What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis




Abstract
1. Introduction
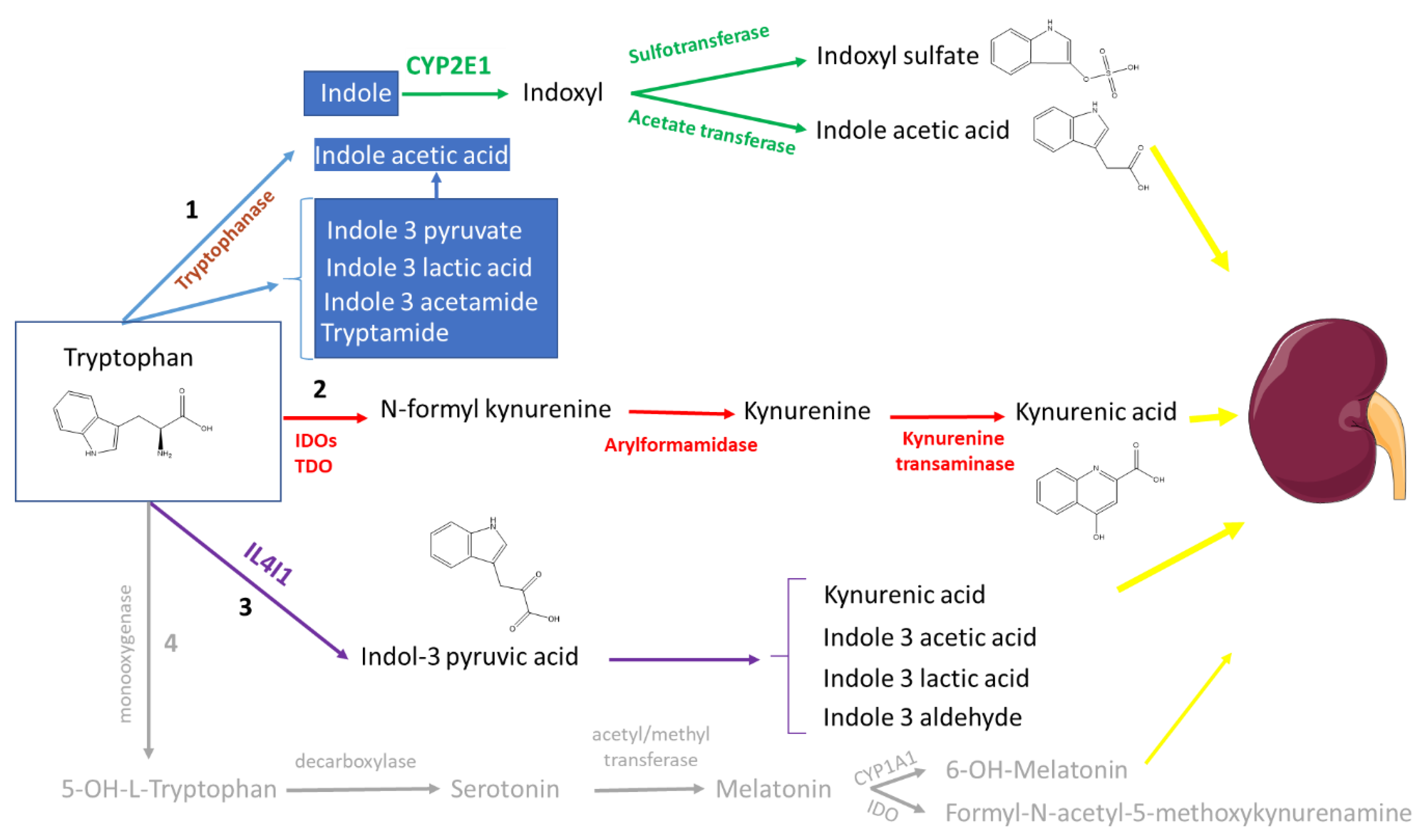
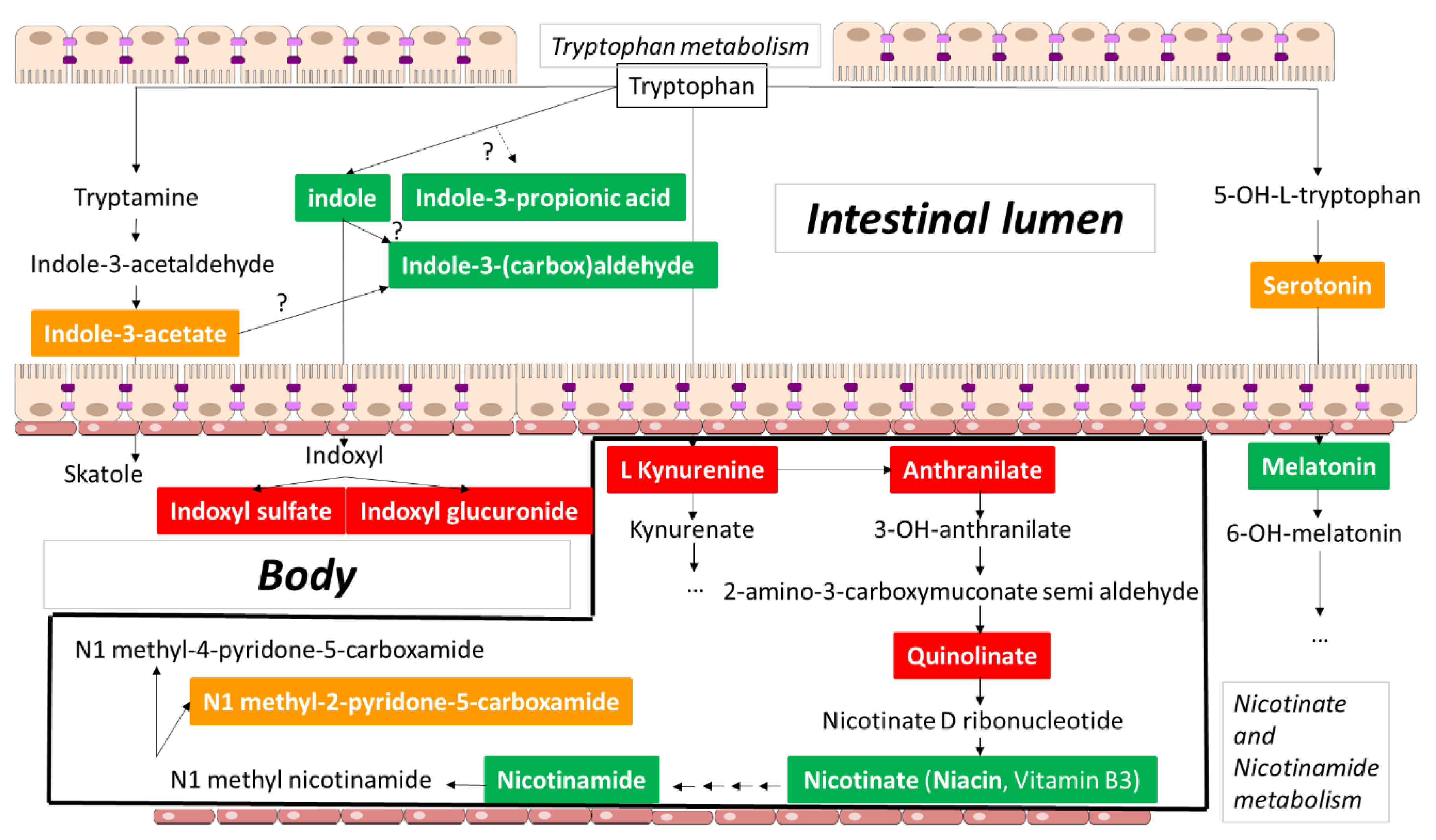
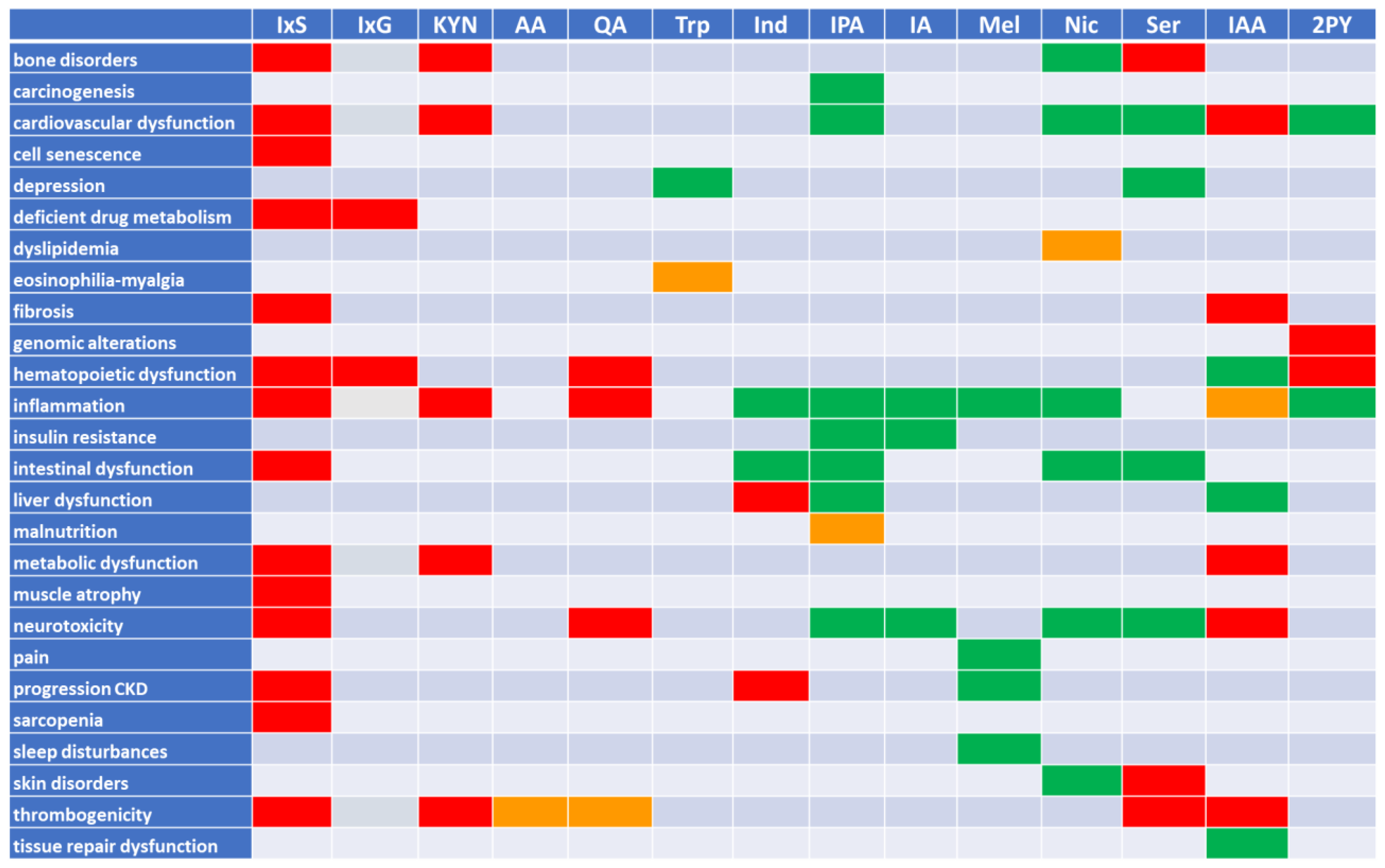
2. Tryptophan Metabolites: The Good, the Bad, and the Ambivalent
2.1. The (Mainly) Toxic Molecules
2.1.1. Indoxyl Sulfate
2.1.2. Indoxyl Glucuronide
2.1.3. Kynurenine and Kynurenic Acid
2.1.4. Anthranilic Acid
2.1.5. Quinolinic Acid
2.2. The (Mainly) Beneficial Molecules
2.2.1. Tryptophan
2.2.2. Indole
2.2.3. Indole-3-propionic Acid
2.2.4. Indole-3-(carbox)aldehyde
2.2.5. Melatonin
2.2.6. Nicotinic Acid and Nicotinamide
2.3. The Ambivalent Molecules
2.3.1. Serotonin
2.3.2. Indole-3-acetic Acid
2.3.3. 1-Methyl-2-pyridone-5-carboxamide
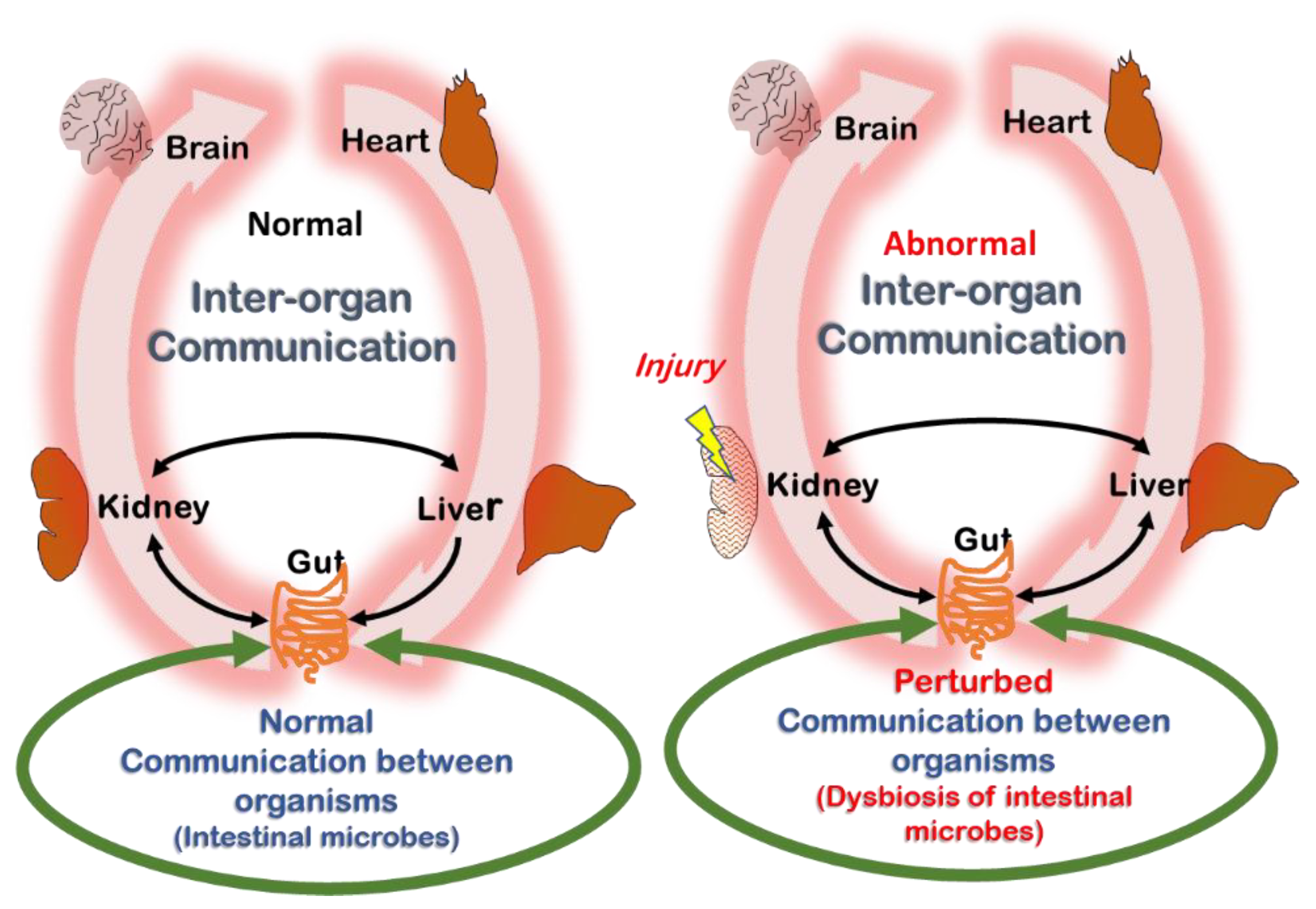
2.4. Remote Sensing and Signaling Theory (RSST) and Its Relation to Uremia
2.5. Summary
3. The Dualism of the Aryl Hydrocarbon Receptor
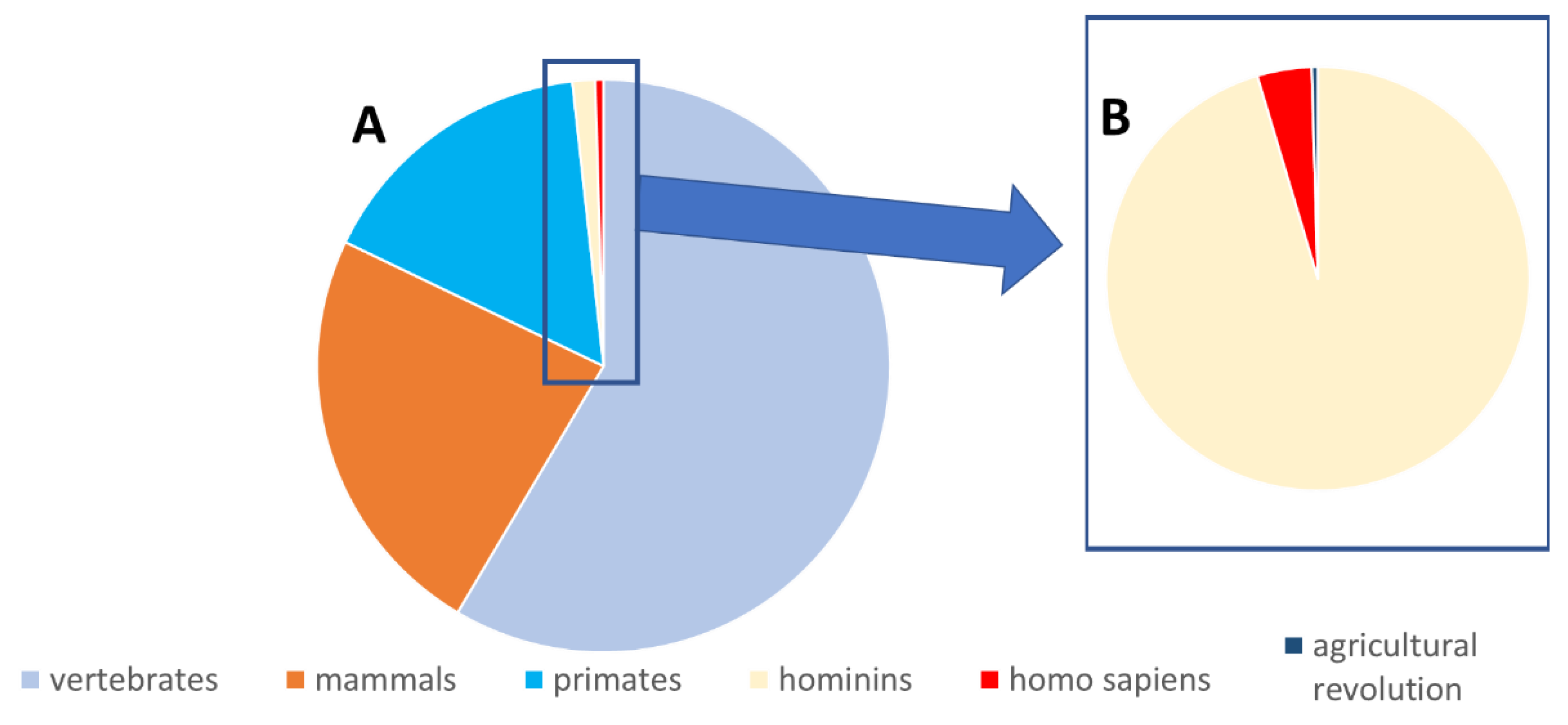
4. Why Did the Body Continue Producing Toxic Tryptophan Metabolites throughout Evolution?
5. Is the Example of Tryptophan Metabolites Representative for Uremic Retention at Large?
6. Summary and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kramer, A.; Boenink, R.; Noordzij, M.; Bosdriesz, J.R.; Stel, V.S.; Beltrán, P.; Ruiz, J.C.; Seyahi, N.; Farnés, J.C.; Stendahl, M.; et al. The ERA-EDTA Registry Annual Report 2017: A summary. Clin. Kidney J. 2020, 13, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Annemans, L.; Bello, A.K.; Bikbov, B.; Gallego, D.; Gansevoort, R.T.; Lameire, N.; A Luyckx, V.; Noruisiene, E.; Oostrom, T.; et al. Fighting the unbearable lightness of neglecting kidney health: The decade of the kidney. Clin. Kidney J. 2021, 14, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Annemans, L.; Brown, E.; Gansevoort, R.; Gout-Zwart, J.J.; Lameire, N.; Morton, R.L.; Oberbauer, R.; Postma, M.J.; Tonelli, M.; et al. Reducing the costs of chronic kidney disease while delivering quality health care: A call to action. Nat. Rev. Nephrol. 2017, 13, 393–409. [Google Scholar] [CrossRef] [PubMed]
- A Silver, S.; Long, J.; Zheng, Y.; Chertow, G.M. Cost of Acute Kidney Injury in Hospitalized Patients. J. Hosp. Med. 2017, 12, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G.L. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Bugnicourt, J.-M.; Godefroy, O.; Chillon, J.-M.; Choukroun, G.; Massy, Z.A. Cognitive disorders and dementia in CKD: The neglected kidney-brain axis. J. Am. Soc. Nephrol. 2013, 24, 353–363. [Google Scholar] [CrossRef]
- Martin, C.E.; Clotet-Freixas, S.; Farragher, J.F.; Hundemer, G.L. Have We Just Scratched the Surface? A Narrative Review of Uremic Pruritus in 2020. Can. J. Kidney Health Dis. 2020, 7, 2054358120954024. [Google Scholar] [CrossRef]
- Vanholder, R.; Argiles, A.; Jankowski, J.; European Uraemic Toxin Work Group. A history of uraemic toxicity and of the European Uraemic Toxin Work Group (EUTox). Clin. Kidney J. 2021, 14, 1514–1523. [Google Scholar]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; de Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. JASN 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Sadik, A.; Patterson, L.F.S.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Sirich, T.; Meyer, T.W. Indoxyl sulfate: Long suspected but not yet proven guilty. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 3–4. [Google Scholar] [CrossRef]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate—Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef]
- Bobot, M.; Thomas, L.; Moyon, A.; Fernandez, S.; McKay, N.; Balasse, L.; Garrigue, P.; Brige, P.; Chopinet, S.; Poitevin, S.; et al. Uremic Toxic Blood-Brain Barrier Disruption Mediated by AhR Activation Leads to Cognitive Impairment during Experimental Renal Dysfunction. J. Am. Soc. Nephrol. 2020, 31, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Li, J.-R.; Wang, Y.-Y.; Lin, S.-Y.; Ou, Y.-C.; Lin, C.-J.; Wang, J.-D.; Liao, S.-L.; Chen, C.-J. Indoxyl sulfate caused behavioral abnormality and neurodegeneration in mice with unilateral nephrectomy. Aging 2021, 13, 6681–6701. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Dal Piaz, F.; Di Iorio, B.R.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef] [PubMed]
- Hamano, H.; Ikeda, Y.; Watanabe, H.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Imanishi, M.; Zamami, Y.; Takechi, K.; Miyamoto, L.; Ishizawa, K.; et al. The uremic toxin indoxyl sulfate interferes with iron metabolism by regulating hepcidin in chronic kidney disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2018, 33, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Dias, G.F.; Bonan, N.B.; Steiner, T.M.; Tozoni, S.S.; Rodrigues, S.; Nakao, L.S.; Kuntsevich, V.; Filho, R.P.; Kotanko, P.; Moreno-Amaral, A.N. Indoxyl Sulfate, a Uremic Toxin, Stimulates Reactive Oxygen Species Production and Erythrocyte Cell Death Supposedly by an Organic Anion Transporter 2 (OAT2) and NADPH Oxidase Activity-Dependent Pathways. Toxins 2018, 10, 280. [Google Scholar] [CrossRef]
