Antibodies against Anthrax Toxins: A Long Way from Benchlab to the Bedside

 , ,
, ,
Abstract
:1. Introduction
2. Results
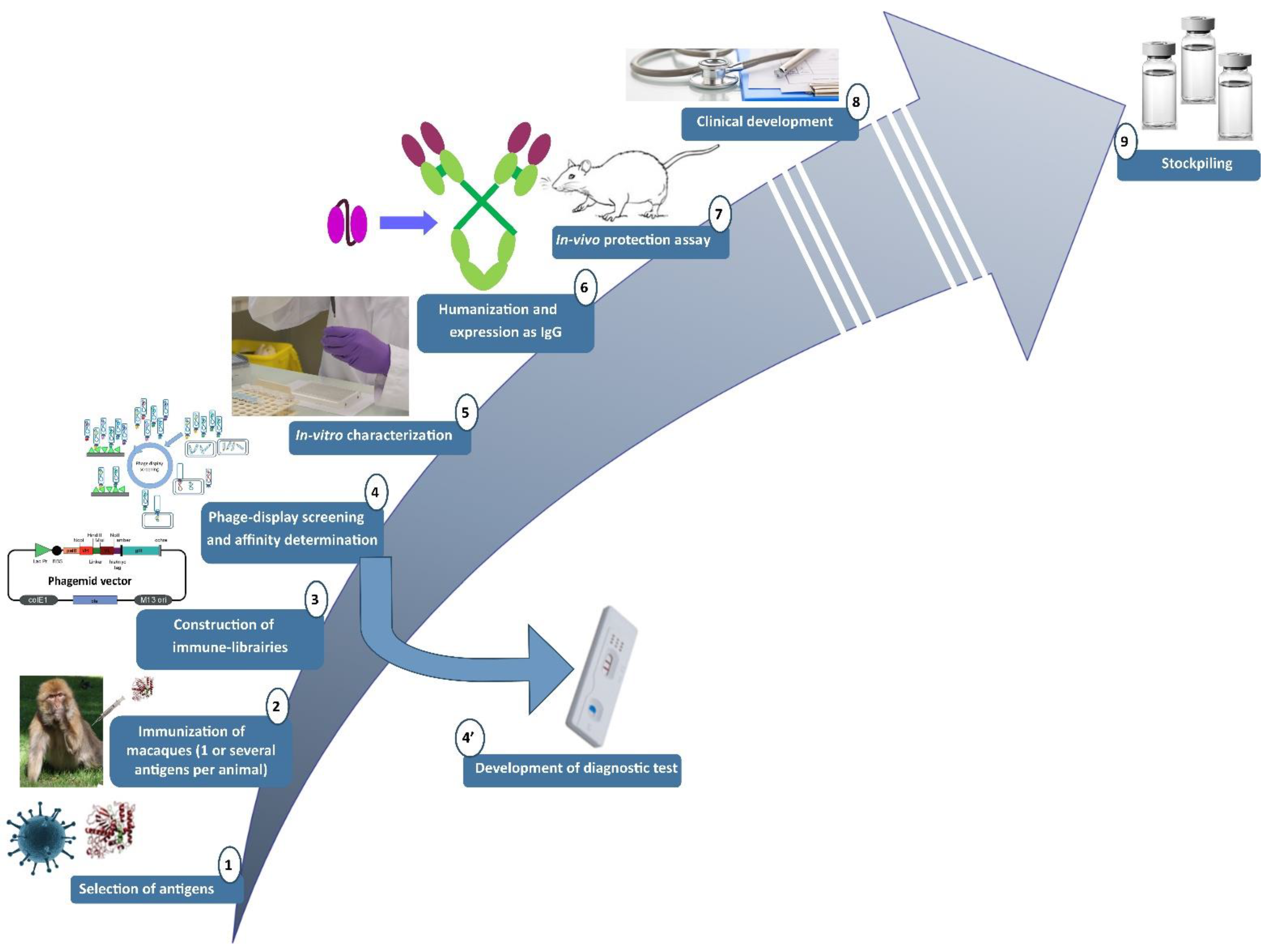
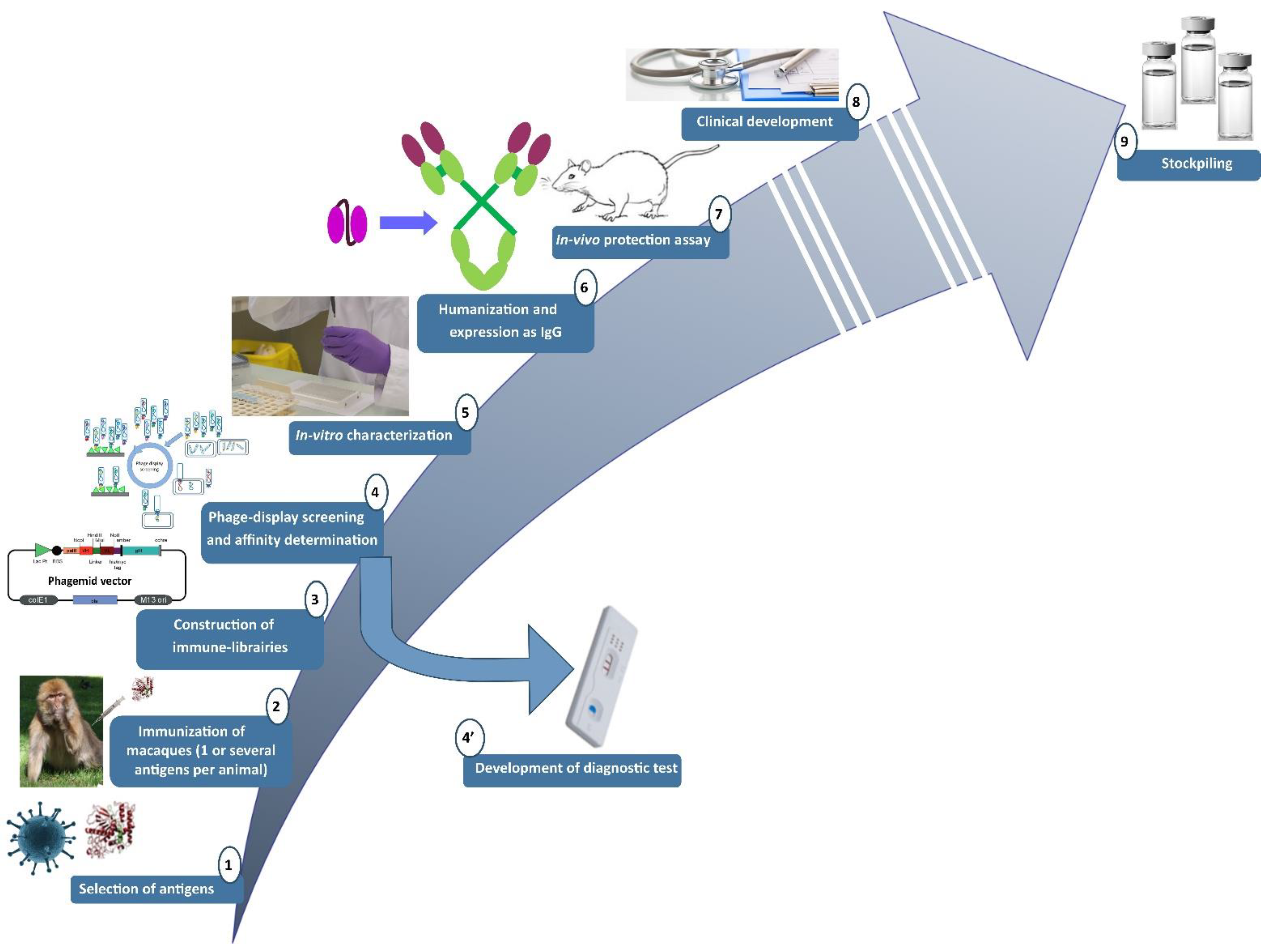
2.1. Pipeline Used for the Research and Development of Human-like Recombinant Antibodies
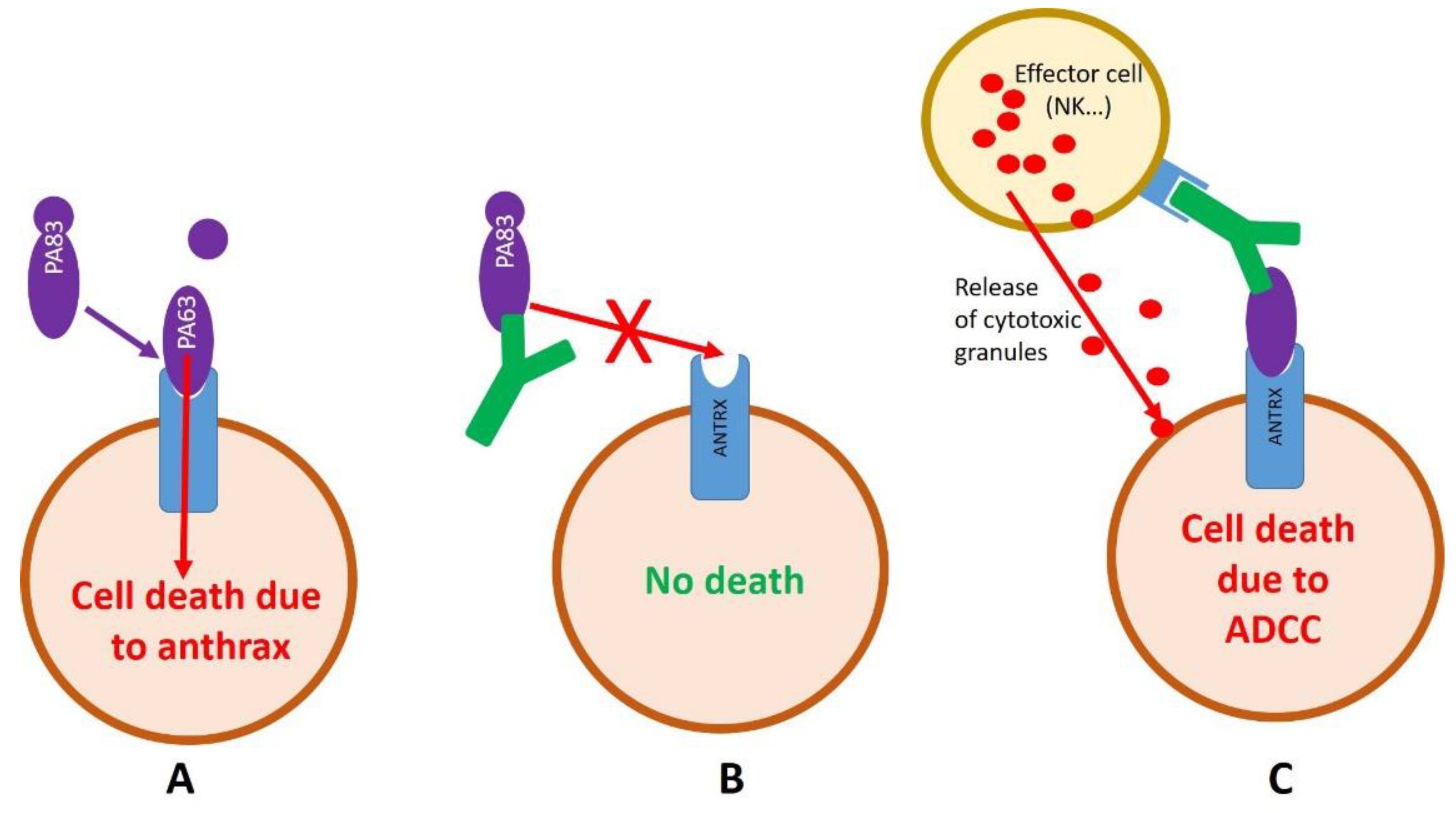
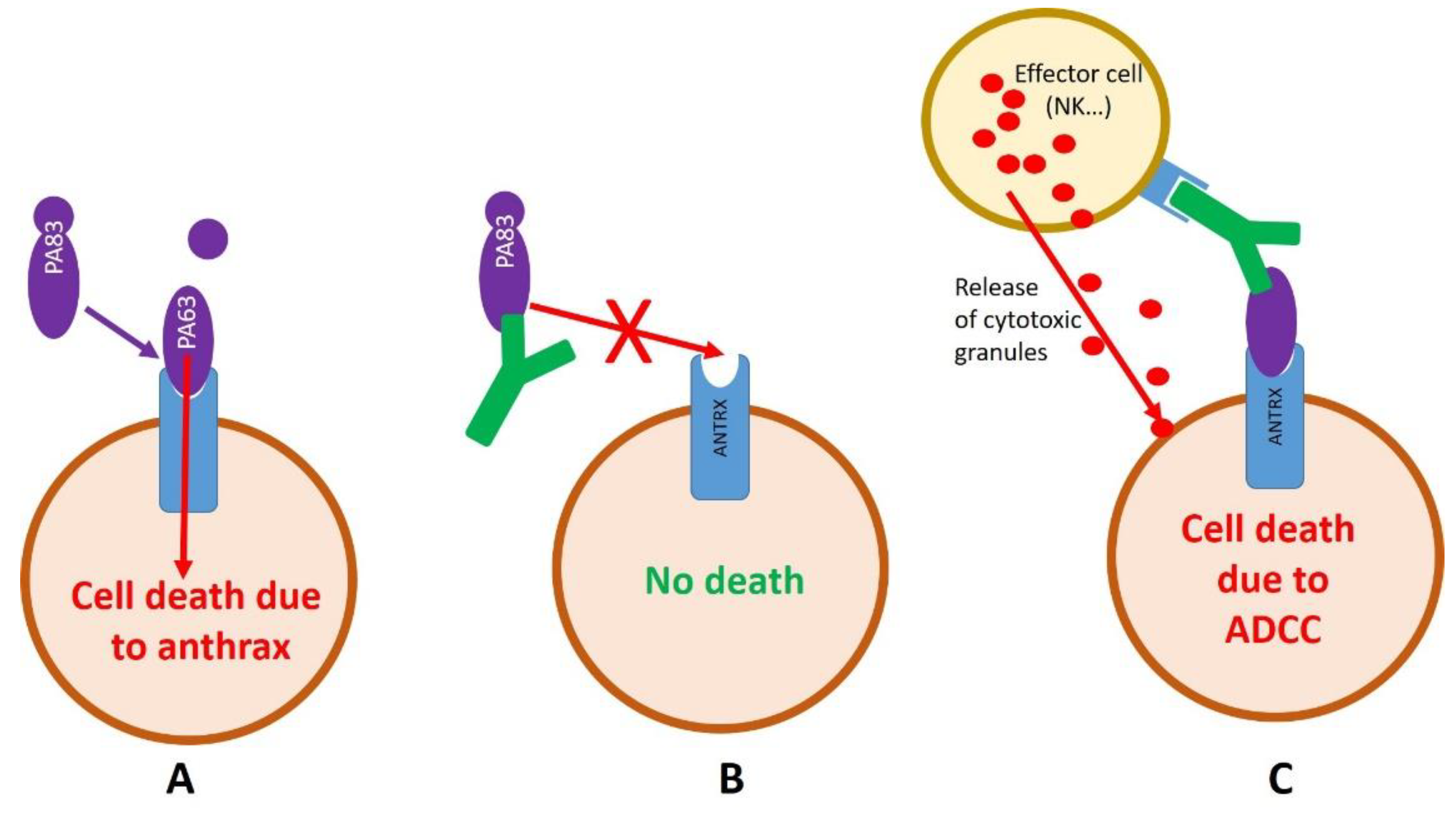
2.2. Development of Anti-Anthrax Antibodies
2.2.1. Development of an Anti-PA Antibody
2.2.2. Development of an Anti-LF Antibody
2.3. Filling the Gap between the Bench and the (Pre-)Clinical Development
2.3.1. Intellectual Property Issue and Affinity Maturation
2.3.2. Humanization of 35PA80 6.20
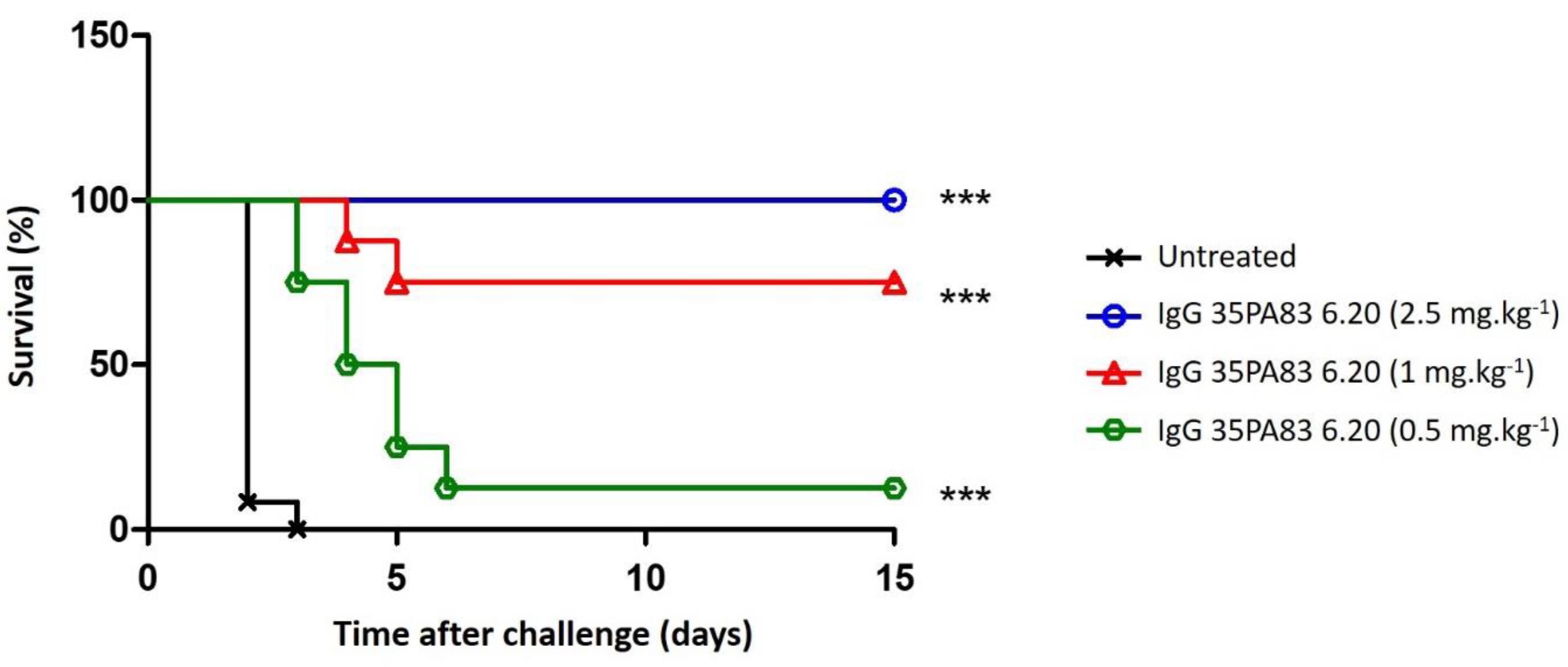
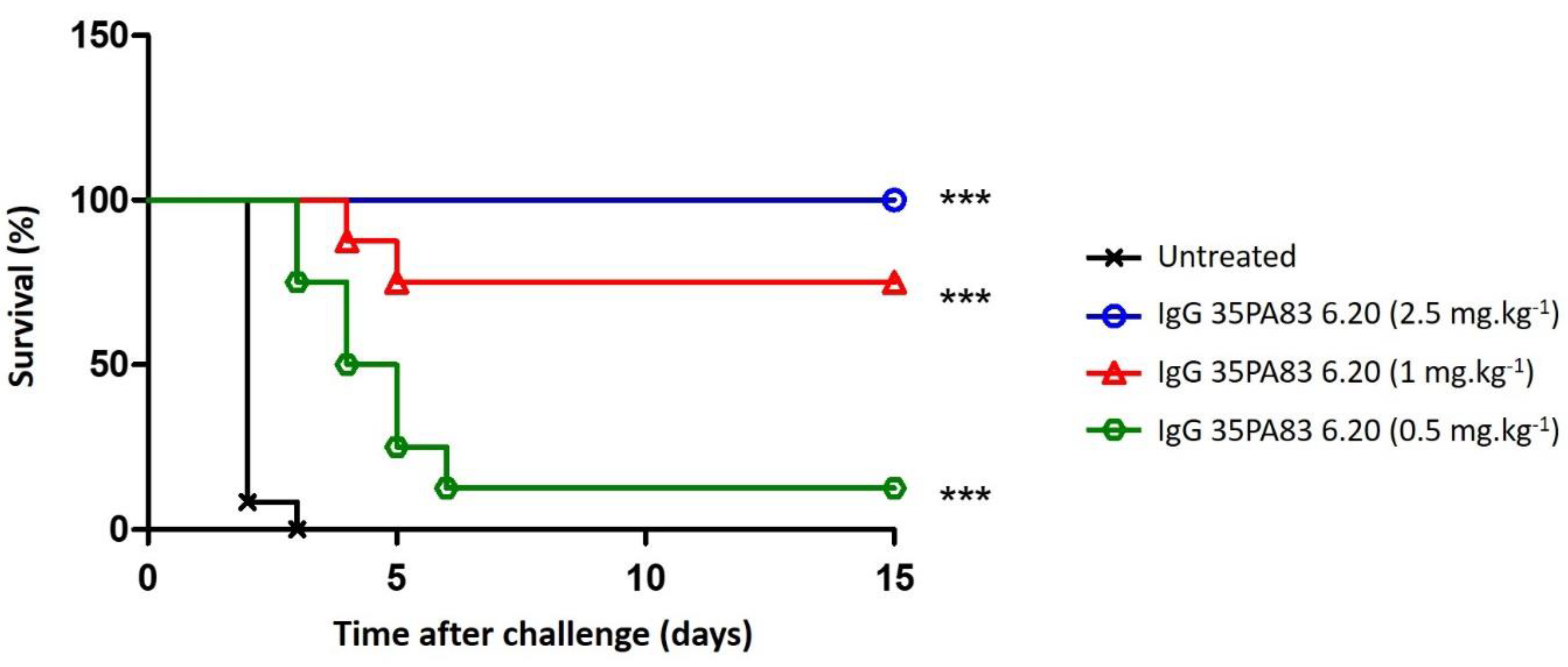
2.3.3. In-Vivo Protection Conferred by 35PA83
Pharmacokinetic Studies
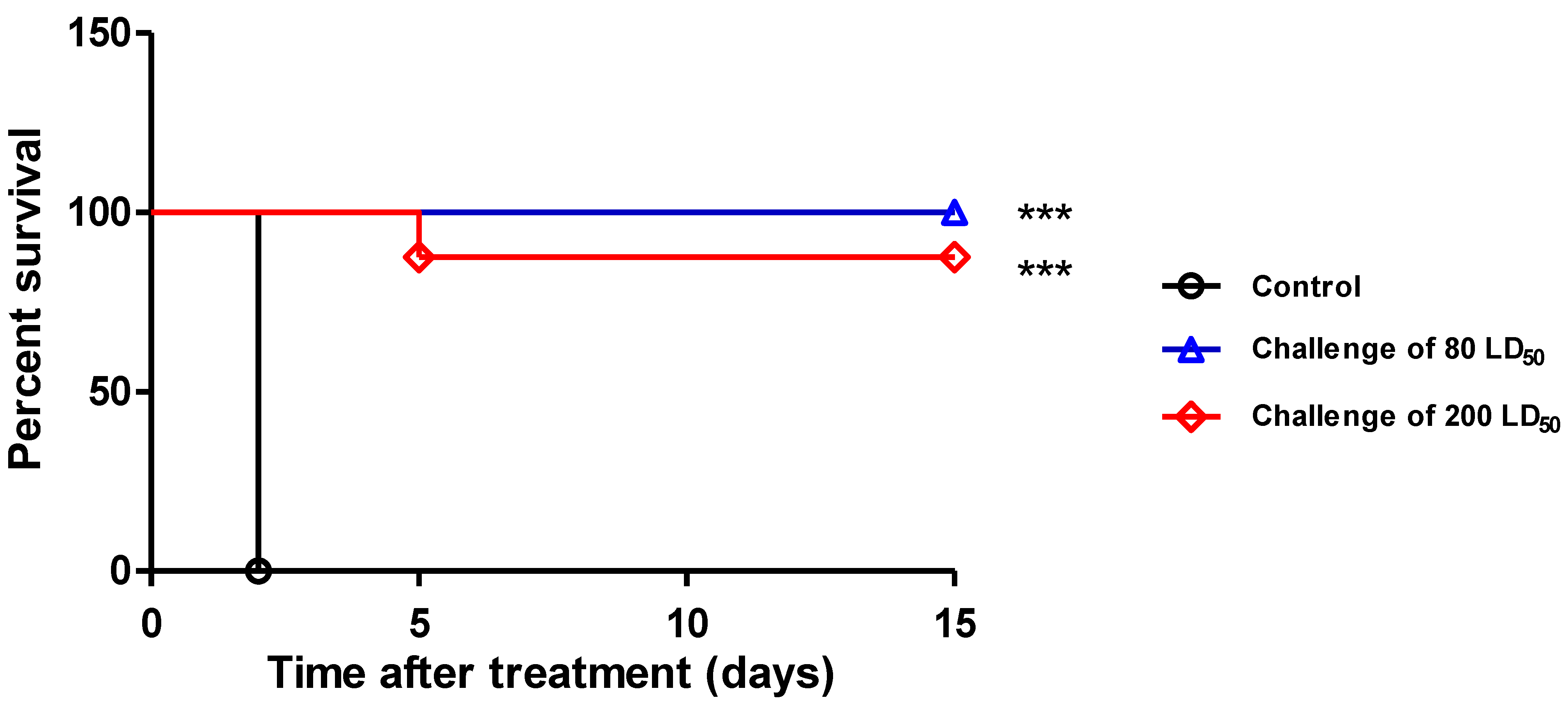
Prophylaxis and Delayed Treatment with IgG 35PA83 Administration Alone in Rabbit Model
2.4. Clinical Development of 35PA83 6.20
2.4.1. Formulation and Stability
2.4.2. Scientific and Industrial Issue
2.4.3. Commercial Issues
3. Discussion
4. Materials and Methods
4.1. Affinity Determination
4.2. Humanization
4.3. Preparation of Bacillus Anthracis Spores
4.4. ELISA
4.5. Pharmacokinetic Studies
4.6. Passive Immunization and Delayed Treatment with IgG 35PA83 Alone in White New Zealand Rabbits Infected by 9602 Spores
4.7. Statistical Analysis of In Vivo Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friebe, S.; van der Goot, F.G.; Bürgi, J. The Ins and Outs of Anthrax Toxin. Toxins 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Goossens, P.L.; Tournier, J.-N. Crossing of the epithelial barriers by Bacillus anthracis: The Known and the Unknown. Front. Microbiol. 2015, 6, 1122. [Google Scholar] [CrossRef] [Green Version]
