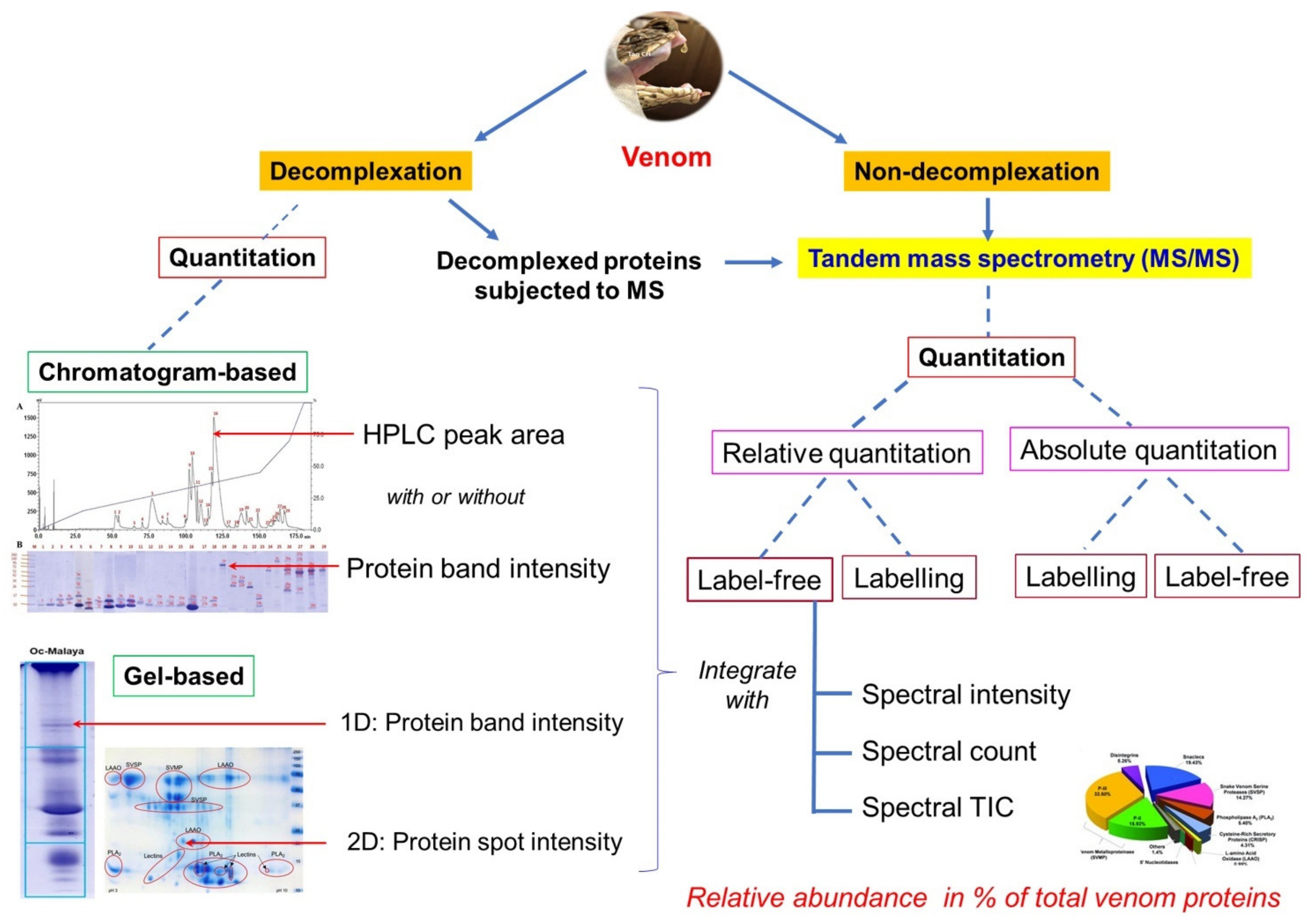
3.1.2. Decomplexation vs. Non-Decomplexation Methods
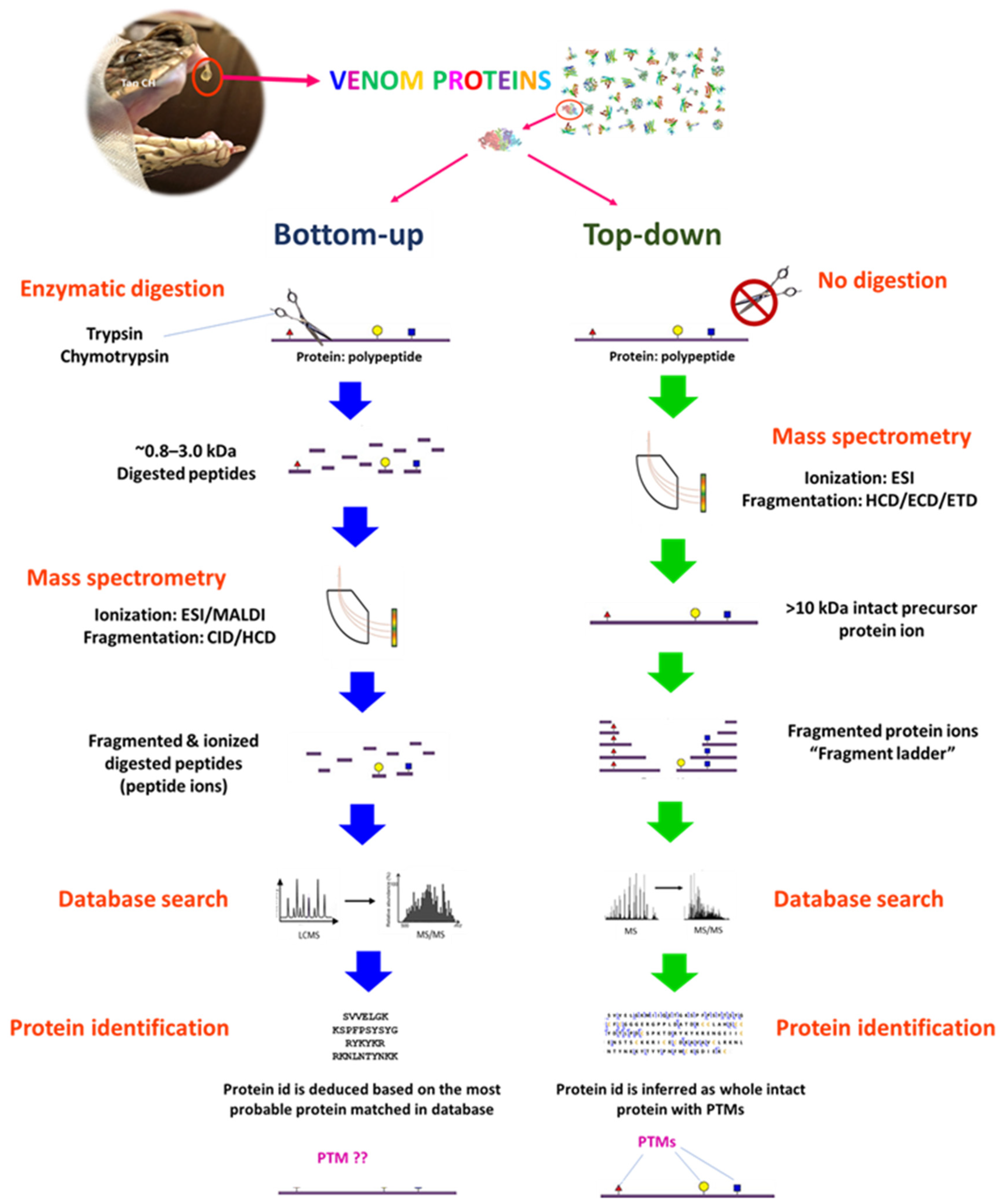
3.1.3. Bottom-Up, Middle-Down (Middle-Up), and Top-Down Sequencing Strategies
3.2. Genomics and Venom-Gland Transcriptomics of Snake
4. Challenges and Recommendations
4.1. Sampling
4.1.1. Availability and Authenticity of the Sample
4.1.2. Batching, Referencing, Storage and Quality Control of Samples
4.1.3. Ethics Approval and Permit Requirement
4.2. Protein Quantitation
4.2.1. Quantity of Protein: How Much, or How Many?
- Method 1 (considering the use of HPLC for venom protein decomplexation):
- Method 2 (regardless of decomplexation or non-decomplexation):
4.2.2. Quantifying a Protein without Model Organism
4.2.3. Reconciling the Divergence
- (i)
- Each protein regarded as a distinct form (proteoform) should be identified based on the presence of unique peptide(s) not otherwise shared with other known proteins or isoforms. While homologous sequences are assigned by search engines to different proteins, these sequences should be inspected meticulously to see if they are indeed representatives of distinct proteoforms, or simply mergeable into one whose full sequence is known. Each distinct proteoform is then quantified accordingly, and the quantity of different proteoforms belonging to a same toxin family should be added up to represent the relative composition of the protein family in a whole venom.