- Wu, C.-J.; Chen, C.-Y.; Lai, T.-S.; Wu, P.-C.; Chuang, C.-K.; Sun, F.-J.; Liu, H.-L.; Chen, H.-H.; Yeh, H.-I.; Lin, C.-S.; et al. The role of indoxyl sulfate in renal anemia in patients with chronic kidney disease. Oncotarget 2017, 8, 83030–83037. [Google Scholar] [CrossRef]
- Rodrigues, G.G.C.; Dellê, H.; Brito, R.B.O.; Cardoso, V.O.; Fernandes, K.; Mesquita-Ferrari, R.A.; Cunha, R.S.; Stinghen, A.; Dalboni, M.A.; Barreto, F.C. Indoxyl Sulfate Contributes to Uremic Sarcopenia by Inducing Apoptosis in Myoblasts. Arch. Med. Res. 2020, 51, 21–29. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Liu, C.-H.; Lai, Y.-H.; Wang, C.-H.; Kuo, C.-H.; Liou, H.-H.; Hsu, B.-G. Association of Serum Indoxyl Sulfate Levels with Skeletal Muscle Mass and Strength in Chronic Hemodialysis Patients: A 2-year Longitudinal Analysis. Calcif. Tissue Res. 2020, 107, 257–265. [Google Scholar] [CrossRef]
- Santana Machado, T.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R. Indoxyl Sulfate Upregulates Liver P-Glycoprotein Expression and Activity through Aryl Hydrocarbon Receptor Signaling. J. Am. Soc. Nephrol. JASN 2018, 29, 906–918. [Google Scholar]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Lee, S.H. Co-Administration of Melatonin Effectively Enhances the Therapeutic Effects of Pioglitazone on Mesenchymal Stem Cells Undergoing Indoxyl Sulfate-Induced Senescence through Modulation of Cellular Prion Protein Expression. Int. J. Mol. Sci. 2018, 19, 1367. [Google Scholar] [CrossRef]
- Sári, Z.; Mikó, E.; Kovács, T.; Boratkó, A.; Ujlaki, G.; Jankó, L.; Kiss, B.; Uray, K.; Bai, P. Indoxylsulfate, a Metabolite of the Microbiome, Has Cytostatic Effects in Breast Cancer via Activation of AHR and PXR Receptors and Induction of Oxidative Stress. Cancers 2020, 12, 2915. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Hirata, J.; Watanabe-Akanuma, M. Indoxyl glucuronide, a protein-bound uremic toxin, inhibits hypoxia-inducible factordependent erythropoietin expression through activation of aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun. 2018, 504, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.W.K.; Hsueh, C.-H.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.-M.; Giacomini, K.M. The Effect of Uremic Solutes on the Organic Cation Transporter 2. J. Pharm. Sci. 2017, 106, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Kalaska, B.; Pawlak, K.; Domaniewski, T.; Oksztulska-Kolanek, E.; Znorko, B.; Roszczenko, A.; Rogalska, J.; Brzóska, M.M.; Lipowicz, P.; Doroszko, M.; et al. Elevated Levels of Peripheral Kynurenine Decrease Bone Strength in Rats with Chronic Kidney Disease. Front. Physiol. 2017, 8, 836. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Pawlak, K.; Kalaska, B.; Domaniewski, T.; Sieklucka, B.; Zieminska, M.; Cylwik, B.; Pawlak, D. Modulation of the Paracrine Kynurenic System in Bone as a New Regulator of Osteoblastogenesis and Bone Mineral Status in an Animal Model of Chronic Kidney Disease Treated with LP533401. Int. J. Mol. Sci. 2020, 21, 5979. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Grzegorzewski, W.; Kuna, J.; Pawlak, D. Association between uremic toxin-anthranilic acid and fibrinolytic system activity in predialysis patients at different stages of chronic kidney disease. Int. Urol. Nephrol. 2017, 50, 127–135. [Google Scholar] [CrossRef]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Kynurenine pathway—A new link between endothelial dysfunction and carotid atherosclerosis in chronic kidney disease patients. Adv. Med. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Leszczyńska, A.; Misztal, T.; Marcińczyk, N.; Kamiński, T.; Kramkowski, K.; Chabielska, E.; Pawlak, D. Effect of quinolinic acid—A uremic toxin from tryptophan metabolism—On hemostatic profile in rat and mouse thrombosis models. Adv. Med. Sci. 2019, 64, 370–380. [Google Scholar] [CrossRef]