- Wilkening, D.A. Sverdlovsk revisited: Modeling human inhalation anthrax. Proc. Natl. Acad. Sci. USA 2006, 103, 7589–7594. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, D.B.; Raghunathan, P.L.; Bell, B.P.; Brechner, R.; Bresnitz, E.A.; Butler, J.C.; Cetron, M.; Cohen, M.; Doyle, T.; Fischer, M.; et al. Investigation of bioterrorism-related anthrax, United States, 2001: Epidemiologic findings. Emerg. Infect. Dis. 2002, 8, 1019–1028. [Google Scholar] [CrossRef]
- Thouret, J.-M.; Rogeaux, O.; Beaudouin, E.; Levast, M.; Ramisse, V.; Biot, F.V.; Valade, E.; Thibault, F.; Gorgé, O.; Tournier, J.-N. Case Report of an Injectional Anthrax in France, 2012. Microorganisms 2020, 8, 985. [Google Scholar] [CrossRef]
- Zacchia, N.A.; Schmitt, K. Medical Spending for the 2001 Anthrax Letter Attacks. Disaster Med. Public Health Prep. 2019, 13, 539–546. [Google Scholar] [CrossRef]
- Froude, J.W.; Thullier, P.; Pelat, T. Antibodies against anthrax: Mechanisms of action and clinical applications. Toxins 2011, 3, 1433–1452. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Nolen, L.D.; Sun, J.; Booth, M.; Donaldson, L.; Quinn, C.P.; Boyer, A.E.; Hendricks, K.; Shadomy, S.; Bothma, P.; et al. Analysis of Anthrax Immune Globulin Intravenous with Antimicrobial Treatment in Injection Drug Users, Scotland, 2009–2010. Emerg. Infect. Dis. 2017, 23, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Glinert, I.; Bar-David, E.; Sittner, A.; Weiss, S.; Schlomovitz, J.; Ben-Shmuel, A.; Mechaly, A.; Altboum, Z.; Kobiler, D.; Levy, H. Revisiting the Concept of Targeting Only Bacillus anthracis Toxins as a Treatment for Anthrax. Antimicrob. Agents Chemother. 2016, 60, 4878–4885. [Google Scholar] [CrossRef] [Green Version]
- Vietri, N.J. Does anthrax antitoxin therapy have a role in the treatment of inhalational anthrax? Curr. Opin. Infect. Dis. 2018, 31, 257–262. [Google Scholar] [CrossRef]