- (ii)
- Protein abundances should be measured quantitatively and not qualitatively, i.e., the expressed percentage should indicate the relative amount instead of relative number of proteins identified. The protein abundances are estimated based on the tandem MS (MS/MS) spectra derived from individual protein in terms of its peptide count or spectral intensity, with or without integrating additional parameters, such as chromatogram peak area and/or gel intensity (for studies involving protein decomplexation prior to MS analysis).
- (iii)
- There should be inclusivity of a label-free protein quantitation approach in the absence of model organism’s genome or transcriptome. Even with the presence of a model organism’s database, one should realize that an individual specimen may not capture the entire genomic (and therefore proteomic) diversity of a species, especially so when it has many populations in which genetic variability is anticipated. The proteomic algorithms for protein search and quantitation should be optimized using a more inclusive database that incorporates sequence information from phylogenetically related snakes and the species itself where available, instead of restricting the search to only one single species and worse still, a single specimen.
- (iv)
- Both top-down and bottom-up sequencing techniques have pros and cons. Acknowledging the differences and understanding the limitations associated with each approach will help to reduce conflicting views among researchers. Both approaches may complement each other, although the top-down method is apparently more attractive as it allows characterization of intact proteins and PTMs. It is, however, high in cost, technically difficult, less established in the venomic field, and is unavailable in most laboratories. Adopting the middle-down sequencing technique may be a promising solution in this regard.
4.3. Interpretation and Application of Findings
5. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mackessy, P.S. Reptile Venoms and Toxins: Unlimited Opportunities for Basic and Applied Research. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; Mackessy, P.S., Ed.; CRC Press: Boca Raton, FL, USA, 2021; pp. 3–18. [Google Scholar]
- Nelsen, D.R.; Nisani, Z.; Cooper, A.M.; Fox, G.A.; Gren, E.C.K.; Corbit, A.G.; Hayes, W.K. Poisons, toxungens, and venoms: Redefining and classifying toxic biological secretions and the organisms that employ them. Biol. Rev. 2014, 89, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.S.W.; Morgenstern, D.; Mofiz, E.; Gombert, S.; Morris, K.M.; Temple-Smith, P.; Renfree, M.; Whittington, C.; King, G.; Warren, W.C.; et al. Proteomics and Deep Sequencing Comparison of Seasonally Active Venom Glands in the Platypus Reveals Novel Venom Peptides and Distinct Expression Profiles. Mol. Cell. Proteom. 2012, 11, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, D.C.; Jeanne, R.L. Venom Source of a sex pheromone in the social wasp Polistes fuscatus (Hymenoptera: Vespidae). J. Chem. Ecol. 1983, 9, 259–266. [Google Scholar] [CrossRef] [PubMed]
- LeBrun, E.G.; Diebold, P.J.; Orr, M.R.; Gilbert, L.E. Widespread Chemical Detoxification of Alkaloid Venom by Formicine Ants. J. Chem. Ecol. 2015, 41, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Saviola, A.J.; Chiszar, D.; Busch, C.; Mackessy, S.P. Molecular basis for prey relocation in viperid snakes. BMC Biol. 2013, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.M.; Calvete, J.; Habib, A.G.; Harrison, R.A.; Williams, D.; Warrell, D.A. Correction: Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 201779. [Google Scholar] [CrossRef]
- Fry, B.G. Snakebite: When the Human Touch Becomes a Bad Touch. Toxins 2018, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef]
- Kalita, B.; Saviola, A.J.; Mukherjee, A.K. From venom to drugs: A review and critical analysis of Indian snake venom toxins envisaged as anticancer drug prototypes. Drug Discov. Today 2021, 26, 993–1005. [Google Scholar] [CrossRef]
- Mackessy, S.P. Evolutionary trends in venom composition in the Western Rattlesnakes (Crotalus viridis sensu lato): Toxicity vs. tenderizers. Toxicon 2010, 55, 1463–1474. [Google Scholar] [CrossRef]
- Fry, B.G.; Vidal, N.; Norman, J.A.; Vonk, F.J.; Scheib, H.; Ramjan, S.F.R.; Kuruppu, S.; Fung, K.; Hedges, S.B.; Richardson, M.K.; et al. Early evolution of the venom system in lizards and snakes. Nature 2005, 439, 584–588. [Google Scholar] [CrossRef]
- Fry, B.G.; Casewell, N.R.; Wüster, W.; Vidal, N.; Young, B.; Jackson, T.N.W. The structural and functional diversification of the Toxicofera reptile venom system. Toxicon 2012, 60, 434–448. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef]
- Weinstein, S.A.; Keyler, D.E.; White, J. Replies to Fry et al. (Toxicon 2012, 60/4, 434–448). Part A. Analyses of squamate reptile oral glands and their products: A call for caution in formal assignment of terminology designating biological function. Toxicon 2012, 60, 954–963. [Google Scholar] [CrossRef]
- Kardong, K.V. Replies to Fry et al. (Toxicon 2012, 60/4, 434–448). Part B. Properties and biological roles of squamate oral products: The “venomous lifestyle” and preadaptation. Toxicon 2012, 60, 964–966. [Google Scholar] [CrossRef]
- Reyes-Velasco, J.; Card, D.C.; Andrew, A.L.; Shaney, K.J.; Adams, R.H.; Schield, D.R.; Casewell, N.; Mackessy, S.; Castoe, T.A. Expression of Venom Gene Homologs in Diverse Python Tissues Suggests a New Model for the Evolution of Snake Venom. Mol. Biol. Evol. 2014, 32, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, A.D.; Swain, M.T.; Logan, D.W.; Mulley, J.F. Testing the Toxicofera: Comparative transcriptomics casts doubt on the single, early evolution of the reptile venom system. Toxicon 2014, 92, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Augusto-De-Oliveira, C.; Stuginski, D.R.; Kitano, E.S.; Andrade-Silva, D.; Liberato, T.; Fukushima, I.; Serrano, S.M.T.; Zelanis, A. Dynamic Rearrangement in Snake Venom Gland Proteome: Insights into Bothrops jararaca Intraspecific Venom Variation. J. Proteome Res. 2016, 15, 3752–3762. [Google Scholar] [CrossRef]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: Elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef] [Green Version]
- Calvete, J.J.; Sanz, L.; Angulo, Y.; Lomonte, B.; Gutierrez, J.M. Venoms, venomics, antivenomics. FEBS Lett. 2009, 583, 1736–1743. [Google Scholar] [CrossRef] [Green Version]
- Calvete, J.J. Snake venomics: From the inventory of toxins to biology. Toxicon 2013, 75, 44–62. [Google Scholar] [CrossRef]
- Lomonte, B.; Calvete, J.J. Strategies in ‘snake venomics’ aiming at an integrative view of compositional, functional, and immunological characteristics of venoms. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.; Daly, N.L. Venomics: A Mini-Review. High-Throughput 2018, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Juárez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar] [CrossRef]