- Shaw, K.; Turner, J.; Del Mar, C. Tryptophan and 5-hydroxytryptophan for depression. Cochrane Database Syst. Rev. 2001, CD003198. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Garrett, R.H. A heretofore undisclosed crux of eosinophilia-myalgia syndrome: Compromised histamine degradation. Agents Actions 2005, 54, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Inubushi, T.; Kamemura, N.; Oda, M.; Sakurai, J.; Nakaya, Y.; Harada, N.; Suenaga, M.; Matsunaga, Y.; Ishidoh, K.; Katunuma, N. L-tryptophan suppresses rise in blood glucose and preserves insulin secretion in type-2 diabetes mellitus rats. J. Nutr. Sci. Vitaminol. 2012, 58, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2009, 107, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Berstad, A.; Raa, J.; Valeur, J. Indole—The scent of a healthy ‘inner soil’. Microb. Ecol. Health Dis. 2015, 26, 27997. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Cargile, C.M.; Cohen, N.D.; Chapkin, R.S.; Weeks, B.R.; Davidson, L.A.; Goldsby, J.S.; Hunt, C.L.; Steinmeyer, S.H.; Menon, R.; Suchodolski, J.S.; et al. The microbiota-derived metabolite indole decreases mucosal inflammation and injury in a murine model of NSAID enteropathy. Gut Microbes 2016, 7, 246–261. [Google Scholar] [CrossRef]
- Beaumont, M.; Neyrinck, A.M.; Olivares, M.; Rodriguez, J.; de Rocca Serra, A.; Roumain, M.; Bindels, L.B.; Cani, P.D.; Evenepoel, P.; Muccioli, G.G.; et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, fj201800544. [Google Scholar] [CrossRef]
- Oh, S.; Go, G.; Mylonakis, E.; Kim, Y. The bacterial signalling molecule indole attenuates the virulence of the fungal pathogen Candida albicans. J. Appl. Microbiol. 2012, 113, 622–628. [Google Scholar] [CrossRef]
- Lee, J.-H.; Cho, H.S.; Kim, Y.; Kim, J.-A.; Banskota, S.; Cho, M.H.; Lee, J. Indole and 7-benzyloxyindole attenuate the virulence of Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2013, 97, 4543–4552. [Google Scholar] [CrossRef]
- Huc, T.; Konop, M.; Onyszkiewicz, M.; Podsadni, P.; Szczepańska, A.; Turło, J.; Ufnal, M. Colonic indole, gut bacteria metabolite of tryptophan, increases portal blood pressure in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R646–R655. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M.; Miyazaki, T. Progression of glomerular sclerosis in experimental uremic rats by administration of indole, a precursor of indoxyl sulfate. Am. J. Nephrol. 1994, 14, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Lin, C.-J.; Pan, H.-C.; Lee, C.-C.; Lu, S.-C.; Hsieh, Y.-T.; Huang, S.-Y.; Huang, H.-Y. Clinical association between the metabolite of healthy gut microbiota, 3-indolepropionic acid and chronic kidney disease. Clin. Nutr. 2019, 38, 2945–2948. [Google Scholar] [CrossRef] [PubMed]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Yoo, K.-Y.; Li, H.; Park, O.K.; Lee, C.H.; Choi, J.H.; Jeong, Y.-G.; Lee, Y.L.; Kim, Y.-M.; Kwon, Y.-G.; et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J. Neurosci. Res. 2009, 87, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z.; et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sári, Z.; Mikó, E.; Kovács, T.; Jankó, L.; Csonka, T.; Lente, G.; Sebő, É.; Tóth, J.; Tóth, D.; Árkosy, P.; et al. Indolepropionic Acid, a Metabolite of the Microbiome, Has Cytostatic Properties in Breast Cancer by Activating AHR and PXR Receptors and Inducing Oxidative Stress. Cancers 2020, 12, 2411. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, L.; Wu, T.; Li, Y.; Zhou, X.; Ruan, Z. Indole-3-propionic Acid Improved the Intestinal Barrier by Enhancing Epithelial Barrier and Mucus Barrier. J. Agric. Food Chem. 2021, 69, 1487–1495. [Google Scholar] [CrossRef]
- Pulakazhi Venu, V.K.; Saifeddine, M.; Mihara, K.; Tsai, Y.; Nieves, K.; Alston, L.; Mani, S.; McCoy, K.D.; Hollenberg, M.D.; Hirota, S.A. The pregnane X receptor and its microbiota-derived ligand indole 3-propionic acid regulate endothelium-dependent vasodilation. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E350–E361. [Google Scholar] [CrossRef]
- de Mello, V.D.; Paananen, J.; Lindstrom, J.; Lankinen, M.A.; Shi, L.; Kuusisto, J.; Pihlajamäki, J.; Auriola, S.; Lehtonen, M.; Rolandsson, O.; et al. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci. Rep. 2017, 7, 46337. [Google Scholar] [CrossRef]
- Konopelski, P.; Konop, M.; Gawrys-Kopczynska, M.; Podsadni, P.; Szczepanska, A.; Ufnal, M. Indole-3-Propionic Acid, a Tryptophan-Derived Bacterial Metabolite, Reduces Weight Gain in Rats. Nutrients 2019, 11, 591. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Ecton, K.E.; Trikha, S.R.; Wrigley, S.D.; Thomas, K.N.; Battson, M.L.; Wei, Y.; Johnson, S.A.; Weir, T.L.; Gentile, C.L. Microbial metabolite indole-3-propionic acid supplementation does not protect mice from the cardiometabolic consequences of a Western diet. Am. J. Physiol. Liver Physiol. 2020, 319, G51–G62. [Google Scholar] [CrossRef] [PubMed]