- Xu, W.; Ohanjanian, L.; Sun, J.; Cui, X.; Suffredini, D.; Li, Y.; Welsh, J.; Eichacker, P.Q. A systematic review and meta-analysis of preclinical trials testing anti-toxin therapies for B. anthracis infection: A need for more robust study designs and results. PLoS ONE 2017, 12, e0182879. [Google Scholar] [CrossRef] [Green Version]
- Pelat, T.; Hust, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.-P.; Dübel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (LF) of Bacillus anthracis by inhibiting protective antigen-LF complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [CrossRef] [Green Version]
- Avril, P.A.; Miethe, S.; Hust, M. Thibaut construction of macaque immune-libraries. In Phage Display; Hust, M., Lim, T.S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1701, pp. 83–112. ISBN 978-1-4939-7446-7. [Google Scholar]
- Avril, A.; Froude, J.W.; Mathieu, J.; Pelat, T.; Thullier, P. Isolation of antibodies from non-human primates for clinical use. Curr. Drug Discov. Technol. 2014, 11, 20–27. [Google Scholar] [CrossRef]
- Avril, A.; Miethe, S.; Popoff, M.R.; Mazuet, C.; Chahboun, S.; Rasetti-Escargueil, C.; Sesardic, D.; Thullier, P.; Hust, M.; Pelat, T. Isolation of nanomolar scFvs of non-human primate origin, cross-neutralizing botulinum neurotoxins A1 and A2 by targeting their heavy chain. BMC Biotechnol. 2015, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Rülker, T.; Voß, L.; Thullier, P.; O’ Brien, L.M.; Pelat, T.; Perkins, S.D.; Langermann, C.; Schirrmann, T.; Dübel, S.; Marschall, H.-J.; et al. Isolation and characterisation of a human-like antibody fragment (scFv) that inactivates VEEV in vitro and in vivo. PLoS ONE 2012, 7, e37242. [Google Scholar] [CrossRef] [Green Version]
- Thullier, P.; Chahboun, S.; Pelat, T. A comparison of human and macaque (Macaca mulatta) immunoglobulin germline V regions and its implications for antibody engineering. MAbs 2010, 2, 528–538. [Google Scholar]
