Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS

 ,
,  ,
,  ,
,  and
and 
Abstract
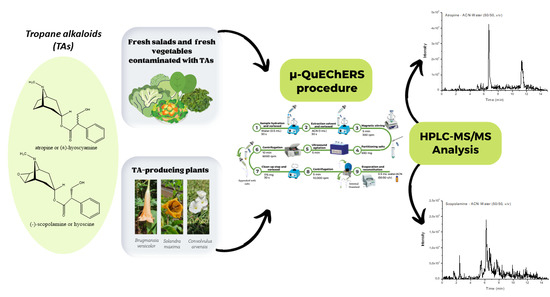
1. Introduction
2. Results and Discussion
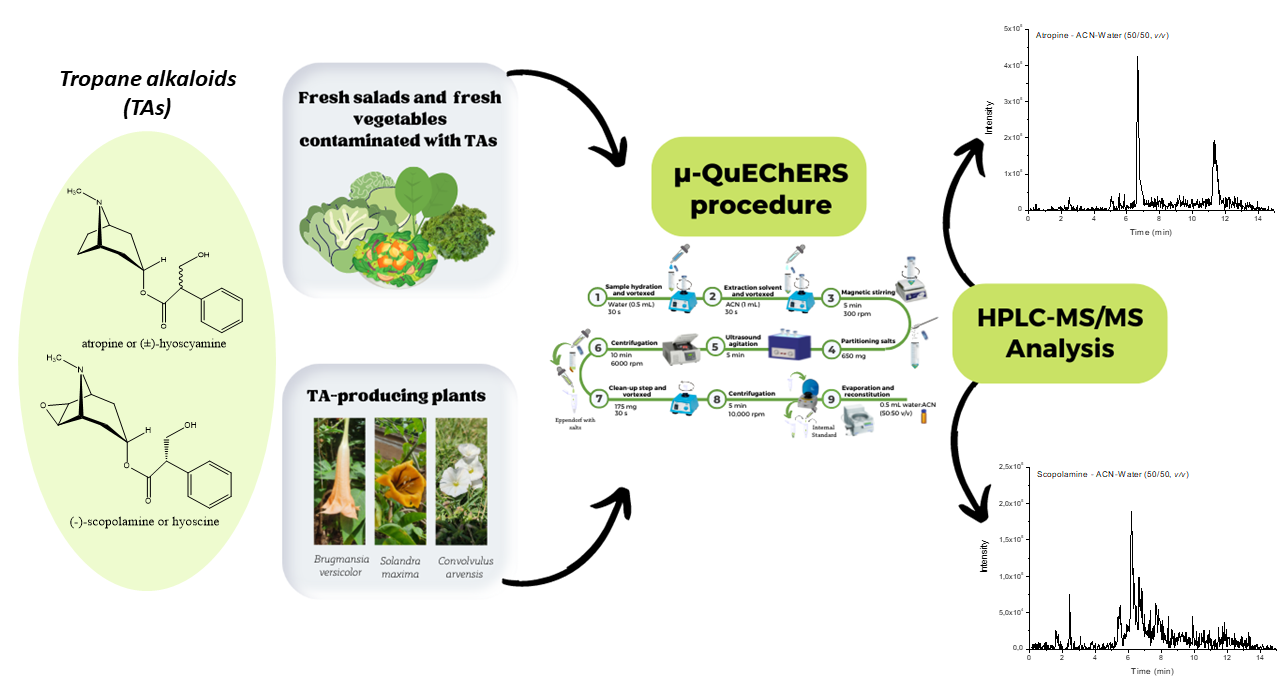
2.1. Optimization of the μ-QuEChERS
2.2. μ-QuEChERS Procedure Validation
2.3. Analysis of TA-Producing Plants and Contamination Assays
2.4. Application of the μ-QuEChERS Procedure to Samples of Fresh Leafy Vegetables
3. Conclusions
4. Materials and Methods
4.1. Chemicals, Reagents, and Standard Solutions
4.2. Samples
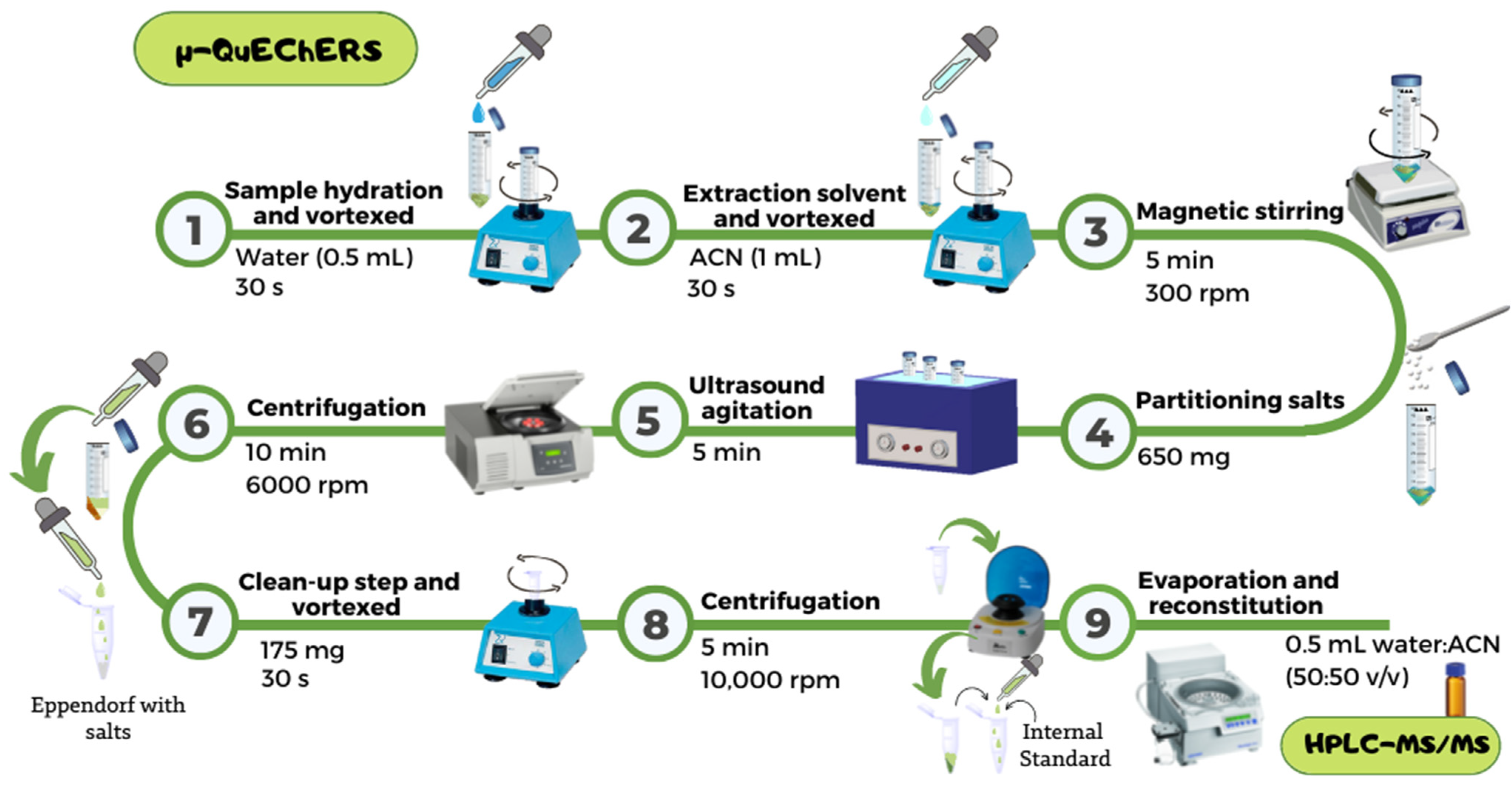
4.3. μ-QuEChERS Procedure
4.4. HPLC-MS/MS Conditions for Analysis of Atropine and Scopolamine
4.5. Samples Quantification
4.6. Method Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority. Scientific Opinion on Tropane alkaloids in food and feed. EFSA J. 2013, 11, 1–113. [Google Scholar] [CrossRef]
- González-Gómez, L.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Occurrence and Chemistry of Tropane Alkaloids in Foods, with a Focus on Sample Analysis Methods: A Review on Recent Trends and Technological Advances. Foods 2022, 11, 407. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.P.J.; de Nijs, M.; Castellari, M.; Hortos, M.; MacDonald, S.; Crews, C.; Hajslova, J.; Stranska, M. Occurrence of tropane alkaloids in food. EFSA Support. Publ. 2016, 13, 1140E. [Google Scholar] [CrossRef]
- European Union. Commission regulation (EU) 2021/1408 of 27 August 2021 amending Regulation (EC) No 1881/2006 as regards maximum levels of tropane alkaloids in certain foodstuffs. Off. J. Eur. Union 2021, 304, 1–4. [Google Scholar]
- RASFF Portal—Food and Feed Safety Alerts. Available online: https://webgate.ec.europa.eu/rasff-window/portal/? (accessed on 24 June 2021).