- Calvete, J.J.; Sanz, L.; Mora-Obando, D.; Lomonte, B.; Tanaka-Azevedo, A.M.; de Morais-Zani, K.; Sant’Anna, S.S.; Caldeira, C.A. What’s in a mass? Biochem. Soc. Trans. 2021, 49, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.Y.; Liew, J.L.; Tan, N.H.; Quah, E.S.; Ismail, A.K.; Tan, C.H. Unlocking the secrets of banded coral snake (Calliophis intestinalis, Malaysia): A venom with proteome novelty, low toxicity and distinct antigenicity. J. Proteom. 2018, 192, 246–257. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Fung, S.Y.; Tan, N.H. Venomics, lethality and neutralization of Naja kaouthia (monocled cobra) venoms from three different geographical regions of Southeast Asia. J. Proteom. 2015, 120, 105–125. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Lomonte, B.; León, G.; Alape-Girón, A.; Flores-Díaz, M.; Sanz, L.; Angulo, Y.; Calvete, J. Snake venomics and antivenomics: Proteomic tools in the design and control of antivenoms for the treatment of snakebite envenoming. J. Proteom. 2009, 72, 165–182. [Google Scholar] [CrossRef]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef]
- Fox, J.W.; Serrano, S.M.T. Exploring snake venom proteomes: Multifaceted analyses for complex toxin mixtures. Proteomics 2008, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Petras, D.; Calderón-Celis, F.; Lomonte, B.; Encinar, J.R.; Sanz-Medel, A. Protein-species quantitative venomics: Looking through a crystal ball. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Calvete, J.J.; Lomonte, B.; Saviola, A.J.; Bonilla, F.; Sasa, M.; Williams, D.J.; Undheim, E.A.B.; Sunagar, K.; Jackson, T.N.W. Mutual enlightenment: A toolbox of concepts and methods for integrating evolutionary and clinical toxinology via snake venomics and the contextual stance. Toxicon X 2021, 9, 100070. [Google Scholar] [CrossRef]
- Au-Garb, J.E. Extraction of Venom and Venom Gland Microdissections from Spiders for Proteomic and Transcriptomic Analyses. JoVE 2014, 93, e51618. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.J.; Ellsworth, S.A.; Rokyta, D.R. Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 2018, 142, 14–29. [Google Scholar] [CrossRef]
- Jouiaei, M.; Casewell, N.R.; Yanagihara, A.A.; Nouwens, A.; Cribb, B.W.; Whitehead, D.; Jackson, T.N.W.; Ali, S.A.; Wagstaff, S.C.; Koludarov, I.; et al. Firing the Sting: Chemically Induced Discharge of Cnidae Reveals Novel Proteins and Peptides from Box Jellyfish (Chironex fleckeri) Venom. Toxins 2015, 7, 936–950. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Yap, M.K.K.; Tan, N.H. Venomics of Tropidolaemus wagleri, the sexually dimorphic temple pit viper: Unveiling a deeply conserved atypical toxin arsenal. Sci. Rep. 2017, 7, 43237. [Google Scholar] [CrossRef] [Green Version]
- Amorim, F.G.; Costa, T.R.; Baiwir, D.; De Pauw, E.; Quinton, L.; Sampaio, S.V. Proteopeptidomic, Functional and Immuno-reactivity Characterization of Bothrops moojeni Snake Venom: Influence of Snake Gender on Venom Composition. Toxins 2018, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Menezes, M.C.; Furtado, M.F.; Travaglia-Cardoso, S.R.; Camargo, A.C.M.; Serrano, S.M.T. Sex-based individual variation of snake venom proteome among eighteen Bothrops jararaca siblings. Toxicon 2006, 47, 304–312. [Google Scholar] [CrossRef]
- Zelanis, A.; Tashima, A.K.; Rocha, M.M.T.; Furtado, M.F.; Camargo, A.C.M.; Ho, P.L.; Serrano, S.M.T. Analysis of the Ontogenetic Variation in the Venom Proteome/Peptidome of Bothrops jararaca Reveals Different Strategies to Deal with Prey. J. Proteome Res. 2010, 9, 2278–2291. [Google Scholar] [CrossRef]
- Wray, K.P.; Margres, M.J.; Seavy, M.; Rokyta, D.R. Early significant ontogenetic changes in snake venoms. Toxicon 2015, 96, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, K. Evolutionary context of venom in animals. In Evolution of Venomous Animals and Their Toxins, 1st ed.; Gopalakrishnakone, P., Malhotra, A., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 3–31. [Google Scholar] [CrossRef]
- Pla, D.; Sanz, L.; Quesada-Bernat, S.; Villalta, M.; Baal, J.; Chowdhury, M.A.W.; León, G.; Gutiérrez, J.M.; Kuch, U.; Calvete, J.J. Phylovenomics of Daboia russelii across the Indian subcontinent. Bioactivities and comparative in vivo neutralization and in vitro third-generation antivenomics of antivenoms against venoms from India, Bangladesh and Sri Lanka. J. Proteom. 2019, 207, 103443. [Google Scholar] [CrossRef] [PubMed]
- Faisal, T.; Tan, K.Y.; Sim, S.M.; Quraishi, N.; Tan, N.H.; Tan, C.H. Proteomics, functional characterization and antivenom neutralization of the venom of Pakistani Russell’s viper (Daboia russelii) from the wild. J. Proteom. 2018, 183, 1–13. [Google Scholar] [CrossRef]
- Patra, A.; Mukherjee, A.K. Proteomic Analysis of Sri Lanka Echis carinatus Venom: Immunological Cross-Reactivity and Enzyme Neutralization Potency of Indian Polyantivenom. J. Proteome Res. 2020, 19, 3022–3032. [Google Scholar] [CrossRef]
- Lee, L.P.; Tan, K.Y.; Tan, C.H. Snake venom proteomics and antivenomics of two Sundaic lance-headed pit vipers: Trimeresurus wiroti (Malaysia) and Trimeresurus puniceus (Indonesia). Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100875. [Google Scholar] [CrossRef]
- Liew, J.L.; Tan, N.H.; Tan, C.H. Proteomics and preclinical antivenom neutralization of the mangrove pit viper (Trimeresurus purpureomaculatus, Malaysia) and white-lipped pit viper (Trimeresurus albolabris, Thailand) venoms. Acta Trop. 2020, 209, 105528. [Google Scholar] [CrossRef]
- Tan, C.H.; Palasuberniam, P.; Tan, K.Y. Snake Venom Proteomics, Immunoreactivity and Toxicity Neutralization Studies for the Asiatic Mountain Pit Vipers, Ovophis convictus, Ovophis tonkinensis, and Hime Habu, Ovophis okinavensis. Toxins 2021, 13, 514. [Google Scholar] [CrossRef]
- Silva, A.; Cristofori-Armstrong, B.; Rash, L.D.; Hodgson, W.C.; Isbister, G.K. Defining the role of post-synaptic α-neurotoxins in paralysis due to snake envenoming in humans. Cell. Mol. Life Sci. 2018, 75, 4465–4478. [Google Scholar] [CrossRef]
- Tan, C.H.; Wong, K.Y.; Chong, H.P.; Tan, N.H.; Tan, K.Y. Proteomic insights into short neurotoxin-driven, highly neurotoxic venom of Philippine cobra (Naja philippinensis) and toxicity correlation of cobra envenomation in Asia. J. Proteom. 2019, 206, 103418. [Google Scholar] [CrossRef]
- Tan, N.H.; Tan, K.Y.; Tan, C.H. Snakebite in Southeast Asia: Envenomation and Clinical Management. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2021; pp. 559–579. [Google Scholar] [CrossRef]
- Tan, N.H.; Wong, K.Y.; Tan, C.H. Venomics of Naja sputatrix, the Javan spitting cobra: A short neurotoxin-driven venom needing improved antivenom neutralization. J. Proteom. 2017, 157, 18–32. [Google Scholar] [CrossRef]
- Wong, K.Y.; Tan, C.H.; Tan, N.H. Venom and Purified Toxins of the Spectacled Cobra (Naja naja) from Pakistan: Insights into Toxicity and Antivenom Neutralization. Am. J. Trop. Med. Hyg. 2016, 94, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wüster, W.; Crookes, S.; Ineich, I.; Mané, Y.; Pook, C.E.; Trape, J.-F.; Broadley, D.G. The phylogeny of cobras inferred from mitochondrial DNA sequences: Evolution of venom spitting and the phylogeography of the African spitting cobras (Serpentes: Elapidae: Naja nigricollis complex). Mol. Phylogenet. Evol. 2007, 45, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, E.; Nazarizadeh, M.; Fatemizadeh, F.; Khani, A.; Kaboli, M. The phylogeny, phylogeography, and diversification history of the westernmost Asian cobra (Serpentes: Elapidae: Naja oxiana) in the Trans-Caspian region. Ecol. Evol. 2020, 11, 2024–2039. [Google Scholar] [CrossRef]