- Gesper, M.; Nonnast, A.B.H.; Kumowski, N.; Stoehr, R.; Schuett, K.; Marx, N.; Kappel, B.A. Gut-Derived Metabolite Indole-3-Propionic Acid Modulates Mitochondrial Function in Cardiomyocytes and Alters Cardiac Function. Front. Med. 2021, 8, 648259. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Puccetti, M.; Giovagnoli, S.; Romani, L. Regulation of host physiology and immunity by microbial indole-3-aldehyde. Curr. Opin. Immunol. 2021, 70, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, X.; Bi, J.; Zhu, A.; He, L. Indole-3-carboxaldehyde regulates RSV-induced inflammatory response in RAW264.7 cells by moderate inhibition of the TLR7 signaling pathway. J. Nat. Med. 2021, 75, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.; Pariano, M.; Solito, V.; Puccetti, M.; Bellet, M.M.; Stincardini, C.; Renga, G.; Vacca, C.; Sellitto, F.; Mosci, P.; et al. Targeting the Aryl Hydrocarbon Receptor With Indole-3-Aldehyde Protects From Vulvovaginal Candidiasis via the IL-22-IL-18 Cross-Talk. Front. Immunol. 2019, 10, 2364. [Google Scholar] [CrossRef]
- Puccetti, M.; Pariano, M.; Borghi, M.; Barola, C.; Moretti, S.; Galarini, R.; Mosci, P.; Ricci, M.; Costantini, C.; Giovagnoli, S. Enteric formulated indole-3-carboxaldehyde targets the aryl hydrocarbon receptor for protection in a murine model of metabolic syndrome. Int. J. Pharm. 2021, 602, 120610. [Google Scholar] [CrossRef]
- Arendt, J. Melatonin, circadian rhythms, and sleep. N. Engl. J. Med. 2000, 343, 1114–1116. [Google Scholar] [CrossRef]
- Li, T.; Jiang, S.; Han, M.; Yang, Z.; Lv, J.; Deng, C.; Reiter, R.J.; Yang, Y. Exogenous melatonin as a treatment for secondary sleep disorders: A systematic review and meta-analysis. Front. Neuroendocr. 2019, 52, 22–28. [Google Scholar] [CrossRef]
- Auld, F.; Maschauer, E.; Morrison, I.; Skene, D.; Riha, R.L. Evidence for the efficacy of melatonin in the treatment of primary adult sleep disorders. Sleep Med. Rev. 2017, 34, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Fatemeh, G.; Sajjad, M.; Niloufar, R.; Neda, S.; Leila, S.; Khadijeh, M. Effect of melatonin supplementation on sleep quality: A systematic review and meta-analysis of randomized controlled trials. J. Neurol. 2021, 269, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Stone, B.M.; Turner, C.; Mills, S.L.; Nicholson, A.N. Hypnotic activity of melatonin. Sleep 2000, 23, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Fernandez-Palanca, P.; San-Miguel, B.; Alvarez, M.; Gonzalez-Gallego, J.; Tunon, M.J. Melatonin modulates mitophagy, innate immunity and circadian clocks in a model of viral-induced fulminant hepatic failure. J. Cell Mol. Med. 2020, 24, 7625–7636. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, Z.; Sun, H.; Song, J.; Chen, X.; Huang, J.; Lin, X.; Zhou, R. Effect of melatonin on T/B cell activation and immune regulation in pinealectomy mice. Life Sci. 2020, 242, 117191. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, M.; Amirian, I.; Reiter, R.J.; Rosenberg, J.; Gögenur, I. Analgesic effects of melatonin: A review of current evidence from experimental and clinical studies. J. Pineal Res. 2011, 51, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Go, G.; Yoon, Y.M.; Yoon, S.; Lee, G.; Lim, J.H.; Han, S.-Y.; Lee, S.H. Melatonin Protects Chronic Kidney Disease Mesenchymal Stem/Stromal Cells against Accumulation of Methylglyoxal via Modulation of Hexokinase-2 Expression. Biomol. Ther. 2021, 30, 28–37. [Google Scholar] [CrossRef]
- Chen, D.Q.; Feng, Y.L.; Chen, L.; Liu, J.R.; Wang, M.; Vaziri, N.D.; Zhao, Y.-Y. Poricoic acid A enhances melatonin inhibition of AKI-to-CKD transition by regulating Gas6/AxlNFkappaB/Nrf2 axis. Free Radic. Biol. Med. 2019, 134, 484–497. [Google Scholar] [CrossRef]
- Marzougui, H.; Hammouda, O.; Ben Dhia, I.; Maaloul, R.; Agrebi, I.; Chaker, H.; Kammoun, K.; Ben Hmida, M.; Ayadi, F.; Kallel, C.; et al. Melatonin ingestion before intradialytic exercise improves immune responses in hemodialysis patients. Int. Urol. Nephrol. 2021, 53, 553–562. [Google Scholar] [CrossRef]
- Creeke, P.I.; Seal, A.J. Quantitation of the niacin metabolites 1-methylnicotinamide and l-methyl-2-pyridone-5-carboxamide in random spot urine samples, by ion-pairing reverse-phase HPLC with UV detection, and the implications for the use of spot urine samples in the assessment of niacin status. J. Chromatogr. B 2005, 817, 247–253. [Google Scholar]
- Hegyi, J.; Schwartz, R.A.; Hegyi, V. Pellagra: Dermatitis, dementia, and diarrhea. Int. J. Dermatol. 2004, 43, 1–5. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, 20B–26B. [Google Scholar] [CrossRef]
- Garg, A.; Sharma, A.; Krishnamoorthy, P.; Garg, J.; Virmani, D.; Sharma, T.; Stefanini, G.; Kostis, J.B.; Mukherjee, D.; Sikorskaya, E. Role of Niacin in Current Clinical Practice: A Systematic Review. Am. J. Med. 2017, 130, 173–187. [Google Scholar] [CrossRef]
- D’Andrea, E.; Hey, S.P.; Ramirez, C.L.; Kesselheim, A.S. Assessment of the Role of Niacin in Managing Cardiovascular Disease Outcomes: A Systematic Review and Meta-analysis. JAMA Netw. Open 2019, 2, e192224. [Google Scholar] [CrossRef]