- Castilla-Fernández, D.; Moreno-González, D.; García-Reyes, J.F.; Ballesteros, E.; Molina-Díaz, A. Determination of atropine and scopolamine in spinach-based products contaminated with genus Datura by UHPLC–MS/MS. Food Chem. 2021, 347, 129020. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commision Recommendation (EU) 2015/976 of 19 June 2015 on the monitoring of the presence of tropane alkaloids in food. Off. J. Eur. Union 2015, 11, 97–98. [Google Scholar]
- Agrawal, A.; Keçili, R.; Ghorbani-Bidkorbeh, F.; Hussain, C.M. Green miniaturized technologies in analytical and bioanalytical chemistry. TrAC Trends Anal. Chem. 2021, 143, 116383. [Google Scholar] [CrossRef]
- Câmara, J.S.; Perestrelo, R.; Berenguer, C.V.; Andrade, C.F.P.; Gomes, T.M.; Olayanju, B.; Kabir, A.; Rocha, C.M.R.; Teixeira, J.A.; Pereira, J.A.M. Green Extraction Techniques as Advanced Sample Preparation Approaches in Biological, Food, and Environmental Matrices: A Review. Molecules 2022, 27, 2953. [Google Scholar] [CrossRef]
- González-Gómez, L.; Pereira, J.A.M.; Morante-Zarcero, S.; Câmara, J.S.; Sierra, I. Green extraction approach based on μSPEed® followed by HPLC-MS/MS for the determination of atropine and scopolamine in tea and herbal tea infusions. Food Chem. 2022, 394, 133512. [Google Scholar] [CrossRef]
- Álvarez, I.; Bermejo, A.M.; Tabernero, M.J.; Fernández, P.; López, P. Determination of cocaine and cocaethylene in plasma by solid-phase microextraction and gas chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 845, 90–94. [Google Scholar] [CrossRef]
- Zeng, J.; Zou, J.; Song, X.; Chen, J.; Ji, J.; Wang, B.; Wang, Y.; Ha, J.; Chen, X. A new strategy for basic drug extraction in aqueous medium using electrochemically enhanced solid-phase microextraction. J. Chromatogr. A 2011, 1218, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Masrournia, M.; haghi, Z.E.; Amini, M. Liquid Chromatographic Determination of Scopolamine in Hair with Suspended Drop Liquid Phase Microextraction Technique. Am. J. Anal. Chem. 2011, 2, 235–242. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Martinello, M.; Borin, A.; Stella, R.; Bovo, D.; Biancotto, G.; Gallina, A.; Mutinelli, F. Development and validation of a QuEChERS method coupled to liquid chromatography and high resolution mass spectrometry to determine pyrrolizidine and tropane alkaloids in honey. Food Chem. 2017, 234, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Romera-Torres, A.; Romero-González, R.; Martínez Vidal, J.L.; Garrido Frenich, A. Comprehensive tropane alkaloids analysis and retrospective screening of contaminants in honey samples using liquid chromatography-high resolution mass spectrometry (Orbitrap). Food Res. Int. 2020, 133, 109130. [Google Scholar] [CrossRef]
- Zheng, W.; Yoo, K.H.; Choi, J.M.; Park, D.H.; Kim, S.K.; Kang, Y.S.; Abd El-Aty, A.M.; Hacımüftüoğlu, A.; Jeong, J.H.; Bekhit, A.E.D.; et al. A modified QuEChERS method coupled with liquid chromatography-tandem mass spectrometry for the simultaneous detection and quantification of scopolamine, L-hyoscyamine, and sparteine residues in animal-derived food products. J. Adv. Res. 2019, 15, 95–102. [Google Scholar] [CrossRef]