- Rasetti-Escargueil, C.; Avril, A.; Miethe, S.; Mazuet, C.; Derman, Y.; Selby, K.; Thullier, P.; Pelat, T.; Urbain, R.; Fontayne, A.; et al. The European AntibotABE Framework Program and Its Update: Development of Innovative Botulinum Antibodies. Toxins 2017, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Pelat, T.; Hust, M.; Hale, M.; Lefranc, M.-P.; Dübel, S.; Thullier, P. Isolation of a human-like antibody fragment (scFv) that neutralizes ricin biological activity. BMC Biotechnol. 2009, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Respaud, R.; Marchand, D.; Pelat, T.; Tchou-Wong, K.-M.; Roy, C.J.; Parent, C.; Cabrera, M.; Guillemain, J.; Mac Loughlin, R.; Levacher, E.; et al. Development of a drug delivery system for efficient alveolar delivery of a neutralizing monoclonal antibody to treat pulmonary intoxication to ricin. J. Control Release 2016, 234, 21–32. [Google Scholar] [CrossRef]
- Orsini Delgado, M.L.; Avril, A.; Prigent, J.; Dano, J.; Rouaix, A.; Worbs, S.; Dorner, B.G.; Rougeaux, C.; Becher, F.; Fenaille, F.; et al. Ricin Antibodies’ Neutralizing Capacity against Different Ricin Isoforms and Cultivars. Toxins 2021, 13, 100. [Google Scholar] [CrossRef]
- Froude, J.W.; Pelat, T.; Miethe, S.; Zak, S.E.; Wec, A.Z.; Chandran, K.; Brannan, J.M.; Bakken, R.R.; Hust, M.; Thullier, P.; et al. Generation and characterization of protective antibodies to Marburg virus. MAbs 2017, 9, 696–703. [Google Scholar] [CrossRef]
- Froude, J.W.; Herbert, A.S.; Pelat, T.; Miethe, S.; Zak, S.E.; Brannan, J.M.; Bakken, R.R.; Steiner, A.R.; Yin, G.; Hallam, T.J.; et al. Post-Exposure Protection in Mice against Sudan Virus by a Two Antibody Cocktail. Viruses 2018, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Laffly, E.; Danjou, L.; Condemine, F.; Vidal, D.; Drouet, E.; Lefranc, M.