- Slominska, E.; Yuen, A.; Osman, L.; Gebicki, J.; Yacoub, M.H.; Smolenski, R. Cytoprotective effects of nicotinamide derivatives in endothelial cells. Nucleosides Nucleotides Nucleic Acids 2008, 27, 863–866. [Google Scholar] [CrossRef]
- Kumakura, S.; Sato, E.; Sekimoto, A.; Hashizume, Y.; Yamakage, S.; Miyazaki, M.; Ito, S.; Harigae, H.; Takahashi, N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins 2021, 13, 50. [Google Scholar] [CrossRef]
- Bugarski, M.; Ghazi, S.; Polesel, M.; Martins, J.R.; Hall, A.M. Changes in NAD and Lipid Metabolism Drive Acidosis-Induced Acute Kidney Injury. J. Am. Soc. Nephrol. 2021, 32, 342–356. [Google Scholar] [CrossRef]
- Faivre, A.; Katsyuba, E.; Verissimo, T.; Lindenmeyer, M.; Rajaram, R.D.; Naesens, M.; Heckenmeyer, C.; Mottis, A.; Feraille, E.; Cippà, P. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2021, 36, 60–68. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tanaka, A.; Nakamura, T.; Fukuwatari, T.; Shibata, K.; Shimada, N.; Ebihara, I.; Koide, H. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004, 65, 1099–1104. [Google Scholar] [CrossRef]
- He, Y.-M.; Feng, L.; Huo, D.-M.; Yang, Z.-H.; Liao, Y.-H. Benefits and harm of niacin and its analog for renal dialysis patients: A systematic review and meta-analysis. Int. Urol. Nephrol. 2014, 46, 433–442. [Google Scholar] [CrossRef]
- Inamadugu, J.K.; Damaramadugu, R.; Mullangi, R.; Ponneri, V. Simultaneous determination of niacin and its metabolites--nicotinamide, nicotinuric acid and N-methyl-2-pyridone-5-carboxamide--in human plasma by LC-MS/MS and its application to a human pharmacokinetic study. Biomed. Chromatogr. 2010, 24, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Liabeuf, S.; El Esper, N.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.-S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The NICOREN study. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2017, 32, 1597. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.A.; Nguyen, J.C.D.; Polglaze, K.E.; Bertrand, P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The Expanded Biology of Serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vilchez, I.; Diaz-Ricart, M.; White, J.G.; Escolar, G.; Galan, A.M. Serotonin enhances platelet procoagulant properties and their activation induced during platelet tissue factor uptake. Cardiovasc. Res. 2009, 84, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Nagasawa, T.; Satoh, M.; Kuraishi, Y. Itch-associated response induced by intradermal serotonin through 5-HT2 receptors in mice. Neurosci. Res. 1999, 35, 77–83. [Google Scholar] [CrossRef]
- Ständer, S.; Böckenholt, B.; Schürmeyer-Horst, F.; Weishaupt, C.; Heuft, G.; Schneider, T. Treatment of chronic pruritus with the selective serotonin re-uptake inhibitors paroxetine and fluvoxamine: Results of an open-labelled, two-arm proof-of-concept study. Acta Derm. Venereol. 2009, 89, 45–51. [Google Scholar] [CrossRef]
- Kerr, P.G.; Argiles, A.; Mion, C. Whole blood serotonin levels are markedly elevated in patients on dialytic therapy. Am. J. Nephrol. 1992, 12, 14–18. [Google Scholar] [CrossRef]
- Pawlak, D.; Oksztulska-Kolanek, E.; Znorko, B.; Domaniewski, T.; Rogalska, J.; Roszczenko, A.; Brzóska, M.M.; Pryczynicz, A.; Kemona, A.; Pawlak, K. The Association between Elevated Levels of Peripheral Serotonin and Its Metabolite—5-Hydroxyindoleacetic Acid and Bone Strength and Metabolism in Growing Rats with Mild Experimental Chronic Kidney Disease. PLoS ONE 2016, 11, e0163526. [Google Scholar] [CrossRef]
- Mehrpooya, M.; Gholyaf, M.; Yasrebifar, F.; Mohammadi, Y.; Sheikh, V. Evaluation of Efficacy of Mirtazapine on Pruritus and Serum Histamine and Serotonin Levels in Patients Undergoing Hemodialysis: A Before—After Pilot Clinical Trial. Int. J. Nephrol. Renov. Dis. 2020, 13, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Balaskas, E.V.; Bamihas, G.I.; Karamouzis, M.; Voyiatzis, G.; Tourkantonis, A. Histamine and serotonin in uremic pruritus: Effect of ondansetron in CAPD-pruritic patients. Nephron 1998, 78, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Wu, P.-H.; Lee, H.-H.; Mubanga, M.; Chen, C.-S.; Kuo, M.-C.; Chiu, Y.-W.; Kuo, P.-L.; Hwang, S.-J. Indole-3 acetic acid increased risk of impaired cognitive function in patients receiving hemodialysis. NeuroToxicology 2019, 73, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.-H.; Lin, Y.-T.; Wu, P.Y.; Lee, H.-H.; Lee, S.-C.; Hung, S.-C.; Chen, S.-C.; Kuo, M.-C.; Chiu, Y.-W. Association between Circulation Indole-3-Acetic Acid Levels and Stem Cell Factor in Maintenance Hemodialysis Patients: A Cross-Sectional Study. J. Clin. Med. 2020, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yin, W.; Liang, Y.; Sun, L.; Yin, Y.; Zhang, W. Anti-Inflammatory and Anti-Oxidative Activity of Indole-3-Acetic Acid Involves Induction of HO-1 and Neutralization of Free Radicals in RAW264.7 Cells. Int. J. Mol. Sci. 2020, 21, 1579. