The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium


Abstract
1. Introduction
2. Results
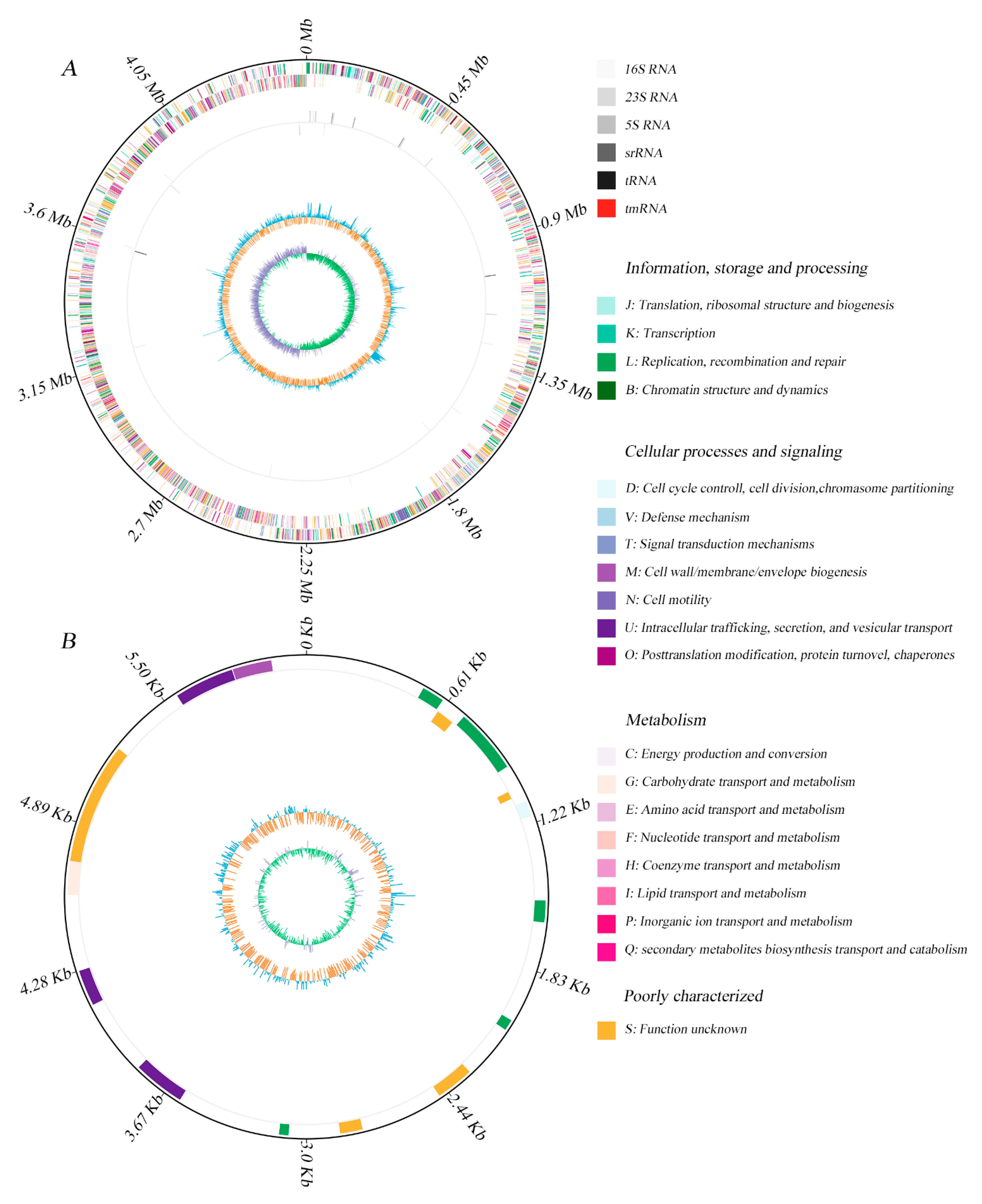
2.1. General Genome Features of Bacillus sp. 1839
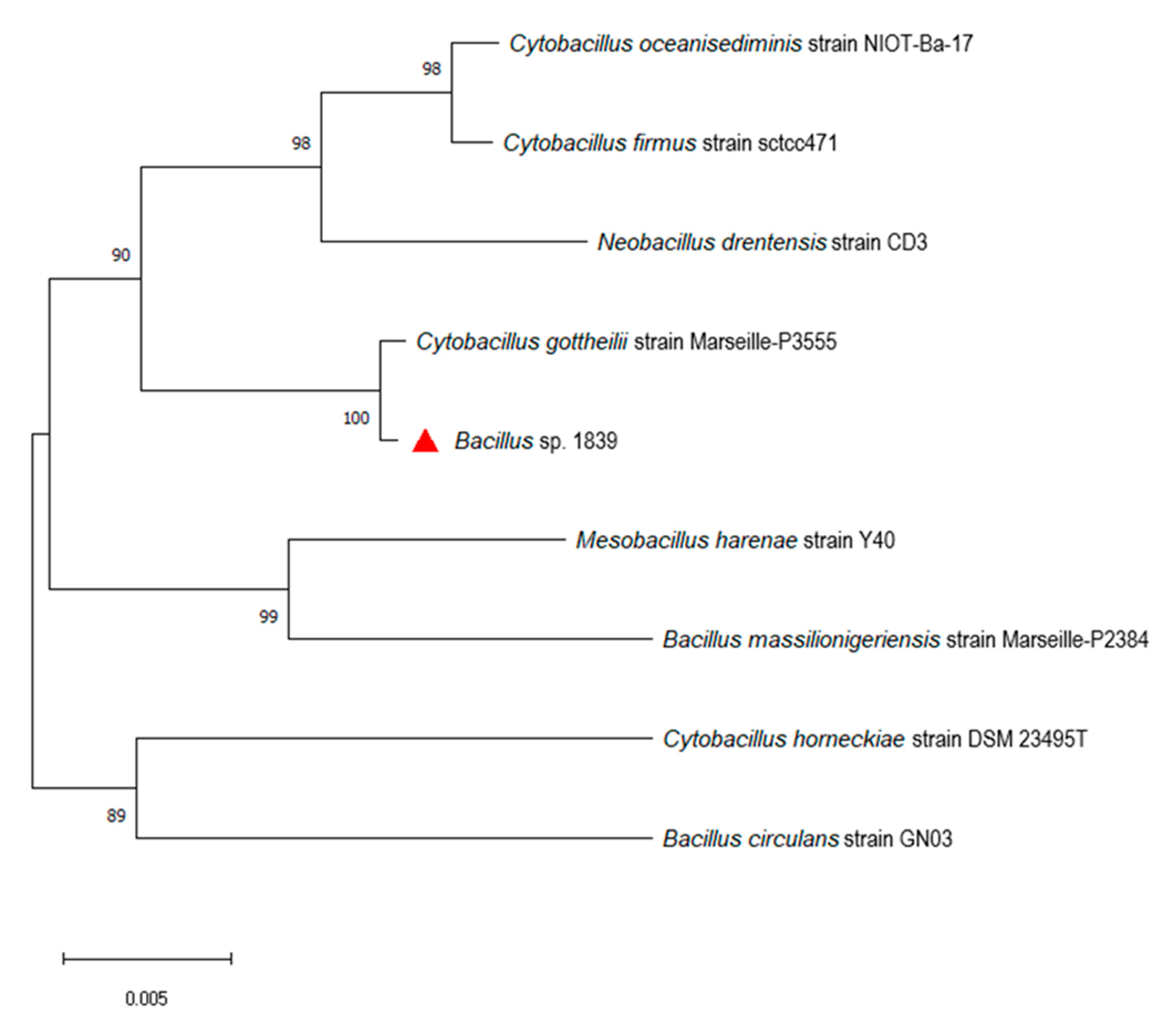
2.2. Phylogenetic Analysis and Genome Similarity Measures
3. Discussion
4. Materials and Methods
4.1. DNA Extraction
4.2. Genome Sequencing
4.3. Genome Assembly
4.4. Genome Annotation
4.5. Phylogenetic Analysis and Genome Similarity Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef]
- Melnikova, D.I.; Khotimchenko, Y.S.; Magarlamov, T.Y. Addressing the issue of tetrodotoxin targeting. Mar. Drugs 2018, 16, 352. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, F.; Lu, Y.; Ma, T.; Zhao, J.; Gong, X.; Bao, B. Production level of tetrodotoxin in Aeromonas is associated with the copy number of a plasmid. Toxicon 2015, 101, 27–34. [Google Scholar] [CrossRef]
- Pratheepa, V.; Alex, A.; Silva, M.; Vasconcelos, V. Bacterial diversity and tetrodotoxin analysis in the viscera of the gastropods from Portuguese coast. Toxicon 2016, 119, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Beleneva, I.A.; Magarlamov, T.Y.; Kukhlevskii, A.D. Characterization, identification, and screening for tetrodotoxin production by bacteria associated with the Cephalotrix simula (Ivata, 1952) proboscis worm. Mikrobiologiia 2014, 83, 312–319. [Google Scholar] [CrossRef]
- Magarlamov, T.Y.; Beleneva, I.A.; Chernyshev, A.V.; Kuhlevsky, A.D. Tetrodotoxin-producing Bacillus sp. from the ribbon worm (Nemertea) Cephalothrix simula (Iwata, 1952). Toxicon 2014, 85, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Shokur, O.A.; Magarlamov, T.Y.; Melnikova, D.I.; Gorobets, E.A.; Beleneva, I.A. Life cycle of tetrodotoxin producing Bacillus sp. on solid and liquid medium: Light and electron microscopy studies. Russ. J. Mar. Biol. 2016, 42, 252–257. [Google Scholar] [CrossRef]