-P.; Bottex, C.; Thullier, P. Selection of a macaque Fab with framework regions like those in humans, high affinity, and ability to neutralize the protective antigen (PA) of Bacillus anthracis by binding to the segment of PA between residues 686 and 694. Antimicrob. Agents Chemother. 2005, 49, 3414–3420. [Google Scholar] [CrossRef] [Green Version]
- Thullier, P.; Avril, A.; Mathieu, J.; Behrens, C.K.; Pellequer, J.-L.; Pelat, T. Mapping the epitopes of a neutralizing antibody fragment directed against the lethal factor of Bacillus anthracis and cross-reacting with the homologous edema factor. PLoS ONE 2013, 8, e65855. [Google Scholar] [CrossRef] [Green Version]
- Pelat, T.; Bedouelle, H.; Rees, A.R.; Crennell, S.J.; Lefranc, M.-P.; Thullier, P. Germline humanization of a non-human primate antibody that neutralizes the anthrax toxin, by in vitro and in silico engineering. J. Mol. Biol. 2008, 384, 1400–1407. [Google Scholar] [CrossRef]
- Sylvestre, J.; Blesa, S.; Marchal, I.; Thullier, P.; Dubreuil, O.; Delcourt, M. Massive Mutagenesis®: The path to smarter genetic libraries. In Modern Biopharmaceuticals; Knäblein, D.J., Ed.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2013; pp. 115–134. ISBN 9783527322831. [Google Scholar]
- Laffly, E.; Pelat, T.; Cédrone, F.; Blésa, S.; Bedouelle, H.; Thullier, P. Improvement of an antibody neutralizing the anthrax toxin by simultaneous mutagenesis of its six hypervariable loops. J. Mol. Biol. 2008, 378, 1094–1103. [Google Scholar] [CrossRef]
- Timmerman, P.; Puijk, W.C.; Meloen, R.H. Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J. Mol. Recognit. 2007, 20, 283–299. [Google Scholar] [CrossRef]
- Wild, M.A.; Xin, H.; Maruyama, T.; Nolan, M.J.; Calveley, P.M.; Malone, J.D.; Wallace, M.R.; Bowdish, K.S. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat. Biotechnol. 2003, 21, 1305–1306. [Google Scholar] [CrossRef]