- Chen, H.; Marín-Sáez, J.; Romero-González, R.; Garrido Frenich, A. Simultaneous determination of atropine and scopolamine in buckwheat and related products using modified QuEChERS and liquid chromatography tandem mass spectrometry. Food Chem. 2017, 218, 173–180. [Google Scholar] [CrossRef]
- Baslé, Q.; Mujahid, C.; Bessaire, T. Application of a streamlined LC-MS/MS methodology for the determination of atropine and scopolamine in cereals from Asian and African countries. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2020, 37, 1744–1754. [Google Scholar] [CrossRef]
- León, N.; Miralles, P.; Yusà, V.; Coscollà, C. A green analytical method for the simultaneous determination of 30 tropane and pyrrolizidine alkaloids and their N-oxides in teas and herbs for infusions by LC-Q-Orbitrap HRMS. J. Chromatogr. A 2022, 1666, 462835. [Google Scholar] [CrossRef]
- Izcara, S.; Casado, N.; Morante-Zarcero, S.; Sierra, I. A miniaturized QuEChERS method combined with ultrahigh liquid chromatography coupled to tandem mass spectrometry for the analysis of pyrrolizidine alkaloids in oregano samples. Foods 2020, 9, 1319. [Google Scholar] [CrossRef]
- QuEChERS CVUA Stuttgart. A Mini-Multiresidue Method for the Analysis of Pesticide Residues in Low-Fat Products. Available online: https://nucleus.iaea.org/sites/fcris/SharedDocuments/SOP/QuEChERS_original_CVUA_Stuttgart.pdf (accessed on 4 July 2022).
- AOAC International AOAC Official Methods of Analysis. Appendix K: Guidelines for Dietary Supplements and Botanicals. AOAC Off. Methods Anal. 2013, 2, 32.
- European Comission Document. N° SANTE 11312/2021. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. 2021, pp. 1–57. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 10 May 2022).
- Adamse, P.; van Egmond, H.P.; Noordam, M.Y.; Mulder, P.P.J.; De Nijs, M. Tropane alkaloids in food: Poisoning incidents. Qual. Assur. Saf. Crop. Foods 2014, 6, 15–24. [Google Scholar] [CrossRef]
- European Commission. Compendium of botanicals reported to contain naturally occuring substances of possible concern for human health when used in food and food supplements. EFSA J. 2012, 10, 2663. [Google Scholar] [CrossRef]
- Todd, F.G.; Stermitz, F.R.; Schultheis, P.; Knight, A.P.; Traub-Dargatz, J. Tropane alkaloids and toxicity of Convolvulus arvensis. Phytochemistry 1995, 39, 301–303. [Google Scholar] [CrossRef]
- Izcara, S.; Casado, N.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Miniaturized and modified QuEChERS method with mesostructured silica as clean-up sorbent for pyrrolizidine alkaloids determination in aromatic herbs. Food Chem. 2022, 380, 132189. [Google Scholar] [CrossRef] [PubMed]
- González-Gómez, L.; Gañán, J.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Sulfonic Acid-Functionalized SBA-15 as Strong Cation-Exchange Sorbent for Solid-Phase Extraction of Atropine and Scopolamine in Gluten-Free Grains and Flours. Foods 2020, 9, 1854. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Moussa, F.; Mokh, S.; Doumiati, S.; Barboni, B.; Bernabò, N.; Al, M. LC-MS/MS method for the determination of hormones: Validation, application and health risk assessment in various bovine matrices. Food Chem. Toxicol. 2020, 138, 111204. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Sample a | Sample Amount (g) | Water Amount (mL) b | Spiked Level (ng/g) c | Atropine Recovery ± SD (%) | Scopolamine Recovery ± SD (%) |
