Computational Approaches to Explore Bacterial Toxin Entry into the Host Cell


Abstract
:
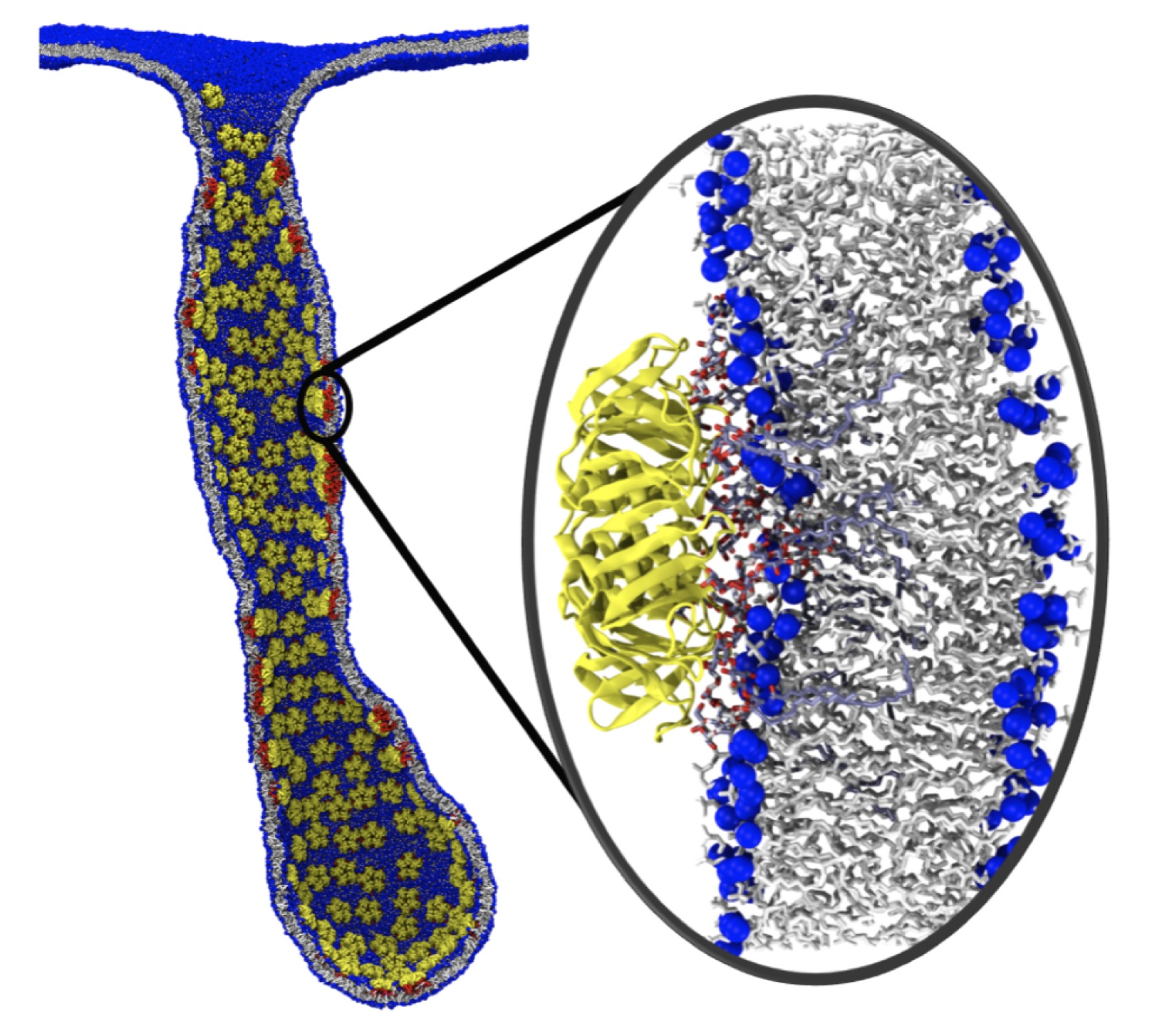{kind=link}
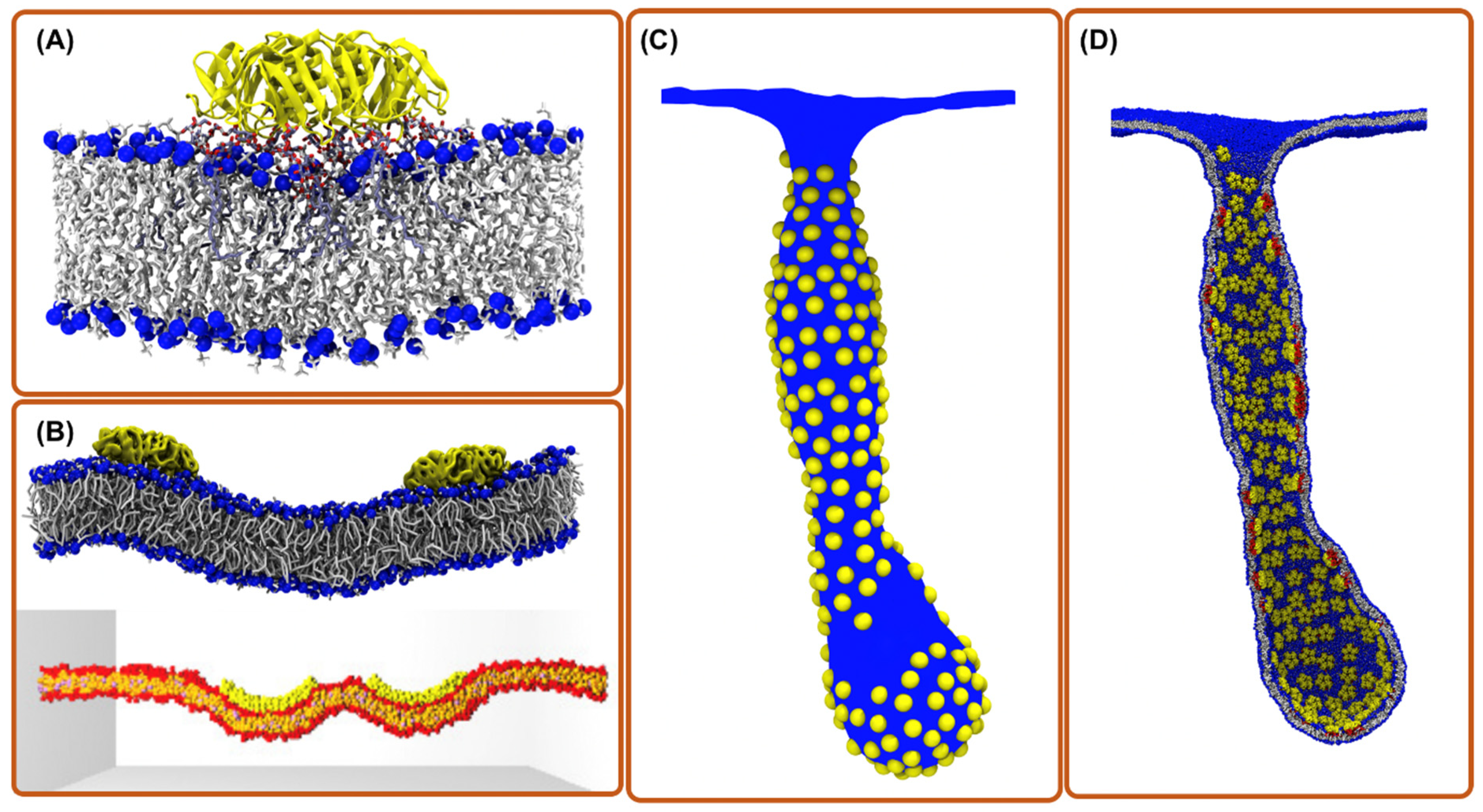
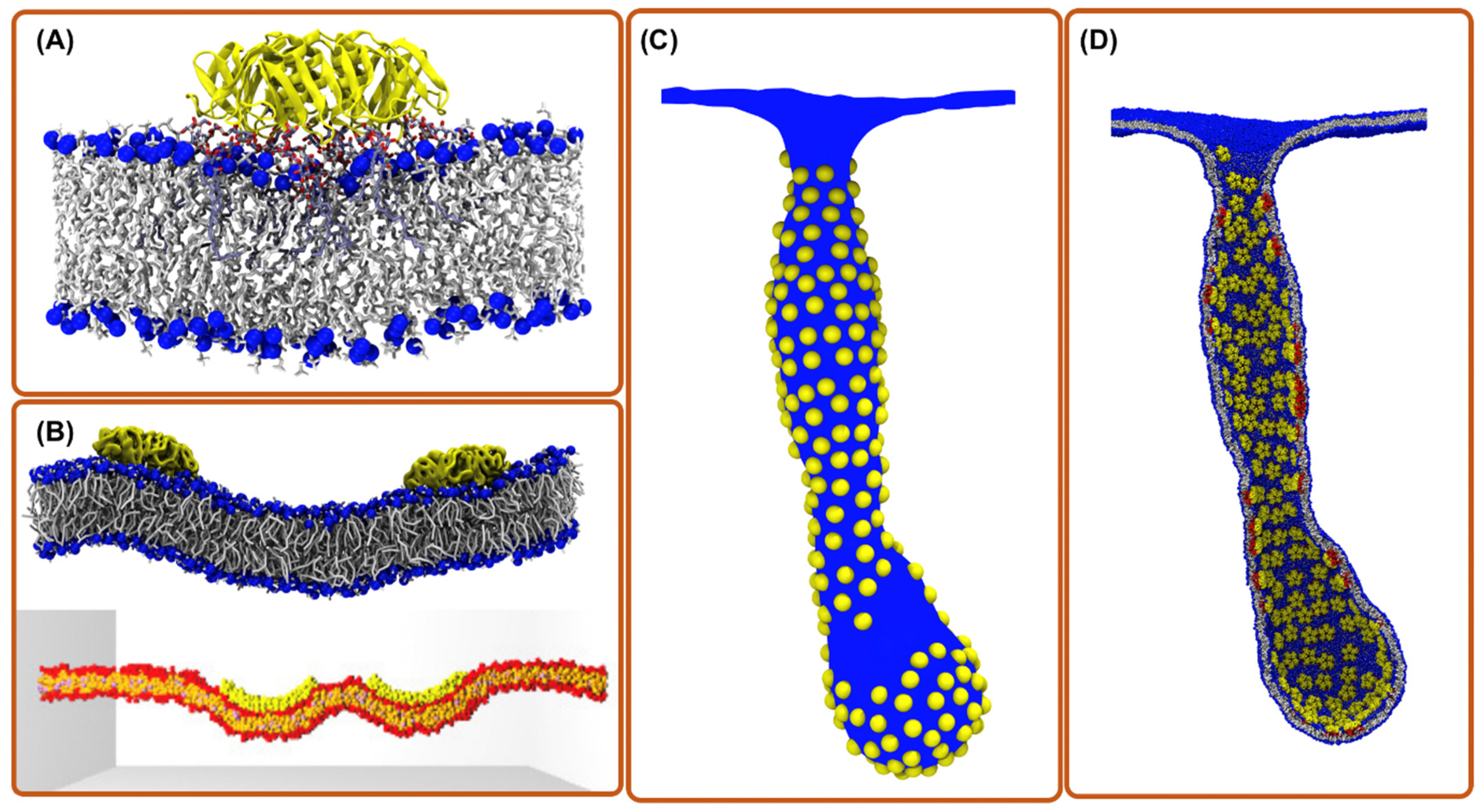{kind=link}
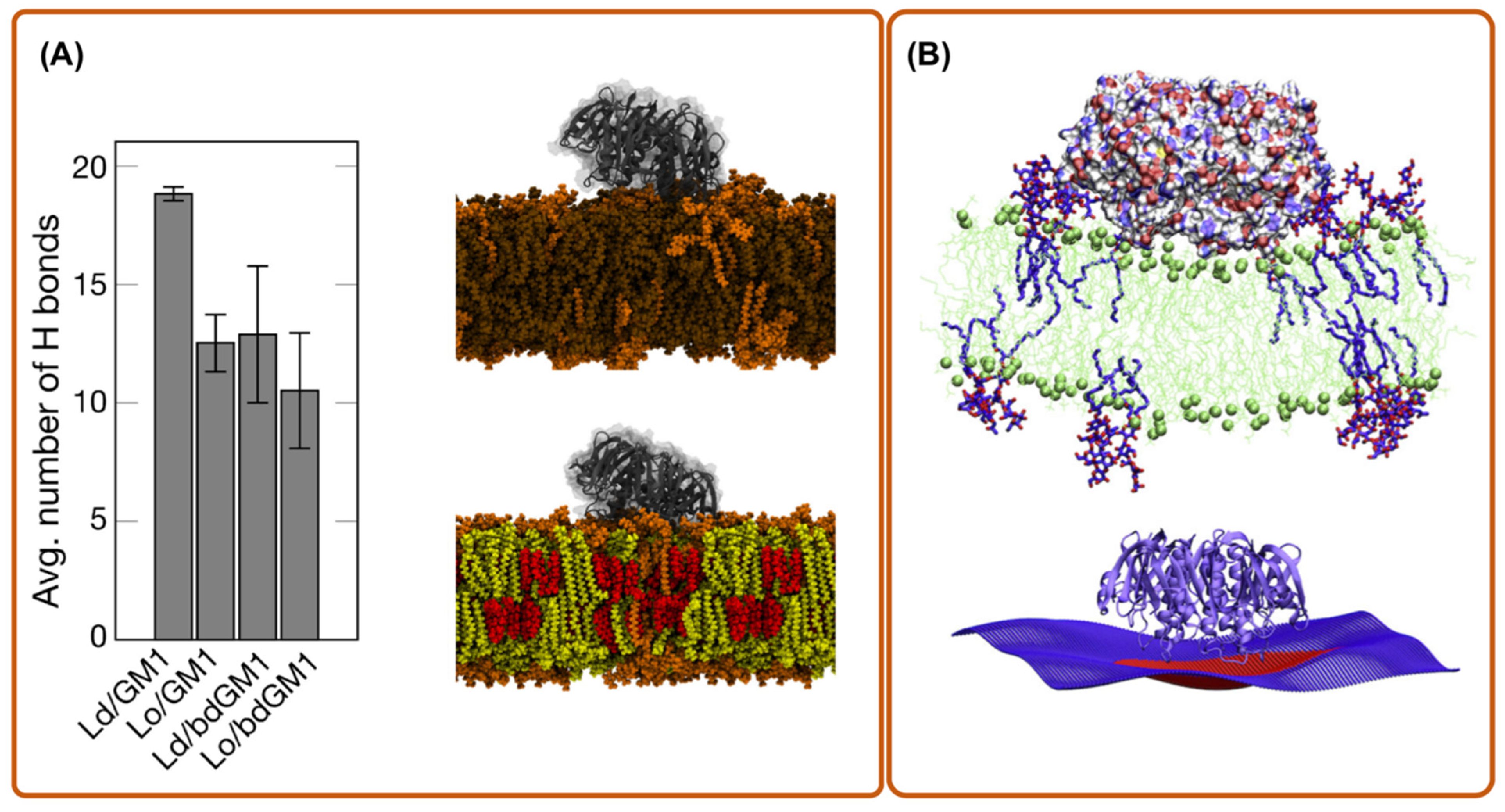{kind=link}
1. Introduction
2. Computational Methods
3. All-Atom Molecular Dynamics Simulations
4. Coarse-Grain Simulations
5. Mesoscopic Models
6. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schiavo, G.; Van Der Goot, F.G. The bacterial toxin toolkit. Nat. Rev. Mol. Cell Biol. 2001, 2, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Klenow, M.B.; Jeppesen, J.C.; Simonsen, A.C. Membrane rolling induced by bacterial toxins. Soft Matter 2020, 16, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Piper, S.J.; Brillault, L.; Rothnagel, R.; I Croll, T.; Box, J.K.; Chassagnon, I.; Scherer, S.; Goldie, K.N.; A Jones, S.; Schepers, F.; et al. Cryo-EM structures of the pore-forming A subunit from the Yersinia entomophaga ABC toxin. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacia, K.; Scherfeld, D.; Kahya, N.; Schwille, P. Fluorescence Correlation Spectroscopy Relates Rafts in Model and Native Membranes. Biophys. J. 2004, 87, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.E.; Fraisier, V.; Florent, J.-C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef]
- Chang, K.J.; Benett, V.; Cuatrecasas, P. Membrane receptors as general markers for plasma membrane isolation procedures. The use of 125-I-labeled wheat germ agglutinin, insulin, and cholera toxin. J. Biol. Chem. 1975, 250, 488–500. [Google Scholar] [CrossRef]