- Sawada-Hirai, R.; Jiang, I.; Wang, F.; Sun, S.M.; Nedellec, R.; Ruther, P.; Alvarez, A.; Millis, D.; Morrow, P.R.; Kang, A.S. Human anti-anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J. Immune Based Ther. Vaccines 2004, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Vitale, L.; Blanset, D.; Lowy, I.; O’Neill, T.; Goldstein, J.; Little, S.F.; Andrews, G.P.; Dorough, G.; Taylor, R.K.; Keler, T. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 2006, 74, 5840–5847. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Moayeri, M.; Zhou, Y.-H.; Leppla, S.; Emerson, S.; Sebrell, A.; Yu, F.; Svitel, J.; Schuck, P.; St Claire, M.; et al. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J. Infect. Dis. 2006, 193, 625–633. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Morris, S. Approval of Raxibacumab for the Treatment of Inhalation Anthrax Under the US Food and Drug Administration “Animal Rule”. Front. Microbiol. 2015, 6, 1320. [Google Scholar] [CrossRef] [Green Version]
- Agency, E.M. Assessment Report Obiltoxaximab SFL, 17/09/2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/obiltoxaximab-sfl-epar-public-assessment-report_en.pdf (accessed on 22 February 2022).
- Migone, T.-S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhu, J.; Lu, H. Antibody glycosylation: Impact on antibody drug characteristics and quality control. Appl. Microbiol. Biotechnol. 2020, 104, 1905–1914. [Google Scholar] [CrossRef]
- Berthier, M.; Fauchere, J.L.; Perrin, J.; Grignon, B.; Oriot, D. Fulminant meningitis due to Bacillus anthracis in 11-year-old girl during Ramadan. Lancet 1996, 347, 828. [Google Scholar] [CrossRef]