- Petras, D.; Sanz, L.; Segura, A.; Herrera, M.; Villalta, M.; Solano, D.; Vargas, M.; León, G.; Warrell, D.A.; Theakston, R.D.G.; et al. Snake Venomics of African Spitting Cobras: Toxin Composition and Assessment of Congeneric Cross-Reactivity of the Pan-African EchiTAb-Plus-ICP Antivenom by Antivenomics and Neutralization Approaches. J. Proteome Res. 2011, 10, 1266–1280. [Google Scholar] [CrossRef]
- Lauridsen, L.P.; Laustsen, A.H.; Lomonte, B.; Gutiérrez, J.M. Exploring the venom of the forest cobra snake: Toxicovenomics and antivenom profiling of Naja melanoleuca. J. Proteom. 2017, 150, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Malih, I.; Rusmili, M.R.A.; Tee, T.Y.; Saile, R.; Ghalim, N.; Othman, I. Proteomic analysis of Moroccan cobra Naja haje legionis venom using tandem mass spectrometry. J. Proteom. 2014, 96, 240–252. [Google Scholar] [CrossRef]
- Kazandjian, T.D.; Petras, D.; Robinson, S.D.; van Thiel, J.; Greene, H.W.; Arbuckle, K.; Barlow, A.; Carter, D.A.; Wouters, R.M.; Whiteley, G.; et al. Convergent evolution of pain-inducing defensive venom components in spitting cobras. Science 2021, 371, 386–390. [Google Scholar] [CrossRef]
- Tan, C.H.; Wong, K.Y.; Tan, N.H.; Ng, T.S.; Tan, K.Y. Distinctive Distribution of Secretory Phospholipases A2 in the Venoms of Afro-Asian Cobras (Subgenus: Naja, Afronaja, Boulengerina and Uraeus). Toxins 2019, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Panagides, N.; Jackson, T.N.W.; Ikonomopoulou, M.P.; Arbuckle, K.; Pretzler, R.; Yang, D.C.; Ali, S.A.; Koludarov, I.; Dobson, J.; Sanker, B.; et al. How the Cobra Got Its Flesh-Eating Venom: Cytotoxicity as a Defensive Innovation and Its Co-Evolution with Hooding, Aposematic Marking, and Spitting. Toxins 2017, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.Y.; Wong, K.Y.; Tan, N.H.; Tan, C.H. Quantitative proteomics of Naja annulifera (sub-Saharan snouted cobra) venom and neutralization activities of two antivenoms in Africa. Int. J. Biol. Macromol. 2020, 158, 605–616. [Google Scholar] [CrossRef]
- Wong, K.; Tan, K.; Tan, N.; Tan, C. A Neurotoxic Snake Venom without Phospholipase A2: Proteomics and Cross-Neutralization of the Venom from Senegalese Cobra, Naja senegalensis (Subgenus: Uraeus). Toxins 2021, 13, 60. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Chan, Y.W.; Tan, K.Y.; Tan, C.H. Snake Venom Proteomics of Samar Cobra (Naja samarensis) from the Southern Philippines: Short Alpha-Neurotoxins as the Dominant Lethal Component Weakly Cross-Neutralized by the Philippine Cobra Antivenom. Front. Pharmacol. 2021, 12, 727756. [Google Scholar] [CrossRef]
- Watt, G.; Laughlin, L.; Padre, L.; Tuazon, M.L.; Theakston, R.D.G. Bites by the Philippine Cobra (Naja naja philippinensis): Prominent Neurotoxicity with Minimal Local Signs. Am. J. Trop. Med. Hyg. 1988, 39, 306–311. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y. Functional Application of Snake Venom Proteomics in In Vivo Antivenom Assessment. Methods Mol. Biol. 2019, 1871, 153–158. [Google Scholar] [CrossRef]
- Ratanabanangkoon, K.; Tan, K.Y.; Pruksaphon, K.; Klinpayom, C.; Gutiérrez, J.M.; Quraishi, N.H.; Tan, C.H. A pan-specific antiserum produced by a novel immunization strategy shows a high spectrum of neutralization against neurotoxic snake venoms. Sci. Rep. 2020, 10, 11261. [Google Scholar] [CrossRef] [PubMed]
- Ratanabanangkoon, K.; Tan, K.Y.; Eursakun, S.; Tan, C.H.; Simsiriwong, P.; Pamornsakda, T.; Wiriyarat, W.; Klinpayom, C.; Tan, N.H. A Simple and Novel Strategy for the Production of a Pan-specific Antiserum against Elapid Snakes of Asia. PLoS Negl. Trop. Dis. 2016, 10, e0004565. [Google Scholar] [CrossRef] [Green Version]
- Wallach, V.; Wüster, W.; G Broadley, D. In Praise Of Subgenera: Taxonomic Status Of Cobras Of The Genus Naja Laurenti (Serpentes: Elapidae). Zootaxa 2009, 2236, 26–36. [Google Scholar] [CrossRef]
- Wong, K.Y.; Tan, C.H.; Tan, K.Y.; Quraishi, N.H.; Tan, N.H. Elucidating the biogeographical variation of the venom of Naja naja (spectacled cobra) from Pakistan through a venom-decomplexing proteomic study. J. Proteom. 2018, 175, 156–173. [Google Scholar] [CrossRef]
- Laxme, R.R.S.; Attarde, S.; Khochare, S.; Suranse, V.; Martin, G.; Casewell, N.R.; Whitaker, R.; Sunagar, K. Biogeographical venom variation in the Indian spectacled cobra (Naja naja) underscores the pressing need for pan-India efficacious snakebite therapy. PLoS Negl. Trop. Dis. 2021, 15, e0009150. [Google Scholar] [CrossRef]
- Wong, K.Y.; Tan, K.Y.; Tan, N.H.; Gnanathasan, C.A.; Tan, C.H. Elucidating the Venom Diversity in Sri Lankan Spectacled Cobra (Naja naja) through De Novo Venom Gland Transcriptomics, Venom Proteomics and Toxicity Neutralization. Toxins 2021, 13, 558. [Google Scholar] [CrossRef]
- Shan, L.-L.; Gao, J.-F.; Zhang, Y.-X.; Shen, S.-S.; He, Y.; Wang, J.; Ma, X.-M.; Ji, X. Proteomic characterization and comparison of venoms from two elapid snakes (Bungarus multicinctus and Naja atra) from China. J. Proteom. 2016, 138, 83–94. [Google Scholar] [CrossRef]
- Liu, C.-C.; Chou, Y.-S.; Chen, C.-Y.; Liu, K.-L.; Huang, G.-J.; Yu, J.-S.; Wu, C.-J.; Liaw, G.-W.; Hsieh, C.-H.; Chen, C.-K. Pathogenesis of local necrosis induced by Naja atra venom: Assessment of the neutralization ability of Taiwanese freeze-dried neurotoxic antivenom in animal models. PLoS Negl. Trop. Dis. 2020, 14, e0008054. [Google Scholar] [CrossRef] [Green Version]
- Calvete, J.J.; Juárez, P.; Sanz, L. Snake venomics. Strategy and applications. Biol. Mass Spectrom. 2007, 42, 1405–1414. [Google Scholar] [CrossRef]
- Tan, C.H.; Wong, K.Y.; Tan, K.Y.; Tan, N.H. Venom proteome of the yellow-lipped sea krait, Laticauda colubrina from Bali: Insights into subvenomic diversity, venom antigenicity and cross-neutralization by antivenom. J. Proteom. 2017, 166, 48–58. [Google Scholar] [CrossRef]
- Lingam, T.M.C.; Tan, K.Y.; Tan, C.H. Proteomics and antivenom immunoprofiling of Russell’s viper (Daboia siamensis) venoms from Thailand and Indonesia. J. Venom. Anim. Toxins Incl. Trop. Dis. 2020, 26, e20190048. [Google Scholar] [CrossRef] [Green Version]
- Petras, D.; Heiss, P.; Süssmuth, R.D.; Calvete, J.J. Venom Proteomics of Indonesian King Cobra, Ophiophagus hannah: Integrating Top-Down and Bottom-Up Approaches. J. Proteome Res. 2015, 14, 2539–2556. [Google Scholar] [CrossRef]
- Al-Shekhadat, R.I.; Lopushanskaya, K.S.; Segura, Á.; Gutiérrez, J.M.; Calvete, J.J.; Pla, D. Vipera berus berus Venom from Russia: Venomics, Bioactivities and Preclinical Assessment of Microgen Antivenom. Toxins 2019, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Baumann, K.; Jackson, T.N.; Wood, K.; Mason, S.; Undheim, E.A.; Nouwens, A.; Koludarov, I.; Hendrikx, I.; Jones, A.; et al. Proteomic comparison of Hypnale hypnale (Hump-Nosed Pit-Viper) and Calloselasma rhodostoma (Malayan Pit-Viper) venoms. J. Proteom. 2013, 91, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.M.T.; Shannon, J.D.; Wang, D.; Camargo, A.C.M.; Fox, J.W. A multifaceted analysis of viperid snake venoms by two-dimensional gel electrophoresis: An approach to understanding venom proteomics. Proteomics 2005, 5, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Fung, S.Y.; Yap, M.K.K.; Leong, P.K.; Liew, J.L.; Tan, N.H. Unveiling the elusive and exotic: Venomics of the Malayan blue coral snake (Calliophis bivirgata flaviceps). J. Proteom. 2016, 132, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, J.; Zhang, X.; Ren, Y.; Wang, N.; Zhao, K.; Chen, X.; Zhao, C.; Li, X.; Shao, J.; et al. Proteomic characterization of two snake venoms: Naja naja atra and Agkistrodon halys. Biochem. J. 2004, 384, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Calvete, J.J. Next-generation snake venomics: Protein-locus resolution through venom proteome decomplexation. Expert Rev. Proteom. 2014, 11, 315–329. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Tan, N.H. A Protein Decomplexation Strategy in Snake Venom Proteomics. In Methods in Molecular Biology; Springer: Clifton, NJ, USA, 2019; Volume 1871, pp. 83–92. [Google Scholar] [CrossRef]