- Shogomori, H.; Futerman, A. Cholera Toxin Is Found in Detergent-insoluble Rafts/Domains at the Cell Surface of Hippocampal Neurons but Is Internalized via a Raft-independent Mechanism. J. Biol. Chem. 2001, 276, 9182–9188. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, S.; Schmieder, S.; Pezeshkian, W.; Becken, U.; Wunder, C.; Chinnapen, D.; Ipsen, J.H.; Kenworthy, A.K.; Lencer, W.; Mayor, S.; et al. Ceramide structure dictates glycosphingolipid nanodomain assembly and function. Nat. Commun. 2021, 12, 3675–3687. [Google Scholar] [CrossRef]
- Hammond, A.T.; Heberle, F.; Baumgart, T.; Holowka, D.; Baird, B.; Feigenson, G.W. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 6320–6325. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Wunder, C.; Shafaq-Zadah, M. Glycolipids and Lectins in Endocytic Uptake Processes. J. Mol. Biol. 2016, 428, 4792–4818. [Google Scholar] [CrossRef]
- Johannes, L. Shiga Toxin—A Model for Glycolipid-Dependent and Lectin-Driven Endocytosis. Toxins 2017, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Doosti, B.A.; Pezeshkian, W.; Bruhn, D.S.; Ipsen, J.; Khandelia, H.; Jeffries, G.D.M.; Lobovkina, T. Membrane Tubulation in Lipid Vesicles Triggered by the Local Application of Calcium Ions. Langmuir 2017, 33, 11010–11017. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkian, W.; Marrink, S.J. Simulating realistic membrane shapes. Curr. Opin. Cell Biol. 2021, 71, 103–111. [Google Scholar] [CrossRef]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Róg, T.; Vattulainen, I. Multiscale Simulations of Biological Membranes: The Challenge To Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [Green Version]
- Joshi, H.; Bhatia, D.; Krishnan, Y.; Maiti, P.K. Probing the structure and in silico stability of cargo loaded DNA icosahedra using MD simulations. Nanoscale 2017, 9, 4467–4477. [Google Scholar] [CrossRef] [Green Version]