- Chakrabarty, K.; Wu, W.; Booth, J.L.; Duggan, E.S.; Coggeshall, K.M.; Metcalf, J.P. Bacillus anthracis spores stimulate cytokine and chemokine innate immune responses in human alveolar macrophages through multiple mitogen-activated protein kinase pathways. Infect. Immun. 2006, 74, 4430–4438. [Google Scholar] [CrossRef] [Green Version]
- Glomski, I.J.; Corre, J.-P.; Mock, M.; Goossens, P.L. Noncapsulated toxinogenic Bacillus anthracis presents a specific growth and dissemination pattern in naive and protective antigen-immune mice. Infect. Immun. 2007, 75, 4754–4761. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
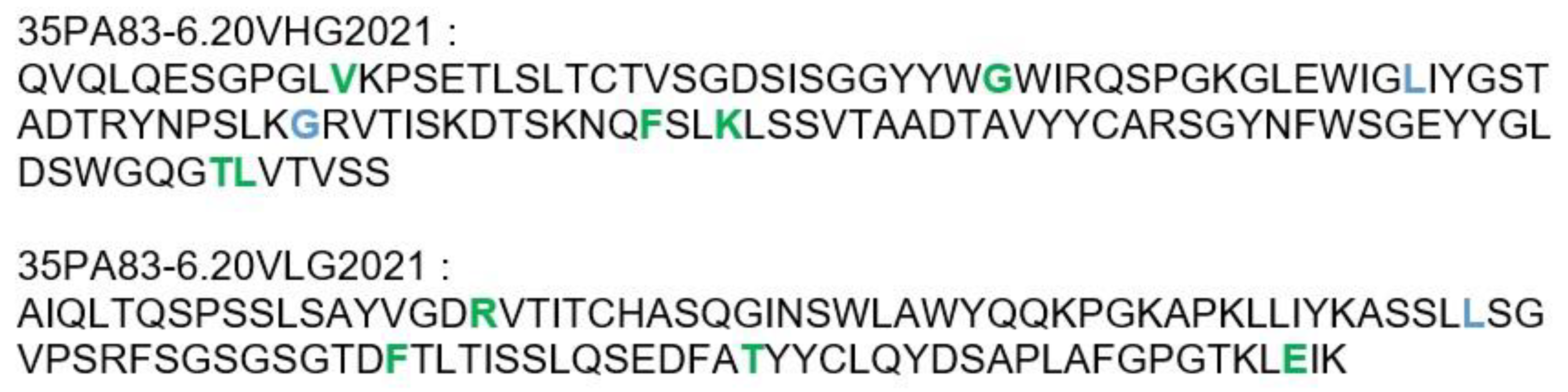{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VH | VL | ||||
|---|---|---|---|---|---|
| Mutation | KD (nM) | Region | Mutation | KD (nM) | Region |
| Q5V | 1.72 | FR1 | V11L | 1.99 | FR1 |
| L11V | 2.09 | FR1 | R18K | 1.64 | FR1 |
| A12V | 2.09 | FR1 | R24H | 1.99 | FR1 |
| K13Q | 2.09 | FR1 | K42N | 2.29 | FR2 |
| G16R | 2.09 | FR1 | I48L | 1.22 | FR2 |
| L34M | 3.09 | FR2 | Y49H | 1.49 | FR2 |
| S49A | 0.8 | FR2 | Q55E | 1.32 | FR3 |
| K73N | 2.17 | FR3 | S56N | 2.6 | FR3 |
| K75N | 7.54 | FR3 | T69A | 1.85 | FR3 |
| V78L | 2.96 | FR3 | F71Y | 2.05 | FR3 |
| S79V | 1.8 | FR3 | P80S | 1.92 | FR3 |
| A87S | 1.5 | FR3 | V106I | 1.53 | FR4 |
| E88D | 2.41 | FR3 | |||
| H94Y | 1.09 | FR3 | |||
| R114Q | 2.09 | FR4 | |||
| V116T | 1.57 | FR4 | |||
| L117M | 2.09 | FR4 | |||
| VH | VL | ||||
|---|---|---|---|---|---|
| Mutation | Region | Mutation | Region | ||
| A29T | 1.2 nM | CDR1 | K56A | 33 nM | CDR2 |
| D35S | 1.3 nM | CDR1 | Y107A | 20 nM | CDR3 |
| T37Y | 1.6 nM | CDR1 | S108N | 0.8 nM | CDR3 |
| T58Y | 1.8 nM | CDR2 | T109S | 0.6 nM | CDR3 |
| G59D | 5.5 nM | CDR2 | S114F | 2.9 nM | CDR3 |
| T64K | 0.8 nM | CDR2 | I116L | 0.8 nM | CDR3 |
| G113D | 19 nM | CDR3 | |||
| P114A | 21 nM | CDR3 | |||
| L115F | 6 nM | CDR3 | |||
| VH | VL | ||||||
|---|---|---|---|---|---|---|---|