|---|---|---|---|---|---|
| Fresh | 1 | - | 5 | 73 ± 3 | 77 ± 9 |
| Lyophilized | 0.1 | 0.9 | 5 | 40 ± 4 | 62 ± 17 |
| Lyophilized | 0.1 | 0.75 | 5 | 95 ± 11 | 101 ± 18 |
| Lyophilized | 0.1 | 0.5 | 5 | 98 ± 5 | 95 ± 2 |
| Lyophilized | 0.1 | 0.5 | 100 | 84 ± 1 | 105 ± 13 |
| Analyte | Linear Range a (ng/mL) | Linearity R2 Slope RSD (%) | MDL b (ng/g) | MQL c (ng/g) | ME d (%) | Spiked Level (ng/g) | Accuracy n = 6 (Recovery ± SD %) | Intra-Day Precision n = 6, 1 Day (RSD %) | Inter-Day Precision n = 9, 3 Days (RSD %) |
|---|---|---|---|---|---|---|---|---|---|
| 0.06x + 0.27 | 5 | 90 ± 10 | 9 | 11 | |||||
| Atropine | 0.5–500 | 0.998 | 0.7 | 2.3 | −38 | 25 | 100 ± 10 | 10 | 12 |
| 1.5 | 200 | 96 ± 4 | 4 | 11 | |||||
| 0.13x + 0.88 | 5 | 93 ± 7 | 8 | 13 | |||||
| Scopolamine | 0.5–500 | 0.997 | 0.6 | 2.2 | −39 | 25 | 96 ± 5 | 5 | 10 |
| 0.3 | 200 | 95 ± 6 | 6 | 7 |
| TA-Producing Plant | Family | Sample Analysed | Atropine (mg/kg) | Scopolamine (mg/kg) | |
|---|---|---|---|---|---|
| Solandra maximaa |  | Solanaceae | Flowers | 10.4 ± 0.1 | 0.11 ± 0.05 |
| Leaves | 0.20 ± 0.07 | 0.023 ± 0.006 | |||
| Brugmansia versicolora |  | Solanaceae | Flowers | 5 ± 1 | 1771 ± 167 |
| Leaves | 0.9 ± 0.3 | 297 ± 36 | |||
| Convolvulus arvensisb |  | Convolvulaceae | Leaves, stems and flowers | 0.0083 ± 0.0012 | c ND |
| Atropine | Scopolamine | |||||
|---|---|---|---|---|---|---|
| Contaminated Sample | Expected Content (ng/g) | Found Content (ng/g ± SD) | Recovery (% ± SD) | Expected Content (ng/g) | Found Content (ng/g ± SD) | Recovery (% ± SD) |
| Mix-1 with 10% S. maxima flowers | 1043 | 874 ± 56 | 84 ± 5 | 11 | 11 ± 1 | 102 ± 9 |
| Mix-1 with 10% S. maxima leaves | 20 | 20 ± 2 | 98 ± 9 | 2 | 2.2 ± 0.5 | 110 ± 3 |
| Mix-1 with 10% B. versicolor flowers | 544 | 444 ± 19 | 82 ± 3 | 177,067 | 156,468 ± 8776 | 88 ± 7 |
| Mix-1 with 10% B. versicolor leaves | 89 | 92 ± 3 | 104 ± 3 | 29,743 | 29,576 ± 3163 | 99 ± 11 |
| Sample | Atropine (ng/g ± SD) | Atropine (ng/g Fresh Weight ± SD) a | Scopolamine (ng/g ± SD) | Scopolamine (ng/g Fresh Weight ± SD) |
|---|---|---|---|---|
| Mix-1 | ≤MQL | - | ND | - |
| Mix-2 | ≤MQL | - | ND | - |
| Mix-3 | 3.2 ± 1.9 | 0.16 ± 0.09 | ND | - |
| Mix-4 | ≤MQL | - | ND | - |
| Mix-5 | 2.7 ± 0.9 | 0.16 ± 0.05 | ND | - |
| Mix-6 | ND | - | ND | - |
| Mix-7 | ND | - | ND | - |
| Mix-8 | ≤MQL | - | ND | - |
| Mix-9 | 3.4 ± 0.8 | 0.20 ± 0.05 | ND | - |
| Mix-10 | ND | - | ND | - |
| Mix-11 | ND | - | ND | - |
| Ice-1 | ≤MQL | - | ND | - |
| Ice-2 | ≤MQL | - | ND | - |
| Kal-1 | ≤MQL | - | ND | - |
| Spi-1 | ND | - | ND | - |
| Cha-1 | ≤MQL | - | ≤MQL | - |
| Cha-2 | ≤MQL | - | ND | - |
| Aru-1 | ND | - | ND | - |
| Code | Sample Description | Origin | Ingredients |
|---|---|---|---|
| Mix-1 | Mixed leafy vegetables | Local supermarket | Curly escarole, red cabbage, lollo rosso lettuce, spinach sprouts and arugula |
| Mix-2 | Mixed leafy vegetables | Local supermarket | Curly escarole, red cabbage, lollo rosso lettuce, spinach sprouts and arugula |
| Mix-3 | Mixed leafy vegetables | Local supermarket | Iceberg lettuce, carrot and red cabbage |