- Patmanidis, I.; De Vries, A.H.; Wassenaar, T.A.; Wang, W.; Portale, G.; Marrink, S.J. Structural characterization of supramolecular hollow nanotubes with atomistic simulations and SAXS. Phys. Chem. Chem. Phys. 2020, 22, 21083–21093. [Google Scholar] [CrossRef]
- Khan, F.I.; Wei, D.; Gu, K.-R.; Hassan, I.; Tabrez, S. Current updates on computer aided protein modeling and designing. Int. J. Biol. Macromol. 2016, 85, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623–1634. [Google Scholar] [CrossRef] [Green Version]
- Florentsen, C.D.; Kamp-Sonne, A.; Moreno-Pescador, G.; Pezeshkian, W.; Zanjani, A.A.H.; Khandelia, H.; Nylandsted, J.; Bendix, P.M. Annexin A4 trimers are recruited by high membrane curvatures in giant plasma membrane vesicles. Soft Matter 2021, 17, 308–318. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Hansen, A.G.; Johannes, L.; Khandelia, H.; Shillcock, J.; Kumar, P.B.S.; Ipsen, J.H. Membrane Invagination Induced by Shiga toxin B-subunit: From Molecular Structure to Tube Formation. Soft Matter 2016, 12, 5164–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezeshkian, W.; Gao, H.; Arumugam, S.; Becken, U.; Bassereau, P.; Florent, J.-C.; Ipsen, J.H.; Johannes, L.; Shillcock, J.C. Mechanism of Shiga Toxin Clustering on Membranes. ACS Nano 2017, 11, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Nåbo, L.J.; Ipsen, J.H. Cholera toxin B subunit induces local curvature on lipid bilayers. FEBS Open Bio 2017, 7, 1638–1645. [Google Scholar] [CrossRef] [Green Version]
- Basu, I.; Mukhopadhyay, C. Insights into Binding of Cholera Toxin to GM1 Containing Membrane. Langmuir 2014, 30, 15244–15252. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Chaban, V.V.; Johannes, L.; Shillcock, J.; Ipsen, J.; Khandelia, H. The effects of globotriaosylceramide tail saturation level on bilayer phases. Soft Matter 2015, 11, 1352–1361. [Google Scholar] [CrossRef] [Green Version]