| Mutation | koff (s−1) | Region | Remark | Mutation | koff (s−1) | Region | Remark |
| L13V | 1.31 × 10−4 | FR1 | Y14S | 4.39 × 10−4 | FR1 | ||
| S40G | 1.10 × 10−4 | FR2 | K18R | 3.45 × 10−4 | FR1 | ||
| S45P | 4.31 × 10−4 | FR2 | Unstable | H24R | 4.35 × 10−4 | FR1 | Unstable |
| K80V | 9.08 × 10−4 | FR3 | L68E | FR3 | Affinity maturation | ||
| L87F | 3.73 × 10−4 | FR3 | Y87F | 1.29 × 10−4 | FR3 | ||
| Q90K | 1.10 × 10−4 | FR3 | S96P | Not determined | FR3 | Unstable | |
| R92S | 3.01 × 10−4 | FR3 | Unstable | S101T | 1.59 × 10−4 | FR3 | |
| A122T | 2.63 × 10−4 | FR4 | D119E | 2.80 × 10−4 | FR4 | ||
| V123L | 2.93 × 10−4 | FR4 | ND | ||||
| Parameters | WNZ Rabbit |
|---|---|
| Dose injected (mg·kg−1) | 5 |
| T ½ (h) | 94.6 |
| MRT (h) | 143 |
| AUC0-∞ (h·µg·mL−1) | 3609 |
| Cmax (µg·mL−1) | 36.9 |
| Cl (mL·h−1·kg−1) | 1.88 |
| Vd (mL·kg−1) | 228 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avril, A.; Tournier, J.-N.; Paucod, J.-C.; Fournes, B.; Thullier, P.; Pelat, T. Antibodies against Anthrax Toxins: A Long Way from Benchlab to the Bedside. Toxins 2022, 14, 172. https://doi.org/10.3390/toxins14030172
Avril A, Tournier J-N, Paucod J-C, Fournes B, Thullier P, Pelat T. Antibodies against Anthrax Toxins: A Long Way from Benchlab to the Bedside. Toxins. 2022; 14(3):172. https://doi.org/10.3390/toxins14030172
Chicago/Turabian StyleAvril, Arnaud, Jean-Nicolas Tournier, Jean-Charles Paucod, Bénédicte Fournes, Philippe Thullier, and Thibaut Pelat. 2022. "Antibodies against Anthrax Toxins: A Long Way from Benchlab to the Bedside" Toxins 14, no. 3: 172. https://doi.org/10.3390/toxins14030172
APA StyleAvril, A., Tournier, J.-N., Paucod, J.-C., Fournes, B., Thullier, P., & Pelat, T. (2022). Antibodies against Anthrax Toxins: A Long Way from Benchlab to the Bedside. Toxins, 14(3), 172. https://doi.org/10.3390/toxins14030172