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; DA Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Ji, Y.; Gao, Y.; Chen, H.; Yin, Y.; Zhang, W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients 2019, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. Suppl. 2003, S19–S21. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, S.B.; Bush, K.T.; Nigam, S.K. A Network of SLC and ABC Transporter and DME Genes Involved in Remote Sensing and Signaling in the Gut-Liver-Kidney Axis. Sci. Rep. 2019, 9, 11879. [Google Scholar] [CrossRef]
- Lowenstein, J.; Nigam, S.K. Uremic Toxins in Organ Crosstalk. Front. Med. 2021, 8, 592602. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Fan, Y.; Yu, Z.; You, G. Regulation of organic anion transporters: Role in physiology, pathophysiology, and drug elimination. Pharmacol. Ther. 2021, 217, 107647. [Google Scholar] [CrossRef]
- Zhu, C.; Nigam, K.B.; Date, R.C.; Bush, K.T.; Springer, S.A.; Saier, M.H.; Wu, W.; Nigam, S.K. Evolutionary Analysis and Classification of OATs, OCTs, OCTNs, and Other SLC22 Transporters: Structure-Function Implications and Analysis of Sequence Motifs. PLoS ONE 2015, 10, e0140569. [Google Scholar] [CrossRef]
- Engelhart, D.C.; Azad, P.; Ali, S.; Granados, J.C.; Haddad, G.G.; Nigam, S.K. Drosophila SLC22 Orthologs Related to OATs, OCTs, and OCTNs Regulate Development and Responsiveness to Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 2002. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The organic anion transporter (OAT) family: A systems biology perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Toraño, J.S.; Zaal, E.A.; Berkers, C.R.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 16105–16110. [Google Scholar] [CrossRef]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef]
- Granados, J.C.; Richelle, A.; Gutierrez, J.M.; Zhang, P.; Zhang, X.; Bhatnagar, V.; Lewis, N.E.; Nigam, S.K. Coordinate regulation of systemic and kidney tryptophan metabolism by the drug transporters OAT1 and OAT3. J. Biol. Chem. 2021, 296, 100575. [Google Scholar] [CrossRef]
- Bush, K.T.; Singh, P.; Nigam, S.K. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight 2020, 5, e133817. [Google Scholar] [CrossRef]
- Bhatnagar, V.; Richard, E.L.; Wu, W.; Nievergelt, C.M.; Lipkowitz, M.S.; Jeff, J.; Maihofer, A.X.; Nigam, S.K. Analysis of ABCG2 and other urate transporters in uric acid homeostasis in chronic kidney disease: Potential role of remote sensing and signaling. Clin. Kidney J. 2016, 9, 444–453. [Google Scholar] [CrossRef]
- Komori, H.; Yamada, K.; Tamai, I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2018, 1860, 973–980. [Google Scholar] [CrossRef]
- Lu, Y.; Nakanishi, T.; Hosomi, A.; Komori, H.; Tamai, I. In-vitro evidence of enhanced breast cancer resistance protein-mediated intestinal urate secretion by uremic toxins in Caco-2 cells. J. Pharm. Pharmacol. 2015, 67, 170–177. [Google Scholar] [CrossRef]
- Torres, A.M.; Dnyanmote, A.V.; Granados, J.C.; Nigam, S.K. Renal and non-renal response of ABC and SLC transporters in chronic kidney disease. Expert Opin. Drug Metab. Toxicol. 2021, 17, 515–542. [Google Scholar] [CrossRef]
- Ravid, J.D.; Kamel, M.H.; Chitalia, V.C. Uraemic solutes as therapeutic targets in CKD-associated cardiovascular disease. Nat. Rev. Nephrol. 2021, 17, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Tian, J.; Feng, Y.; Fu, H.; Xie, H.Q.; Jiang, J.X.; Zhao, B. The Aryl Hydrocarbon Receptor: A Key Bridging Molecule of External and Internal Chemical Signals. Environ. Sci. Technol. 2015, 49, 9518–9531. [Google Scholar] [CrossRef]
- Hahn, M.E. Aryl hydrocarbon receptors: Diversity and evolution. Chem. Interact. 2002, 141, 131–160. [Google Scholar] [CrossRef]
- Poland, A.P.; Glover, E.; Robinson, J.R.; Nebert, D.W. Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1-450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons. J. Biol. Chem. 1974, 249, 5599–5606. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. The aryl hydrocarbon receptor and the gut–brain axis. Cell. Mol. Immunol. 2021, 18, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A Molecular Mechanism To Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef] [PubMed]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; De Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Sullivan, A.P.; Sebastian, A.; Perry, G.H.; Jablonski, N.G.; Perdew, G.H. Divergent Ah Receptor Ligand Selectivity during Hominin Evolution. Mol. Biol. Evol. 2016, 33, 2648–2658. [Google Scholar] [CrossRef]