- Flores-Canales, J.C.; Kurnikova, M.; Maria, K. Microsecond Simulations of the Diphtheria Toxin Translocation Domain in Association with Anionic Lipid Bilayers. J. Phys. Chem. B 2015, 119, 12074–12085. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, C.; Rodnin, M.V.; Vargas-Uribe, M.; McCluskey, A.J.; Flores-Canales, J.C.; Kurnikova, M.; Ladokhin, A.S. Role of Acidic Residues in Helices TH8–TH9 in Membrane Interactions of the Diphtheria Toxin T Domain. Toxins 2015, 7, 1303–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Canales, J.C.; Simakov, N.A.; Kurnikova, M. Microsecond Molecular Dynamics Simulations of Diphtheria Toxin Translocation T-Domain pH-Dependent Unfolding in Solution. bioRxiv 2019, 572040. [Google Scholar] [CrossRef]
- Ladokhin, A.S. pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 2013, 5, 1362–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissanen, S.; Grzybek, M.; Orłowski, A.; Róg, T.; Cramariuc, O.; Levental, I.; Eggeling, C.; Sezgin, E.; Vattulainen, I. Phase Partitioning of GM1 and Its Bodipy-Labeled Analog Determine Their Different Binding to Cholera Toxin. Front. Physiol. 2017, 8, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Binnington, B.; Róg, T.; Vattulainen, I.; Grzybek, M.; Ünal, C.; Lingwood, C.A.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef]
- Manna, M.; Javanainen, M.; Monne, H.M.-S.; Gabius, H.-J.; Rog, T.; Vattulainen, I. Long-chain GM1 gangliosides alter transmembrane domain registration through interdigitation. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 870–878. [Google Scholar] [CrossRef]
- Merritt, E.A.; Kuhn, P.; Sarfaty, S.; Erbe, J.L.; Holmes, R.K.; Hol, W.G.J. The 1.25 Å resolution refinement of the cholera toxin B-pentamer: Evidence of peptide backbone strain at the receptor-binding site. J. Mol. Biol. 1998, 282, 1043–1059. [Google Scholar] [CrossRef]
