Analytical Separation of Carcinogenic and Genotoxic Alkenylbenzenes in Foods and Related Products (2010–2020)


Abstract
1. Introduction
2. Gas Chromatography
2.1. Analysis

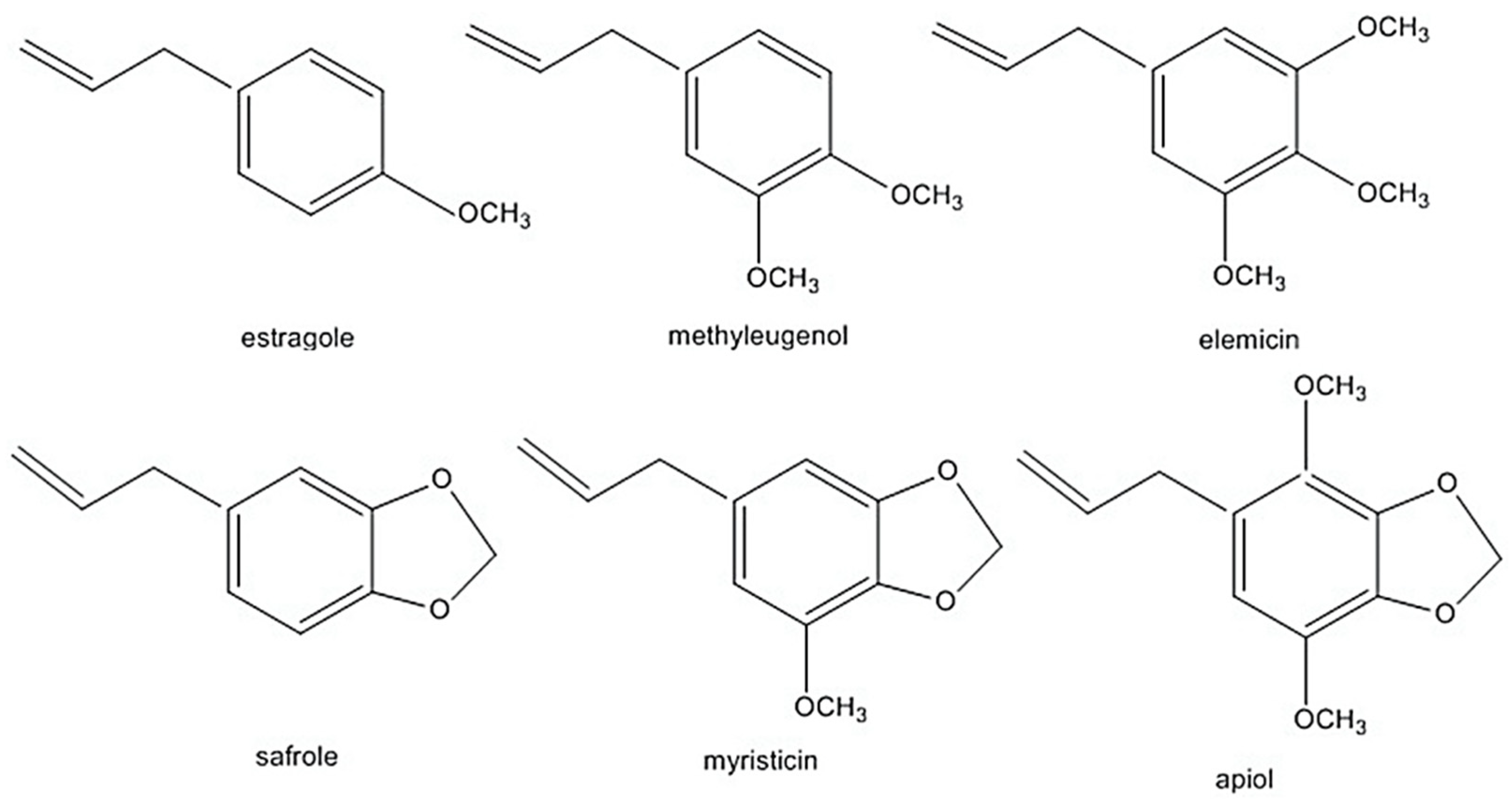{kind=link}