- Tan, C.H.; Liew, J.L.; Navanesan, S.; Sim, K.S.; Tan, N.H.; Tan, K.Y. Cytotoxic and anticancer properties of the Malaysian mangrove pit viper (Trimeresurus purpureomaculatus) venom and its disintegrin (purpureomaculin). J. Venom. Anim. Toxins Incl. Trop. Dis. 2020, 26, e20200013. [Google Scholar] [CrossRef]
- Chong, H.P.; Tan, K.Y.; Tan, C.H. Cytotoxicity of Snake Venoms and Cytotoxins from Two Southeast Asian Cobras (Naja sumatrana, Naja kaouthia): Exploration of Anticancer Potential, Selectivity, and Cell Death Mechanism. Front. Mol. Biosci. 2020, 7, 583587. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Ng, T.S.; Sim, S.M.; Tan, N.H. Venom Proteome of Spine-Bellied Sea Snake (Hydrophis curtus) from Penang, Malaysia: Toxicity Correlation, Immunoprofiling and Cross-Neutralization by Sea Snake Antivenom. Toxins 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.H.; Tan, K.Y.; Lim, S.E.; Tan, N.H. Venomics of the beaked sea snake, Hydrophis schistosus: A minimalist toxin arsenal and its cross-neutralization by heterologous antivenoms. J. Proteom. 2015, 126, 121–130. [Google Scholar] [CrossRef]
- Lauridsen, L.P.; Laustsen, A.H.; Lomonte, B.; Gutiérrez, J.M. Toxicovenomics and antivenom profiling of the Eastern green mamba snake (Dendroaspis angusticeps). J. Proteom. 2016, 136, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Aird, S.D.; Watanabe, Y.; Villar-Briones, A.; Roy, M.C.; Terada, K.; Mikheyev, A.S. Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genom. 2013, 14, 790. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.H.; Tan, K.Y.; Ng, T.S.; Quah, E.S.H.; Ismail, A.K.; Khomvilai, S.; Sitprija, V.; Tan, N.H. Venomics of Trimeresurus (Popeia) nebularis, the Cameron Highlands Pit Viper from Malaysia: Insights into Venom Proteome, Toxicity and Neutralization of Antivenom. Toxins 2019, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Oh, A.M.F.; Tan, K.Y.; Tan, N.H.; Tan, C.H. Proteomics and neutralization of Bungarus multicinctus (Many-banded Krait) venom: Intra-specific comparisons between specimens from China and Taiwan. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 247, 109063. [Google Scholar] [CrossRef]
- Kalita, B.; Patra, A.; Das, A.; Mukherjee, A.K. Proteomic Analysis and Immuno-Profiling of Eastern India Russell’s Viper (Daboia russelii) Venom: Correlation between RVV Composition and Clinical Manifestations Post RV Bite. J. Proteome Res. 2018, 17, 2819–2833. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Tsai, W.-C.; Ureña-Diaz, J.M.; Sanz, L.; Mora-Obando, D.; Sánchez, E.E.; Fry, B.G.; Gutiérrez, J.M.; Gibbs, H.L.; Sovic, M.G.; et al. Venomics of New World pit vipers: Genus-wide comparisons of venom proteomes across Agkistrodon. J. Proteom. 2013, 96, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genom. 2015, 16, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, A.T.C.; Leme, A.F.P.; Zelanis, A.; Rocha, M.M.T.; Furtado, M.D.F.D.; Silva, D.A.; Trugilho, M.R.O.; da Rocha, S.L.G.; Perales, J.; Ho, P.L.; et al. Venomics Profiling of Thamnodynastes strigatus Unveils Matrix Metalloproteinases and Other Novel Proteins Recruited to the Toxin Arsenal of Rear-Fanged Snakes. J. Proteome Res. 2012, 11, 1152–1162. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, N.H.; Tan, C.H. Venom proteomics and antivenom neutralization for the Chinese eastern Russell’s viper, Daboia siamensis from Guangxi and Taiwan. Sci. Rep. 2018, 8, 8545. [Google Scholar] [CrossRef]
- Chapeaurouge, A.; Reza, A.; Mackessy, S.; Carvalho, P.C.; Valente, R.H.; Teixeira-Ferreira, A.; Perales, J.; Lin, Q.; Kini, M. Interrogating the Venom of the Viperid Snake Sistrurus catenatus edwardsii by a Combined Approach of Electrospray and MALDI Mass Spectrometry. PLoS ONE 2015, 10, e0092091. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.H.; Tan, K.Y.; Tan, N.H. Revisiting Notechis scutatus venom: On shotgun proteomics and neutralization by the “bivalent” Sea Snake Antivenom. J. Proteom. 2016, 144, 33–38. [Google Scholar] [CrossRef]
- Pandeswari, P.B.; Sabareesh, V. Middle-down approach: A choice to sequence and characterize proteins/proteomes by mass spectrometry. RSC Adv. 2019, 9, 313–344. [Google Scholar] [CrossRef] [Green Version]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [Green Version]
- Ghezellou, P.; Garikapati, V.; Kazemi, S.M.; Strupat, K.; Ghassempour, A.; Spengler, B. A perspective view of top-down proteomics in snake venom research. Rapid Commun. Mass Spectrom. 2018, 33, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-C.; Tsai, T.-S.; Tsai, I.-H. Functional proteomic approach to discover geographic variations of king cobra venoms from Southeast Asia and China. J. Proteom. 2013, 89, 141–153. [Google Scholar] [CrossRef]
- Petras, D.; Heiss, P.; Harrison, R.; Süssmuth, R.D.; Calvete, J.J. Top-down venomics of the East African green mamba, Dendroaspis angusticeps, and the black mamba, Dendroaspis polylepis, highlight the complexity of their toxin arsenals. J. Proteom. 2016, 146, 148–164. [Google Scholar] [CrossRef]
- Melani, R.D.; Nogueira, F.C.S.; Domont, G.B. It is time for top-down venomics. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 44. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, A.; Marino, F.; Post, H.; Toorn, H.W.P.V.D.; Mohammed, S.; Heck, A.J.R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Tran, J.C.; Zamdborg, L.; Durbin, K.R.; Li, M.; Ahlf, D.R.; Early, B.P.; Thomas, P.; Sweedler, J.; Kelleher, N.L. A protease for ‘middle-down’ proteomics. Nat. Methods 2012, 9, 822–824. [Google Scholar] [CrossRef] [Green Version]
- Tasoulis, T.; Pukala, T.L.; Isbister, G.K. Comments on Proteomic Investigations of Two Pakistani Naja Snake Venoms Species Unravel the Venom Complexity, Posttranslational Modifications, and Presence of Extracellular Vesicles. Toxins 2020, 12, 780. [Google Scholar] [CrossRef]
- Barua, A.; Mikheyev, A.S. An ancient, conserved gene regulatory network led to the rise of oral venom systems. Proc. Natl. Acad. Sci. USA 2021, 118, e2021311118. [Google Scholar] [CrossRef]
- Kerkkamp, H.M.I.; Kini, R.M.; Pospelov, A.S.; Vonk, F.J.; Henkel, C.V.; Richardson, M.K. Snake Genome Sequencing: Results and Future Prospects. Toxins 2016, 8, 360. [Google Scholar] [CrossRef] [Green Version]
- Holding, M.L.; Margres, M.J.; Mason, A.J.; Parkinson, C.L.; Rokyta, D.R. Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics. Toxins 2018, 10, 249. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.H.; Tan, K.Y. De Novo Venom-Gland Transcriptomics of Spine-Bellied Sea Snake (Hydrophis curtus) from Penang, Malaysia—Next-Generation Sequencing, Functional Annotation and Toxinological Correlation. Toxins 2021, 13, 127. [Google Scholar] [CrossRef]