- Johannes, L.; Parton, R.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [Google Scholar] [CrossRef]
- Kabbani, A.M.; Raghunathan, K.; Lencer, W.I.; Kenworthy, A.K.; Kelly, C.V. Structured clustering of the glycosphingolipid GM1 is required for membrane curvature induced by cholera toxin. Proc. Natl. Acad. Sci. USA 2020, 117, 14978–14986. [Google Scholar] [CrossRef]
- Groza, R.; Ewers, H. Membrane deformation by the cholera toxin beta subunit requires more than one binding site. Proc. Natl. Acad. Sci. USA 2020, 117, 17467–17469. [Google Scholar] [CrossRef]
- Ewers, H.; Römer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2009, 12, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ivashenka, A.; Wunder, C.; Chambon, V.; Sandhoff, R.; Jennemann, R.; Dransart, E.; Podsypanina, K.; Lombard, B.; Loew, D.; Lamaze, C.; et al. Glycolipid-dependent and lectin-driven transcytosis in mouse enterocytes. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Lakshminarayan, R.; Wunder, C.; Becken, U.; Howes, M.; Benzing, C.; Arumugam, S.; Sales, S.; Ariotti, N.; Chambon, V.; Lamaze, C.; et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol. 2014, 16, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Renard, H.-F.; Tyckaert, F.; Giudice, C.L.; Hirsch, T.; Valades-Cruz, C.A.; Lemaigre, C.; Shafaq-Zadah, M.; Wunder, C.; Wattiez, R.; Johannes, L.; et al. Endophilin-A3 and Galectin-8 control the clathrin-independent endocytosis of CD166. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingólfsson, H.I.; Lopez, C.A.; Uusitalo, J.J.; De Jong, D.H.; Gopal, S.M.; Periole, X.; Marrink, S.J. The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 225–248. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-Graining Methods for Computational Biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Lipowsky, R.; Brinkmann, M.; Dimova, R.; Haluska, C.; Kierfeld, J.; Shillcock, J. Wetting, budding, and fusion—morphological transitions of soft surfaces. J. Phys. Condens. Matter 2005, 17, S2885–S2902. [Google Scholar] [CrossRef]
- Johannes, L.; Pezeshkian, W.; Ipsen, J.H.; Shillcock, J.C. Clustering on Membranes: Fluctuations and More. Trends Cell Biol. 2018, 28, 405–415. [Google Scholar] [CrossRef]
- Reynwar, B.J.; Illya, G.; Harmandaris, V.A.; Müller, M.M.; Kremer, K.; Deserno, M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature 2007, 447, 461–464. [Google Scholar] [CrossRef]
- Cooke, I.; Kremer, K.; Deserno, M. Tunable generic model for fluid bilayer membranes. Phys. Rev. E 2005, 72 Pt 1, 011506. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Badaczewska-Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Barnoud, J.; Marrink, S.J. Gangliosides Destabilize Lipid Phase Separation in Multicomponent Membranes. Biophys. J. 2019, 117, 1215–1223. [Google Scholar] [CrossRef]
- Sridhar, A.; Kumar, A.; DasMahapatra, A.K. Multi-scale molecular dynamics study of cholera pentamer binding to a GM1-phospholipid membrane. J. Mol. Graph. Model. 2016, 68, 236–251. [Google Scholar] [CrossRef]
- Kociurzynski, R.; Makshakova, O.N.; Knecht, V.; Römer, W. Multiscale Molecular Dynamics Studies Reveal Different Modes of Receptor Clustering by Gb3-Binding Lectins. J. Chem. Theory Comput. 2021, 17, 2488–2501. [Google Scholar] [CrossRef]
- Flores-Canales, J.C.; Vargas-Uribe, M.; Ladokhin, A.S.; Kurnikova, M. Membrane Association of the Diphtheria Toxin Translocation Domain Studied by Coarse-Grained Simulations and Experiment. J. Membr. Biol. 2015, 248, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef] [Green Version]