- Magarlamov, T.Y.; Melnikova, D.I.; Shokur, O.A.; Gorobets, E.A. Rapid production of tetrodotoxin-like compounds during sporulation in a marine isolate Bacillus sp. 1839. Microbiology 2017, 86, 192–196. [Google Scholar] [CrossRef]
- Melnikova, D.I.; Vlasenko, A.E.; Magarlamov, T.Y. Stable tetrodotoxin production by Bacillus sp. strain 1839. Mar. Drugs 2019, 17, 704. [Google Scholar] [CrossRef]
- Chau, R.; Kalaitzis, J.A.; Neilan, B.A. On the origins and biosynthesis of tetrodotoxin. Aquat. Toxicol. 2011, 104, 61–72. [Google Scholar] [CrossRef]
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. J. Comput. Biol. 2013, 10, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Galardini, M.; Biondi, E.G.; Bazzicalupo, M.; Mengoni, A. CONTIGuator: A bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNammer: Consistent annotation of ribosomal RNA genes. Nucl. Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Pérochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucl. Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucl. Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucl. Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucl. Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucl. Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucl. Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Feature | Value | |
|---|---|---|
| Chromosome | Plasmid | |
| Genome size (bp) | 4,523,455 | 61,233 |
| G + C content (mol %) | 39 | 34 |
| Genes number (total) | 4458 | 69 |
| Coding sequences total number | 4339 | 69 |
| Coding sequences with protein number | 4300 | 69 |
| Protein-coding genes number | 4300 | 69 |
| rRNAs number | 11, 10, 10 (5S, 16S, 23S) | 0 |
| tRNAs number | 83 | 0 |
| ncRNAs number | 5 | 0 |
| Pseudogenes number | 39 | 0 |
| Insertion sequences number | 92 | 0 |
| Genomic islands number | 9 | 0 |
| Clustered regularly interspaced short palindromic repeats number | 2 | 0 |
| Prophage number | 4 | 0 |
| Code | Value | % 1 | Function Description |
|---|---|---|---|