- Aarts, J.M.M.J.G.; Alink, G.M.; Franssen, H.J.; Roebroeks, W. Evolution of Hominin Detoxification: Neanderthal and Modern Human Ah Receptor Respond Similarly to TCDD. Mol. Biol. Evol. 2021, 38, 1292–1305. [Google Scholar] [CrossRef]
- Furue, M.; Tsuji, G. Chloracne and Hyperpigmentation Caused by Exposure to Hazardous Aryl Hydrocarbon Receptor Ligands. Int. J. Environ. Res. Public Health 2019, 16, 4864. [Google Scholar] [CrossRef]
- Available online: https://www.publichealth.va.gov/exposures/agentorange/index.asp (accessed on 13 February 2022).
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-S.; Cao, F.; Zhang, Y.; Pan, H.-F. Therapeutic potential of aryl hydrocarbon receptor in autoimmunity. Inflammopharmacology 2020, 28, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Bosch, I.; Polonio, C.M.; Torti, M.F.; Wheeler, M.A.; Li, Z.; Romorini, L.; Varela, M.S.R.; Rothhammer, V.; Barroso, A.; et al. AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat. Neurosci. 2020, 23, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. From TCDD-mediated toxicity to searches of physiologic AHR functions. Biochem. Pharmacol. 2018, 155, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.H.; Bradfield, C.A. Rodent genetic models of Ah receptor signaling. Drug Metab. Rev. 2021, 53, 350–374. [Google Scholar] [CrossRef]
- Sonowal, R.; Swimm, A.; Sahoo, A.; Luo, L.; Matsunaga, Y.; Wu, Z.; Bhingarde, J.A.; Ejzak, E.A.; Ranawade, A.; Qadota, H.; et al. Indoles from commensal bacteria extend healthspan. Proc. Natl. Acad. Sci. USA 2017, 114, E7506–E7515. [Google Scholar] [CrossRef] [PubMed]
- Serna, E.; Cespedes, C.; Vina, J. Anti-Aging Physiological Roles of Aryl Hydrocarbon Receptor and Its Dietary Regulators. Int. J. Mol. Sci. 2020, 22, 374. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.-L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749.e4. [Google Scholar] [CrossRef]
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl hydrocarbon receptor ligand production by the gut microbiota is decreased in celiac disease leading to intestinal inflammation. Sci. Transl. Med. 2020, 12, eaba0624. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, L.; Ciocan, D.; Hugot, C.; Spatz, M.; Dupeux, M.; Houron, C.; Moal, V.L.-L.; Puchois, V.; Ferrere, G.; Trainel, N.; et al. Microbiota tryptophan metabolism induces aryl hydrocarbon receptor activation and improves alcohol-induced liver injury. Gut 2020, 70, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Perdew, G.H. How Ah Receptor Ligand Specificity Became Important in Understanding Its Physiological Function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. Human AHR functions in vascular tissue: Pro- and anti-inflammatory responses of AHR agonists in atherosclerosis. Biochem. Pharmacol. 2019, 159, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://arsindustrialis.org/pharmakon (accessed on 13 February 2022).
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Bosch, T.C.G.; Ledón-Rettig, C. Eco-Evo-Devo: Developmental symbiosis and developmental plasticity as evolutionary agents. Nat. Rev. Genet. 2015, 16, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Alnouti, Y.; Klaassen, C.D. Tissue distribution and ontogeny of sulfotransferase enzymes in mice. Toxicol. Sci. 2006, 93, 242–255. [Google Scholar] [CrossRef]
- Coughtrie, M.W.H. Sulfation through the looking glass—Recent advances in sulfotransferase research for the curious. Pharm. J. 2002, 2, 297–308. [Google Scholar] [CrossRef]
- Yamazoe, Y.; Nagata, K.; Ozawa, S.; Kato, R. Structural similarity and diversity of sulfotransferases. Chem. Interact. 1994, 92, 107–117. [Google Scholar] [CrossRef]
- Davenport, E.R.; Sanders, J.G.; Song, S.J.; Amato, K.R.; Clark, A.G.; Knight, R. The human microbiome in evolution. BMC Biol. 2017, 15, 127. [Google Scholar] [CrossRef]
- Weyrich, L.S.; Duchene, S.; Soubrier, J.; Arriola, L.; Llamas, B.; Breen, J.; Morris, A.G.; Alt, K.W.; Caramelli, D.; Dresely, V.; et al. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 2017, 544, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxidative Med. Cell. Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; de Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. JASN 2016, 27, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Huys, G.R.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Wood, B.M.; Raichlen, D.A. Hunter-gatherers as models in public health. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2018, 19 (Suppl. 1), 24–35. [Google Scholar] [CrossRef]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L. Hypertension. Nat. Rev. Dis. Primers. 2018, 4, 18014. [Google Scholar] [CrossRef]
- Weller, O.D.G. Available online: http://www.antiquity.ac.uk/projgall/weller306/ (accessed on 13 February 2022).