- Chong, H.P.; Tan, K.Y.; Tan, N.H.; Tan, C.H. Exploring the Diversity and Novelty of Toxin Genes in Naja sumatrana, the Equatorial Spitting Cobra from Malaysia through De Novo Venom-Gland Transcriptomics. Toxins 2019, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2015, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Greenleaf, W.J.; Sidow, A. The future of sequencing: Convergence of intelligent design and market Darwinism. Genome Biol. 2014, 15, 303. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Bleidorn, C. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 2015, 14, 1–8. [Google Scholar] [CrossRef]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Weinstock, G.M.; Robinson, G.E.; Gibbs, R.A.; Weinstock, G.M.; Weinstock, G.M.; Robinson, G.E.; Worley, K.C.; Evans, J.D.; Maleszka, R.; Robertson, H.M.; et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef]
- Vicoso, B.; Emerson, J.J.; Zektser, Y.; Mahajan, S.; Bachtrog, D. Comparative Sex Chromosome Genomics in Snakes: Differentiation, Evolutionary Strata, and Lack of Global Dosage Compensation. PLoS Biol. 2013, 11, e1001643. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; de Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [Green Version]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [Green Version]
- Ullate-Agote, A.; Milinkovitch, M.C.; Tzika, A.C. The genome sequence of the corn snake (Pantherophis guttatus), a valuable resource for EvoDevo studies in squamates. Int. J. Dev. Biol. 2014, 58, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, B.W.; Card, D.C.; McGlothlin, J.W.; Pasquesi, G.I.M.; Adams, R.H.; Schield, D.R.; Hales, N.R.; Corbin, A.B.; Demuth, J.P.; Hoffmann, F.G.; et al. Molecular Adaptations for Sensing and Securing Prey and Insight into Amniote Genome Diversity from the Garter Snake Genome. Genome Biol. Evol. 2018, 10, 2110–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Wang, Z.-J.; Li, Q.; Lian, J.; Zhou, Y.; Lu, B.-Z.; Jin, L.-J.; Qiu, P.-X.; Zhang, P.; Zhu, W.-B.; et al. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat. Commun. 2016, 7, 13107. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N.; Nakamura, H.; Yamaguchi, K.; Hattori, S.; Matsubara, K.; Matsuda, Y.; Yamashita, A.; Isomoto, A.; et al. The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 2018, 8, 11300. [Google Scholar] [CrossRef]
- Margres, M.J.; Rautsaw, R.M.; Strickland, J.L.; Mason, A.J.; Schramer, T.D.; Hofmann, E.P.; Stiers, E.; Ellsworth, S.A.; Nystrom, G.S.; Hogan, M.P.; et al. The Tiger Rattlesnake genome reveals a complex genotype underlying a simple venom phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2014634118. [Google Scholar] [CrossRef]
- Li, A.; Wang, J.; Sun, K.; Wang, S.; Zhao, X.; Wang, T.; Xiong, L.; Xu, W.; Qiu, L.; Shang, Y.; et al. Two Reference-Quality Sea Snake Genomes Reveal Their Divergent Evolution of Adaptive Traits and Venom Systems. Mol. Biol. Evol. 2021, 38, 4867–4883. [Google Scholar] [CrossRef]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.J.; Li, C.; Li, Q.Y.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2011, 40, D130–D135. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, S.K.; et al. The Pfam protein families database. Nucleic Acids Res. 2009, 38, D211–D222. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Tan, K.Y.; Tan, C.H. De novo venom gland transcriptomics of Calliophis bivirgata flaviceps: Uncovering the complexity of toxins from the Malayan blue coral snake. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20210024. [Google Scholar] [CrossRef]
- Aird, S.D.; Da Silva, N.J.; Qiu, L.; Villar-Briones, A.; Saddi, V.A.; Telles, M.P.D.C.; Grau, M.L.; Mikheyev, A.S. Coralsnake Venomics: Analyses of Venom Gland Transcriptomes and Proteomes of Six Brazilian Taxa. Toxins 2017, 9, 187. [Google Scholar] [CrossRef]
- Margres, M.; McGivern, J.J.; Seavy, M.; Wray, K.P.; Facente, J.; Rokyta, D.R. Contrasting Modes and Tempos of Venom Expression Evolution in Two Snake Species. Genetics 2014, 199, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 312. [Google Scholar] [CrossRef] [Green Version]
- Spielman, S.J.; Wan, S.; Wilke, C.O. A Comparison of One-Rate and Two-Rate Inference Frameworks for Site-Specific dN/dS Estimation. Genetics 2016, 204, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Nobuhisa, I.; Deshimaru, M.; Nakai, M.; Ogawa, T.; Shimohigashi, Y.; Fukumaki, Y.; Hattori, M.; Sakaki, Y.; Hattori, S. Accelerated evolution in the protein-coding regions is universal in crotalinae snake venom gland phospholipase A2 isozyme genes. Proc. Natl. Acad. Sci. USA 1995, 92, 5605–5609. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Ogawa, T.; Oda, N.; Hattori, M.; Sakaki, Y.; Kihara, H.; Ohno, M. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc. Natl. Acad. Sci. USA 1993, 90, 5964–5968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casewell, N.R. On the ancestral recruitment of metalloproteinases into the venom of snakes. Toxicon 2012, 60, 449–454. [Google Scholar] [CrossRef]
- Castoe, T.A.; Hall, K.T.; Mboulas, M.L.G.; Gu, W.; de Koning, A.J.; Fox, S.E.; Poole, A.W.; Vemulapalli, V.; Daza, J.M.; Mockler, T.; et al. Discovery of Highly Divergent Repeat Landscapes in Snake Genomes Using High-Throughput Sequencing. Genome Biol. Evol. 2011, 3, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Aird, S.D. Ophidian envenomation strategies and the role of purines. Toxicon 2001, 40, 335–393. [Google Scholar] [CrossRef]
- Li, M.; Fry, B.G.; Kini, R.M. Putting the Brakes on Snake Venom Evolution: The Unique Molecular Evolutionary Patterns of Aipysurus eydouxii (Marbled Sea Snake) Phospholipase A2 Toxins. Mol. Biol. Evol. 2005, 22, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Tekaia, F. Inferring Orthologs: Open Questions and Perspectives. Genom. Insights 2016, 9, GEI.S37925-28. [Google Scholar] [CrossRef]
- Vonk, F.J.; Admiraal, J.F.; Jackson, K.; Reshef, R.; de Bakker, M.; Vanderschoot, K.; Berge, I.V.D.; Van Atten, M.; Burgerhout, E.; Beck, A.; et al. Evolutionary origin and development of snake fangs. Nature 2008, 454, 630–633. [Google Scholar] [CrossRef]
- Dowell, N.L.; Giorgianni, M.W.; Kassner, V.A.; Selegue, J.E.; Sanchez, E.E.; Carroll, S.B. The Deep Origin and Recent Loss of Venom Toxin Genes in Rattlesnakes. Curr. Biol. 2016, 26, 2434–2445. [Google Scholar] [CrossRef] [Green Version]
- Almeida, D.D.; Viala, V.L.; Nachtigall, P.G.; Broe, M.; Gibbs, H.L.; Serrano, S.M.D.T.; Moura-Da-Silva, A.M.; Ho, P.L.; Nishiyama, M.Y., Jr.; Junqueira-De-Azevedo, I.L.M. Tracking the recruitment and evolution of snake toxins using the evolutionary context provided by the Bothrops jararaca genome. Proc. Natl. Acad. Sci. USA 2021, 118, e2015159118. [Google Scholar] [CrossRef]
- Telford, M.J.; Copley, R.R. Improving animal phylogenies with genomic data. Trends Genet. 2011, 27, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Maria Gutiérrez, J.; Knudsen, C.; Johansen, K.H.; Bermúdez-Méndez, E.; Cerni, F.A.; Jürgensen, J.A.; Ledsgaard, L.; Martos-Esteban, A.; Øhlenschlæger, M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018, 146, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Lingam, T.M.C.; Tan, K.Y. Varespladib (LY315920) rescued mice from fatal neurotoxicity caused by venoms of five major Asiatic kraits (Bungarus spp.) in an experimental envenoming and rescue model. Acta Trop 2022, 227, 106289. [Google Scholar] [CrossRef] [PubMed]