| INFORMATION STORAGE AND PROCESSING | |||
| B | 1 | 0.02 | Chromatin structure and dynamics |
| J | 171 | 3.9 | Translation, ribosomal structure, and biogenesis |
| K | 269 | 6.2 | Transcription |
| L | 197 | 4.5 | Replication, recombination, and repair |
| METABOLISM | |||
| C | 189 | 4.3 | Energy production and conversion |
| E | 255 | 5.8 | Amino acid transport and metabolism |
| F | 88 | 2 | Nucleotide transport and metabolism |
| G | 213 | 4.9 | Carbohydrate transport and metabolism |
| H | 100 | 2.3 | Coenzyme transport and metabolism |
| I | 88 | 2 | Lipid transport and metabolism |
| P | 248 | 5.7 | Inorganic ion transport and metabolism |
| Q | 29 | 0.7 | Secondary metabolites biosynthesis, transport, and catabolism |
| CELLULAR PROCESSES AND SIGNALING | |||
| D | 36 | 0,8 | Cell cycle control, Cell division, and chromosome partitioning |
| M | 170 | 3.9 | Cell wall/membrane/envelope biogenesis |
| N | 43 | 1 | Cell motility |
| O | 113 | 2.6 | Posttranslational modification, protein turnover, chaperones |
| T | 191 | 4.4 | Signal transduction mechanisms |
| U | 38 | 0.9 | Intracellular trafficking, secretion, and vesicular transport |
| V | 75 | 1.7 | Defense mechanisms |
| POORLY CHARACTERIZED | |||
| S | 994 | 22.8 | Function unknown |
| - | 848 | 19.5 | Not in COGs |
| Type | From (bp) | To (bp) | Most Similar Known Cluster | % Similarity | Accession Number |
|---|---|---|---|---|---|
| Terpene | 348,477 | 367,415 | Cytobacillus gottheilii strain FJAT-2394 | 88 | NZ_KV440945 |
| Thiopeptide-Linear azol(in)e-containing peptides | 447,791 | 476,894 | 82 | ||
| Terpene | 973,108 | 1,001,908 | 89 | ||
| Type III polyketide synthase cluster | 2,777,409 | 2,818,491 | 100 |
| Organism | NCBI Accession No. | Assembly Level | Size (bp) | GC (%) | Predicted Coding Sequences | No. of Genes | No. of Proteins | No. of RNAs |
|---|---|---|---|---|---|---|---|---|
| Cytobacillus gottheilii Marseille-P3555 | NZ_FUVC00000000.1 | Scaffold | 4,719,939 | 39 | 4492 | 4621 | 4452 | 129 |