- Eknoyan, G. A history of obesity, or how what was good became ugly and then bad. Adv. Chronic Kidney Dis. 2006, 13, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mc Guigan, K.; Nishimura, N.; Currey, M.; Hurwit, D.; Cresko, W.A. Cryptic genetic variation and body size evolution in threespine stickleback. Evolution 2010, 65, 1203–1211. [Google Scholar] [CrossRef]
- Scoville, A.G.; Pfrender, M.E. Phenotypic plasticity facilitates recurrent rapid adaptation to introduced predators. Proc. Natl. Acad. Sci. USA 2010, 107, 4260–4263. [Google Scholar] [CrossRef] [PubMed]
- Aubret, F.; Shine, R. Genetic assimilation and the postcolonization erosion of phenotypic plasticity in island tiger snakes. Curr. Biol. 2009, 19, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Dobber, R.; Hertogh-Huijbregts, A.; Rozing, J.; Bottomly, K.; Nagelkerken, L. The involvement of the intestinal microflora in the expansion of CD4+ T cells with a-naive phenotype in the periphery. Dev. Immunol. 1992, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: Microbial endocrinology in the design and use of probiotics. BioEssays 2011, 33, 574–581. [Google Scholar] [CrossRef]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut microbes and the brain: Paradigm shift in neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef]
- Zhao, Y. Auxin biosynthesis: A simple two-step pathway converts tryptophan to indole-3-acetic acid in plants. Mol. Plant 2012, 5, 334–338. [Google Scholar] [CrossRef]
- Fu, S.-F.; Wei, J.-Y.; Chen, H.-W.; Liu, Y.-Y.; Lu, H.-Y.; Chou, J.-Y. Indole-3-acetic acid: A widespread physiological code in interactions of fungi with other organisms. Plant Signal. Behav. 2015, 10, e1048052. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodriguez, E.; Pizarro-Sanchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Fujimiya, M.; Sakamaki, R.; Shinfuku, N.; Ueta, Y.; Meguid, M.M.; Kasuga, M. Stomach regulates energy balance via acylated ghrelin and desacyl ghrelin. Gut 2005, 54, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Pazos, I.M.; Gai, F. Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO). Proc. Natl. Acad. Sci. USA 2014, 111, 8476–8481. [Google Scholar] [CrossRef]
- Vanholder, R.; Gryp, T.; Glorieux, G. Urea and chronic kidney disease: The comeback of the century? (in uraemia research). Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2018, 33, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Lee, H.A.; Sadler, P.J. Uraemia: Is urea more important than we think? Lancet 1991, 338, 1438–1440. [Google Scholar] [CrossRef]
- Spiga, R.; Marini, M.A.; Mancuso, E.; di Fatta, C.; Fuoco, A.; Perticone, F.; Andreozzi, F.; Mannino, G.C.; Sesti, G. Uric Acid Is Associated With Inflammatory Biomarkers and Induces Inflammation via Activating the NF-kappaB Signaling Pathway in HepG2 Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1241–1249. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef]
- London, J.A.; Wang, E.C.S.; Barsukov, I.L.; Yates, E.A.; Stachulski, A.V. Synthesis and toxicity profile in 293 human embryonic kidney cells of the beta D-glucuronide derivatives of ortho-, meta- and para-cresol. Carbohydr. Res. 2021, 499, 108225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Blaze, J.; Haghighi, F.; Kim-Schulze, S.; Raval, U.; Trageser, K.J.; Pasinetti, G.M. Characterization of 3(3,4-dihydroxy-phenyl) propionic acid as a novel microbiome-derived epigenetic modifier in attenuation of immune inflammatory response in human monocytes. Mol. Immunol. 2020, 125, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Siribamrungwong, M.; Jaber, B.L. Convective therapies versus low-flux hemodialysis for chronic kidney failure: A meta-analysis of randomized controlled trials. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2013, 28, 2859–2874. [Google Scholar] [CrossRef]
- Nistor, I.; Palmer, S.C.; Craig, J.; Saglimbene, V.; Vecchio, M.; Covic, A.; Strippoli, G.F. Convective versus diffusive dialysis therapies for chronic kidney failure: An updated systematic review of randomized controlled trials. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2014, 63, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Salminen, S.; Collado, M.C.; Endo, A.; Hill, C.; Lebeer, S.; Quigley, E.M.M.; Sanders, M.E.; Shamir, R.; Swann, J.R.; Szajewska, H.; et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of postbiotics. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 649–667. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, G.; Vanholder, R.; Van Biesen, W.; Pletinck, A.; Schepers, E.; Neirynck, N.; Speeckaert, M.; de Bacquer, D.; Verbeke, F. Free p-cresyl sulfate shows the highest association with cardiovascular outcome in chronic kidney disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2021, 36, 998–1005. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Structure Formula | Enzymes Involved |
|---|---|---|
| Tryptophan |  | |
| Indole pathway | ||
| Indole |  | Tryptophanase |
| Indoxyl sulfate |  | Sulfotransferase/CYP2E1 |
| Indoxyl glucuronide |  | Glucuronyltransferase |
| Indole-3-propionic acid |  | Tryptophanase (?) |
| Indole-3-acetic acid |  | Aldehyde dehydrogenase (NAD+)/indole-3-acetaldehyde oxidase/IL4I1 |
| Indole-3-(carbox)aldehyde |  | Aromatic-L-amino-acid/L-tryptophan decarboxylase |
| Kynurenine pathway | ||
| L-Kynurenine |  | Tryptophan 2,3-dioxygenase or indoleamine 2,3-dioxygenase/arylformamidase |
| Kynurenic acid |  | Kynurenine-oxoglutarate transaminase/cysteine-S-conjugate beta-lyase/glutamine-phenylpyruvate transaminase |
| Anthranilic acid |  | Kynureninase |
| Quinolinic acid |  | Anthranilate 3-monooxygenase (FAD)/4-hydroxyphenylacetate 3-monooxygenase/3-hydroxyanthranilate 3,4-dioxygenase |
| Nicotinic acid (niacin) (Nicotinate and Nicotinamide metabolism) |  | Nicotinate-nucleotide pyrophosphorylase (carboxylating)/nicotinate phosphoribosyltransferase |
| Nicotinamide (Nicotinate and Nicotinamide metabolism) |  | Nicotinate-nucleotide adenylyltransferase/nicotinamide adenine dinucleotide (NAD)+ synthase/NAD+ diphosphatase/5′-nucleotidase |
| N1-Methyl-2-pyridone-5-carboxamide (Nicotinate and Nicotinamide metabolism) |  | Nicotinamide methyl transferase/aldehyde oxidase |
| Serotonin Pathway | ||
| Serotonin |  | Tryptophan 5-monooxygenase/aromatic-L-amino-acid/L-tryptophan decarboxylase |
| Melatonin |  | Arylalkylamine N-acetyltransferase/acetylserotonin O-methyltransferase |
| Positive | Negative |
|---|---|
| Detoxification | Pro-inflammatory effect |
| Preservation vascular structure | Chloracne |
| Closure ductus venosus (post-partum) | Lymphoma |
| Normalization blood pressure | Malformative syndrome |
| Preservation liver function | Carcinogenicity |
| Prevention fibrosis | Prothrombotic effect |
| Prolongation healthy life | Vascular toxicity |
| Forestalls ageing | Neurotoxicity |
| Preservation digestive epithelial function | Auto-immune diseases |
| Prevention inflammatory bowel disease | Zika infection |
| Prevention metabolic syndrome | |
| Prevention gluten enteropathy | |
| Prevention alcohol toxicity |
| Toxic | Neutral or Non-Toxic |
|---|---|
| Complement factor D | Complement factor Ba |
| Interleukin-1β | Interleukin-1 receptor antagonist |
| Tumor necrosis factor-α | Soluble tumor necrosis factor receptor |
| Interleukin-6 | Interleukin-10 |
| Cholecystokinin | Ghrelin |
| Desacyl Ghrelin | Ghrelin |
| Leptin | Orexin A |
| Peptide YY | Neuropeptide Y |
| Tryptophan metabolites |
|---|
| Provide a complete mapping of the evolution of compounds with positive and negative biological impacts throughout all CKD stages. |
| Extend biological research to a broader array of compounds than the usual suspects |
| Assess the effect of therapeutic strategies on molecules with as well positive as negative biological effects. |
| Consider choosing therapies that maintain or restore the balance between components with positive and negative impact, rather than removing toxins as well as beneficial compounds. |
| Promote the publication of suitable studies showing atypical results of uremic toxin actions. |
| Researchers should not shelve uremic toxin research with atypical results. |
| Other than tryptophan metabolites |
| Develop extensive reviews and studies on a broader array of metabolites than the ones frequently considered now. |
| Based on this knowledge, extend the analysis and development of therapeutic options. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanholder, R.; Nigam, S.K.; Burtey, S.; Glorieux, G. What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis. Toxins 2022, 14, 221. https://doi.org/10.3390/toxins14030221
Vanholder R, Nigam SK, Burtey S, Glorieux G. What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis. Toxins. 2022; 14(3):221. https://doi.org/10.3390/toxins14030221
Chicago/Turabian StyleVanholder, Raymond, Sanjay K. Nigam, Stéphane Burtey, and Griet Glorieux. 2022. "What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis" Toxins 14, no. 3: 221. https://doi.org/10.3390/toxins14030221
APA StyleVanholder, R., Nigam, S. K., Burtey, S., & Glorieux, G. (2022). What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis. Toxins, 14(3), 221. https://doi.org/10.3390/toxins14030221