- Salvador, G.H.M.; Gomes, A.A.S.; Bryan-Quiros, W.; Fernandez, J.; Lewin, M.R.; Gutierrez, J.M.; Lomonte, B.; Fontes, M.R.M. Structural basis for phospholipase A2-like toxin inhibition by the synthetic compound Varespladib (LY315920). Sci Rep 2019, 9, 17203. [Google Scholar] [CrossRef]
- Rockman, M.V.; Kruglyak, L. Genetics of global gene expression. Nat. Rev. Genet. 2006, 7, 862–872. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Op den Brouw, B.; Coimbra, F.C.P.; Bourke, L.A.; Huynh, T.M.; Vlecken, D.H.W.; Ghezellou, P.; Visser, J.C.; Dobson, J.S.; Fernandez-Rojo, M.A.; Ikonomopoulou, M.P.; et al. Extensive Variation in the Activities of Pseudocerastes and Eristicophis Viper Venoms Suggests Divergent Envenoming Strategies Are Used for Prey Capture. Toxins 2021, 13, 112. [Google Scholar] [CrossRef]
- Powell, D.W.; Weaver, C.M.; Jennings, J.L.; McAfee, K.J.; He, Y.; Weil, P.A.; Link, A.J. Cluster Analysis of Mass Spectrometry Data Reveals a Novel Component of SAGA. Mol. Cell. Biol. 2004, 24, 7249–7259. [Google Scholar] [CrossRef] [Green Version]
- Old, W.; Meyer-Arendt, K.; Aveline-Wolf, L.; Pierce, K.G.; Mendoza, A.; Sevinsky, J.R.; Resing, K.A.; Ahn, N.G. Comparison of Label-free Methods for Quantifying Human Proteins by Shotgun Proteomics. Mol. Cell. Proteom. 2005, 4, 1487–1502. [Google Scholar] [CrossRef]
- Heck, M.; Neely, B. Proteomics in non-model organisms: A new analytical frontier. J. Proteome Res. 2020, 19, 3595–3606. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Decomplexation | Non-Decomplexation | |
|---|---|---|
| Sample requirement | Moderate to large amount especially if chromatography is involved, typically in milligrams of protein. | Minute amount, typically in as low as micrograms of protein. |
| Techniques | Protein separation methods applying chromatography, e.g., reverse-phase/ion-exchange/size-exclusion HPLC, and gel electrophoresis techniques (1D or 2D). | Proteins in venom sample are subjected to mass spectrometry (including the preparative work of protein digestion) without prior biochemical separation *. |
| Downstream experiment | Proteins eluted from chromatography can be readily collected for further purification and characterization. | Limited. |
| Advantages | Provides additional information regarding protein characteristics, e.g., hydrophobicity, pI, and molecular size. Further downstream studies, e.g., toxin-specific neutralization and antivenomics, are possible. | Technically less demanding. Time-saving and profiling can be achieved fast with fewer resources. Useful when venom sample is limited. |
| Disadvantages | Laborious and time-consuming. Large amount of sample and more resources are required. | Limited information of protein characteristics. |
| Examples | HPLC [94,95] HPLC and 1D SDS-PAGE [29,96] 1D SDS-PAGE [83,97] 2D SDS-PAGE [81,98] | [49,92,93,99] |
| Bottom-Up | Top-Down | |
|---|---|---|
| Protein truncation | Yes, achieved by proteolytic digestion with enzymes, e.g., trypsin and chymotrypsin. Commonly performed as in-solution or in-gel digestion. | Venom proteins are not subjected to proteolytic digestion. |
| Protein/peptide size | Peptides of ~7–20 amino acid residues (0.8–2 kDa) are analyzed. | The intact protein is analyzed whole. |
| Ionization and Fragmentation | Peptides from proteolytic cleavage are ionized by ESI/MALDI techniques. | Intact protein is fragmented in the mass spectrometer. Fragmentation is accomplished by ECD or ETD. |
| Advantages | Technique is mature, commonly used, and widely available. Less sophisticated instrumentation and expertise are required. High-resolution separations can be achieved. | Technique avoids time-consuming protein digestion (typically overnight). Full amino acid sequence can be recovered. Protein isoforms can be determined.PTMs can be located and characterized. |
| Disadvantages | A low percentage coverage of the amino acid sequence. Information of PTMs is limited. | Instrumentation is expensive and operation is technically sophisticated. Not commonly available. The favored dissociation techniques (ECT, ETC) are less efficient. Resolution is low, especially for high molecular weight proteins. |
| No. | Date of Submission | Common Name | Scientific Name | Family | Sex | Assembly Type | Genome Representation | Notes |
|---|---|---|---|---|---|---|---|---|
| 1 | 15/09/2013 | Burmese python | Python bivittatus | Pythonidae | Female | Scaffold | Full | GCA_000186305.2 |
| 2 | 11/12/2013 | King Cobra | Ophiophagus hannah | Elapidae | Male | Scaffold | Full | GCA_000516915.1 |
| 3 | 01/08/2014 | Southwestern Speckled Rattlesnake | Crotalus pyrrhus | Viperidae | Female | Scaffold | Full | GCA_000737285.1 |
| 4 | 10/12/2014 | European Adder | Vipera berus berus | Viperidae | Female | Scaffold | Full | GCA_000800605.1 |
| 5 | 26/06/2015 | Common Garter Snake | Thamnophis sirtalis | Colubridae | Female | Scaffold | Full | GCA_001077635.2 |
| 6 | 22/01/2016 | Brown Spotted Pitviper; Taiwanese Habu | Protobothrops mucrosquamatus | Viperidae | Not stated | Scaffold | Full | GCA_001527695.3 |
| 7 | 21/04/2016 | Timber Rattlesnake | Crotalus horridus | Viperidae | Female | Scaffold | Full | GCA_001625485.1 |
| 8 | 02/08/2018 | Okinawa Habu | Protobothrops flavoviridis | Viperidae | Female | Scaffold | Full | GCA_003402635.1 |
| 9 | 05/09/2018 | Xizang Hot-spring Keel-back | Thermophis baileyi Ecotype: Yangbajing | Colubridae | Female | Scaffold | Full | GCA_003457575.1 |
| 10 | 24/09/2018 | Eastern Brown Snake | Pseudonaja textilis | Elapidae | Not stated | Scaffold | Full | GCA_900518735.1 |
| 11 | 24/09/2018 | Mainland Tiger Snake | Notechis scutatus | Elapidae | Not stated | Scaffold | Full | GCA_900518725.1 |
| 12 | 08/01/2019 | Prairie Rattlesnake | Crotalus viridis viridis | Viperidae | Male | Chromosome | Full | GCA_003400415.2 |
| 13 | 09/01/2019 | Ijima’s Turtleheaded Sea Snake | Emydocephalus ijimae | Elapidae | Not stated | Scaffold | Full | GCA_004319985.1 |
| 14 | 09/01/2019 | Yellow-Lipped Sea Krait | Laticauda colubrina | Elapidae | Not stated | Scaffold | Full | GCA_004320045.1 |
| 15 | 09/01/2019 | Blue-ringed Sea Krait | Laticauda laticaudata | Elapidae | Not stated | Scaffold | Full | GCA_004320025.1 |
| 16 | 15/01/2019 | Asian Annulated Sea Snake | Hydrophis cyanocinctus | Elapidae | Not stated | Scaffold | Full | GCA_004023725.1 |
| 17 | 15/01/2019 (latest) | Hardwick’s Sea Snake | Hydrophis hardwickii | Elapidae | Not stated | Scaffold | Full | GCA_004023765.1 |
| 18 | 13/02/2019 | Slender-necked Sea Snake | Hydrophis melanocephalus | Elapidae | Not stated | Scaffold | GCA_004320005.1 | |
| 19 | 11/12/2019 | Indian Cobra | Naja naja | Elapidae | male | Chromosome | Full | GCA_009733165.1 |
| 20 | 19/12/2019 | Western Terrestrial Garter Snake | Thamnophis elegans | Colubridae | Female | Type: alternate-pseudohaplotype Level: Scaffold | Full | GCA_009769695.1 |
| 21 | 23/12/2019 | Western Terrestrial Garter Snake | Thamnophis elegans | Colubridae | Female | Assembly type: haploid (principal pseudohaplotype of diploid) Assembly level: Chromosome | Full | GCA_009769535.1 |
| 22 | 13/04/2020 | Corn Snake | Pantherophis guttatus | Colubridae | Male | Scaffold | Full | GCA_001185365.2 |
| 23 | 22/04/2020 | Western Rat Snake | Pantherophis obsoletus | Colubridae | Female | Scaffold | Full | GCA_012654085.1 |