| Cytobacillus gottheilii FJAT-2394 | NZ_KV440945 | Genome | 4,584,535 | 39 | 4370 | 4475 | 4310 | 105 |
| Bacillus sp. 1839 | Cytobacillus gottheilii Marseille-P3555 | Cytobacillus gottheilii FJAT-2394 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ANIb | Aligned | Tetra | ANIb | Aligned | Tetra | ANIb | Aligned | Tetra | |
| Bacillus sp. 1839 | * | 97.55 | 85.95 | 0.99891 | 97.55 | 85.95 | 0.99892 | ||
| Cytobacillusgottheilii Marseille-P3555 | 97.32 | 86.16 | 0.99891 | * | 100.00 | 99.49 | 1.0 | ||
| Cytobacillus gottheilii FJAT-2394 | 97.34 | 86.18 | 0.99892 | 100.00 | 99.47 | 1.0 | * | ||
| Query Genome | Reference Genome | Formula 1 | Formula 2 | Formula 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DDH (%) | Model C.I. (%) | Distance | DDH (%) | Model C.I. (%) | Distance | DDH (%) | Model C.I. (%) | Distance | ||
| Bacillus sp. 1839 | Cytobacillus gottheilii Marseille-P3555 | 83.7 | 79.8–86.9 | 0.1140 | 79.6 | 76.6–82.2 | 0.0239 | 85.9 | 82.8–88.5 | 0.1351 |
| Bacillus sp. 1839 | Cytobacillus gottheilii JAT-2394 | 83.7 | 79.9–86.9 | 0.1137 | 79.6 | 76.6–82.2 | 0.0239 | 85.9 | 82.8–88.6 | 0.1349 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnikova, D.I.; Nijland, R.; Magarlamov, T.Y. The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium. Toxins 2021, 13, 410. https://doi.org/10.3390/toxins13060410
Melnikova DI, Nijland R, Magarlamov TY. The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium. Toxins. 2021; 13(6):410. https://doi.org/10.3390/toxins13060410
Chicago/Turabian StyleMelnikova, Daria I., Reindert Nijland, and Timur Yu. Magarlamov. 2021. "The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium" Toxins 13, no. 6: 410. https://doi.org/10.3390/toxins13060410
APA StyleMelnikova, D. I., Nijland, R., & Magarlamov, T. Y. (2021). The First Data on the Complete Genome of a Tetrodotoxin-Producing Bacterium. Toxins, 13(6), 410. https://doi.org/10.3390/toxins13060410