- Cornet, J.; Destainville, N.; Manghi, M. Domain formation in bicomponent vesicles induced by composition-curvature coupling. J. Chem. Phys. 2020, 152, 244705. [Google Scholar] [CrossRef]
- Pezeshkian, W.; König, M.; Marrink, S.; Ipsen, J.H. A Multi-Scale Approach to Membrane Remodeling Processes. Front. Mol. Biosci. 2019, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Paraschiv, A.; Lagny, T.J.; Campos, C.V.; Coudrier, E.; Bassereau, P.; Šarić, A. Influence of membrane-cortex linkers on the extrusion of membrane tubes. Biophys. J. 2021, 120, 598–606. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Ipsen, J.H. Fluctuations and conformational stability of a membrane patch with curvature inducing inclusions. Soft Matter 2019, 15, 9974–9981. [Google Scholar] [CrossRef]
- Pezeshkian, W.; König, M.; Wassenaar, T.A.; Marrink, S.J. Backmapping triangulated surfaces to coarse-grained membrane models. Nat. Commun. 2020, 11, 2296–2305. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezeshkian, W.; Shillcock, J.C.; Ipsen, J.H. Computational Approaches to Explore Bacterial Toxin Entry into the Host Cell. Toxins 2021, 13, 449. https://doi.org/10.3390/toxins13070449
Pezeshkian W, Shillcock JC, Ipsen JH. Computational Approaches to Explore Bacterial Toxin Entry into the Host Cell. Toxins. 2021; 13(7):449. https://doi.org/10.3390/toxins13070449
Chicago/Turabian StylePezeshkian, Weria, Julian C. Shillcock, and John H. Ipsen. 2021. "Computational Approaches to Explore Bacterial Toxin Entry into the Host Cell" Toxins 13, no. 7: 449. https://doi.org/10.3390/toxins13070449
APA StylePezeshkian, W., Shillcock, J. C., & Ipsen, J. H. (2021). Computational Approaches to Explore Bacterial Toxin Entry into the Host Cell. Toxins, 13(7), 449. https://doi.org/10.3390/toxins13070449