| Mix-4 | Mixed leafy vegetables | Local supermarket | Green sprout lettuce and red sprout lettuce |
| Mix-5 | Mixed leafy vegetables | Local supermarket | Curly endive, red lettuce sprouts, red radicchio and mizuna |
| Mix-6 | Mixed leafy vegetables | Local supermarket | Green lettuce sprouts, red lettuce sprouts and wild arugula |
| Mix-7 | Mixed leafy vegetables | Local supermarket | Arugula and lamb’s lettuce |
| Mix-8 | Mixed leafy vegetables | Local supermarket | Red lettuce sprouts, lamb’s lettuce, arugula and watercress |
| Mix-9 | Mixed leafy vegetables | Local supermarket | Red lettuce sprouts, spinach sprouts, green lettuce sprouts, lamb’s lettuce, arugula and tatsoi |
| Mix-10 | Mixed leafy vegetables | Local supermarket | Green baby leaves, red baby leaves and lamb’s lettuce |
| Mix-11 | Mixed leafy vegetables | Local supermarket | Spinach sprouts, red spinach and arugula |
| Ice-1 | Chopped iceberg lettuce | Local supermarket | Iceberg lettuce (Lactuca sativa var. capitata) |
| Ice-2 | Whole iceberg lettuce | Local supermarket | Iceberg lettuce (Lactuca sativa var. capitata) |
| Kal-1 | Chopped kale | Local supermarket | Green curly kale (Brassica oleracea var. sabellica L.) |
| Spi-1 | Spinach sprouts | Local supermarket | Spinach sprouts (Spinacia oleracea L.) |
| Cha-1 | Chopped Swiss chard | Local supermarket | Swiss chard (Beta vulgaris var. cycla) |
| Cha-2 | Whole Swiss chard | Vegetable patch | Swiss chard (Beta vulgaris var. cycla) |
| Aru-1 | Arugula salad | Local supermarket | Arugula (Eruca vesicaria) |
| Analyte | tR (min) | Ionization Mode | Precursor Ions (Q1, m/z, [M+H]+) | Capillary (V) | MS2 Product Ions (Q3, m/z) | CE (V) | Dwell Time (s) | Chemical Structures and Fragment Ions |
|---|---|---|---|---|---|---|---|---|
| Atropine | 6.7 | ESI (+) | 290.2 | 70 | 90.9 | 34 | 0.25 |  |
| 93.0 | 29 | 0.25 | ||||||
| 124.1 * | 20.5 | 0.25 | ||||||
| (±)-Atropine-D3 | 6.7 | ESI (+) | 293.4 | 70 | 92.9 | 25 | 0.25 |  |
| 102.7 | 42 | 0.25 | ||||||
| 121.3 | 28 | 0.25 | ||||||
| 127.2 * | 21 | 0.25 | ||||||
| Scopolamine | 6.3 | ESI (+) | 304.1 | 70 | 121.0 | 16 | 0.25 |  |
| 138.1 * | 12 | 0.25 | ||||||
| 156.0 | 9.5 | 0.25 | ||||||
| (-)-Scopolamine-D3 | 6.3 | ESI (+) | 307.4 | 70 | 121.1 | 24.5 | 0.25 |  |
| 141.2 * | 24.5 | 0.25 | ||||||
| 159.0 | 9 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Gómez, L.; Morante-Zarcero, S.; Pereira, J.A.M.; Câmara, J.S.; Sierra, I. Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS. Toxins 2022, 14, 650. https://doi.org/10.3390/toxins14100650
González-Gómez L, Morante-Zarcero S, Pereira JAM, Câmara JS, Sierra I. Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS. Toxins. 2022; 14(10):650. https://doi.org/10.3390/toxins14100650
Chicago/Turabian StyleGonzález-Gómez, Lorena, Sonia Morante-Zarcero, Jorge A. M. Pereira, José S. Câmara, and Isabel Sierra. 2022. "Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS" Toxins 14, no. 10: 650. https://doi.org/10.3390/toxins14100650
APA StyleGonzález-Gómez, L., Morante-Zarcero, S., Pereira, J. A. M., Câmara, J. S., & Sierra, I. (2022). Improved Analytical Approach for Determination of Tropane Alkaloids in Leafy Vegetables Based on µ-QuEChERS Combined with HPLC-MS/MS. Toxins, 14(10), 650. https://doi.org/10.3390/toxins14100650