| 24 | 22/04/2020 | Dhaman; Oriental Ratsnake | Ptyas mucosa | Colubridae | Female | Scaffold | Full | GCA_012654045.1 |
| 25 | 04/11/2020 (latest) | Yellow-Lipped Sea Krait | Laticauda colubrina | Elapidae | Not stated | Scaffold | Full | GCA_015471245.1 |
| 26 | 22/11/2020 | Eastern Brown Snake | Pseudonaja textilis | Elapidae | Not stated | Scaffold | Full | GCA_900608585.1 |
| 27 | 22/11/2020 (latest) | Mainland Tiger Snake | Notechis scutatus | Elapidae | Not stated | Scaffold | Full | GCA_900608555.1 |
| 28 | 06/01/2021 | Tiger Rattlesnake | Crotalus tigris Infraspecific name: Ecotype: Arizon | Viperidae | Not stated | Contig | Full | GCA_016545835.1 |
| 29 | 01/04/2021 | Mud Snake | Myanophis thanlyinensis | Homalopsidae | Male | Scaffold | Full | GCA_017656035.1 |
| 30 | 19/04/2021 | Indian Cobra | Naja naja | Elapidae | Female | Scaffold | Full | GCA_018093825.1 |
| 31 | 2021/05/11/05/2021 | Jararaca | Bothrops jararaca | Viperidae | Female | Scaffold | Full | GCA_018340635.1 |
| 32 | 25/05/2021 | Eastern Diamondback Rattlesnake | Crotalus adamanteus | Viperidae | Female | Scaffold | Full | GCA_018446365.1 |
| 33 | 06/08/2021 | Golden Tree Snake | Chrysopelea ornata | Colubridae | Not stated | Scaffold | Full | GCA_019457695.1 |
| 34 | 09/08/2021 | Shaw’s Sea Snake | Hydrophis curtus | Elapidae | Male | Chromosome | Full | GCA_019472885.1 |
| 35 | 09/08/2021 | Annulated Sea Snake | Hydrophis cyanocinctus | Elapidae | Male | Chromosome | Full | GCA_019473425.1 |
| 36 | 18/08/2021 | Gopher Snake | Pituophis catenifer pumilus Infraspecific name: Ecotype: Santa Cruz Island | Colubridae | Female | Scaffold | Full | GCA_019677565.1 |
| 37 | 23/02/2022 | Prong-snouted blind snake | Anilios bituberculatus | Typhlopidae | Not stated | Sca | Full | GCA_022379055.1 |
| 38 | 15/03/2022 | Glossy snake | Arizona elegans Infraspecific name: Ecotype: subspecies occidentalis | Colubridae | Not stated | Scaffold (alternate-pseudohaplotype) | Full | GCA_022578425.1 |
| 39 | 15/03/2022 | Glossy snake | Arizona elegans Infraspecific name: Ecotype: subspecies occidentalis | Colubridae | Not stated | Scaffold (haploid, principal pseudohaplotype of diploid) | Full | GCA_022577455.1 |
| Protein Family/Subtype a | Accession No. b,c | Relative Protein Abundance d (%) | |
|---|---|---|---|
| Method 1 | Method 2 | ||
| Snake Venom Serine Protease (15) | 19.03 | 25.42 | |
| Alpha-fibrinogenase albofibrase | P0CJ41 | 3.74 | 1.69 |
| Alpha-fibrinogenase shedaoenase | Q6T5L0 | 1.20 | 1.69 |
| Beta-fibrinogenase mucrofibrase-2 | Q91508 | 1.08 | 1.69 |
| Snake venom serine protease 1 | Unigene42520_TWM | 1.46 | 1.69 |
| Snake venom serine protease 2 | CL403.contig2_TWM | 0.58 | 1.69 |
| Snake venom serine protease 2A homolog | O13060 | 1.01 | 1.69 |
| Snake venom serine protease 2C | O13062 | 0.53 | 1.69 |
| Snake venom serine protease KN14 | Q71QH9 | 1.70 | 1.69 |
| Snake venom serine protease KN8 | Q71QH5 | 0.87 | 1.67 |
| Snake venom serine protease pallase | O93421 | 1.34 | 1.69 |
| Snake venom serine protease salmonase | Q9PTL3 | 1.16 | 1.69 |
| Thrombin-like enzyme 1 | A7LAC6 | 0.80 | 1.69 |
| Thrombin-like enzyme 2 | A7LAC7 | 1.19 | 1.69 |
| Thrombin-like enzyme calobin-1 | Q91053 | 0.65 | 1.69 |
| Venom plasminogen activator TSV-PA | Q91516 | 1.71 | 1.69 |
| Snake Venom Metalloproteinase (11) | 17.29 | 18.64 | |
| P-II SVMP | |||
| Zinc metalloproteinase/disintegrin | Unigene5053_TWM | 0.34 | 1.69 |
| Zinc metalloproteinase homolog-disintegrin albolatin | P0C6B6 | 0.92 | 1.69 |
| P-III SVMP | |||
| Zinc metalloproteinase/disintegrin | P0C6E8 | 1.30 | 1.69 |
| Zinc metalloproteinase-disintegrin-like ACLD | CL1397.contig5_TWM | 1.73 | 1.69 |
| Zinc metalloproteinase-disintegrin-like ACLD | O42138 | 0.17 | 1.69 |
| Zinc metalloproteinase-disintegrin-like HF3 | Q98UF9 | 1.81 | 1.69 |
| Zinc metalloproteinase-disintegrin-like stejnihagin-A | Q3HTN1 | 1.70 | 1.69 |
| Zinc metalloproteinase-disintegrin-like stejnihagin-B | Q3HTN2 | 0.57 | 1.69 |
| Zinc metalloproteinase-disintegrin-like TSV-DM | J3RYA3 | 0.16 | 1.69 |
| Zinc metalloproteinase-disintegrin-like VMP-III | CL1397.contig1_TWM | 8.41 | 1.69 |
| Zinc metalloproteinase-disintegrin-like VMP-III | C9E1S0 | 0.18 | 1.69 |
| Disintegrin (2) | 15.82 | 3.39 | |
| Disintegrin albolabrin | P62384 | 4.74 | 1.69 |
| Disintegrin trigramin-gamma | P62383 | 11.08 | 1.69 |
| C-type Lectin (9) | 8.92 | 15.25 | |
| C-type lectin 6 | Unigene46336_TWM | 1.01 | 1.69 |
| C-type lectin TsL | Q9YGP1 | 1.85 | 1.69 |
| Snaclec alboaggregin-D subunit alpha | P0DM38 | 0.88 | 1.69 |
| Snaclec clone 2100755 | Q8JIV8 | 0.85 | 1.69 |
| Snaclec coagulation factor IX/factor X-binding protein subunit A | Q71RR4 | 0.70 | 1.69 |
| Snaclec convulxin subunit alpha | Unigene46337_TWM | 0.35 | 1.69 |
| Snaclec purpureotin subunit alpha | P0DJL2 | 1.48 | 1.69 |
| Snaclec purpureotin subunit beta | P0DJL3 | 1.27 | 1.69 |
| Snaclec stejaggregin-A subunit alpha | CL746.contig2_TWM | 0.53 | 1.69 |
| Phospholipase A2 (7) | 27.54 | 11.86 | |
| Acidic phospholipase A2 | P20249 | 2.54 | 1.69 |
| Acidic phospholipase A2 6 | P70088 | 2.77 | 1.69 |
| Acidic phospholipase A2 Tpu-E6c | P0DJP4 | 4.77 | 1.69 |
| Basic phospholipase A2 daboxin P | C0HK16 | 1.18 | 1.69 |
| Basic phospholipase A2 homolog Tpu-K49a | Q2YHJ9 | 7.50 | 1.69 |
| Basic phospholipase A2 homolog Tpu-K49b | Q2YHJ8 | 4.45 | 1.69 |
| Basic phospholipase A2 Tpu-G6D49 | Q2YHJ7 | 4.33 | 1.69 |
| Cysteine-rich Secretory Protein (3) | 1.69 | 5.08 | |
| Cysteine-rich secretory protein | P60623 | 0.91 | 1.69 |
| Cysteine-rich secretory protein | Unigene30615_TWM | 0.26 | 1.69 |
| Cysteine-rich secretory protein triflin | Q8JI39 | 0.52 | 1.69 |
| L-Amino Acid Oxidase (4) | 4.82 | 6.78 | |
| L-amino-acid oxidase | B0VXW0 | 0.66 | 1.69 |
| L-amino-acid oxidase | Q6WP39 | 1.72 | 1.69 |
| L-amino-acid oxidase | Q90W54 | 0.87 | 1.69 |
| L-amino-acid oxidase | Unigene40029_TWM | 1.57 | 1.69 |
| Snake Venom Phosphodiesterase (3) | 2.27 | 5.08 | |
| Phosphodiesterase | Unigene5177_TWM | 0.65 | 1.69 |
| Venom phosphodiesterase 1 | J3SEZ3 | 0.71 | 1.69 |
| Venom phosphodiesterase 2 | J3SBP3 | 0.90 | 1.69 |
| Snake Venom Vascular Endothelial Growth Factor (1) | 2.49 | 1.69 | |
| Snake venom vascular endothelial growth factor toxin | Unigene25068_TWM | 1.54 | 1.69 |
| Snake Venom 5′-Nucleotidase (2) | 0.71 | 3.39 | |
| Snake venom 5′-nucleotidase | Unigene721_TWM | 0.38 | 1.69 |
| Snake venom 5′-nucleotidase | B6EWW8 | 0.33 | 1.69 |
| Snake Venom Nerve Growth Factor (1) | 0.18 | 1.69 | |
| Nerve growth factor | CL2590.contig1_TWM | 0.18 | 1.69 |
| Phospholipase B (1) | 0.18 | 1.69 | |
| Phospholipase B | Unigene25350_TWM | 0.18 | 1.69 |
| Total number of proteins: 59 | Total protein abundance (%) | 100 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.H. Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade. Toxins 2022, 14, 247. https://doi.org/10.3390/toxins14040247
Tan CH. Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade. Toxins. 2022; 14(4):247. https://doi.org/10.3390/toxins14040247
Chicago/Turabian StyleTan, Choo Hock. 2022. "Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade" Toxins 14, no. 4: 247. https://doi.org/10.3390/toxins14040247
APA StyleTan, C. H. (2022). Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade. Toxins, 14(4), 247. https://doi.org/10.3390/toxins14040247