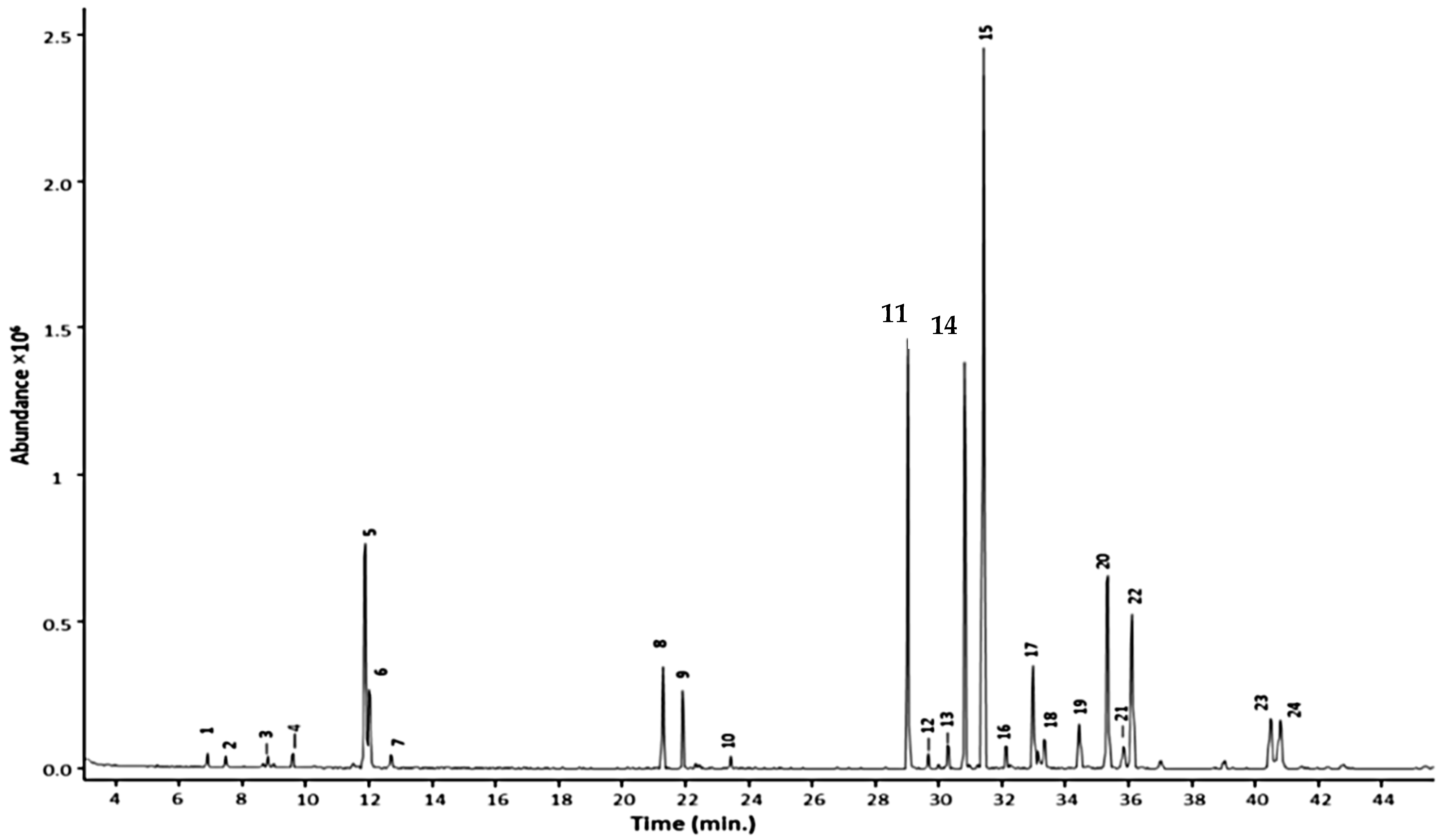{kind=link}
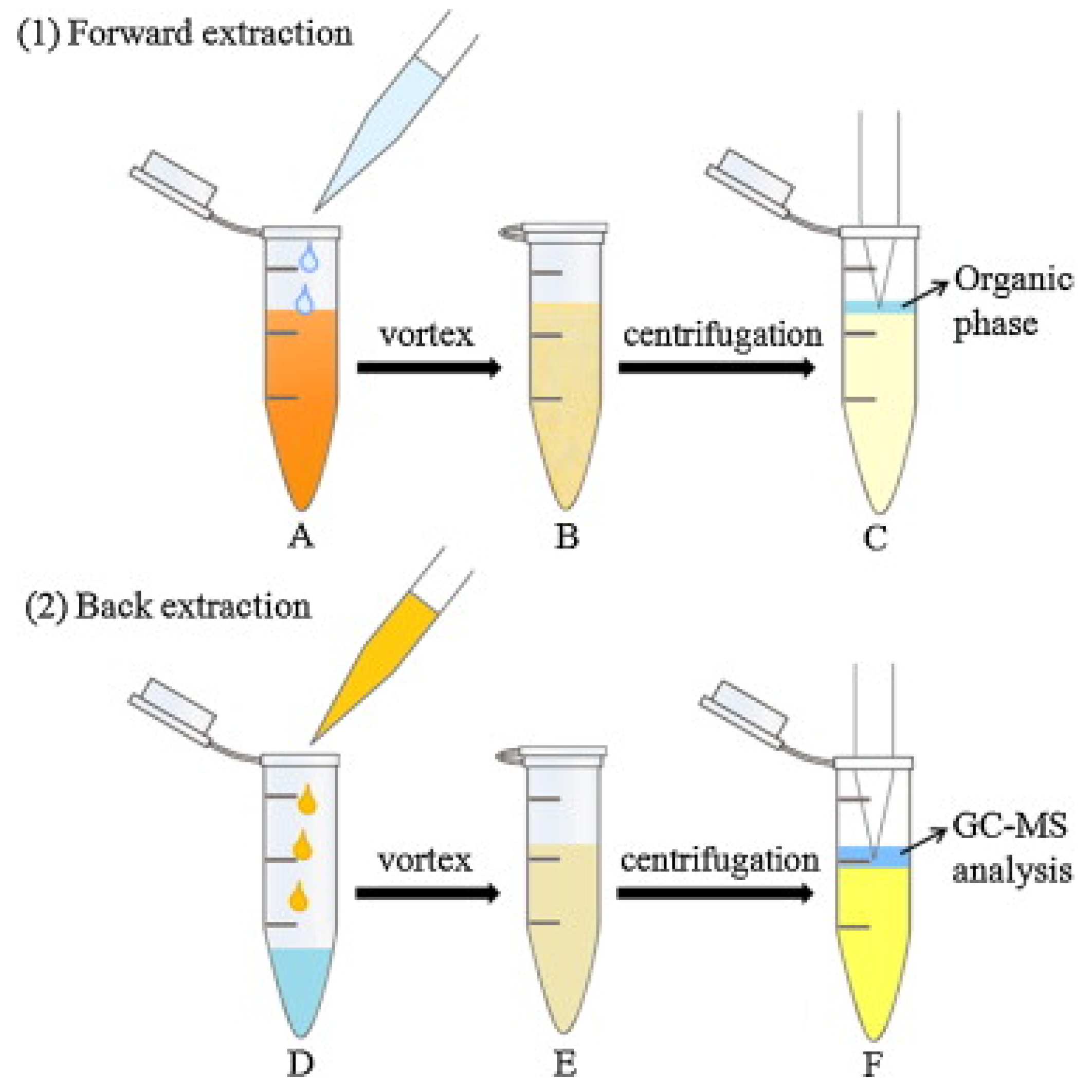{kind=link}
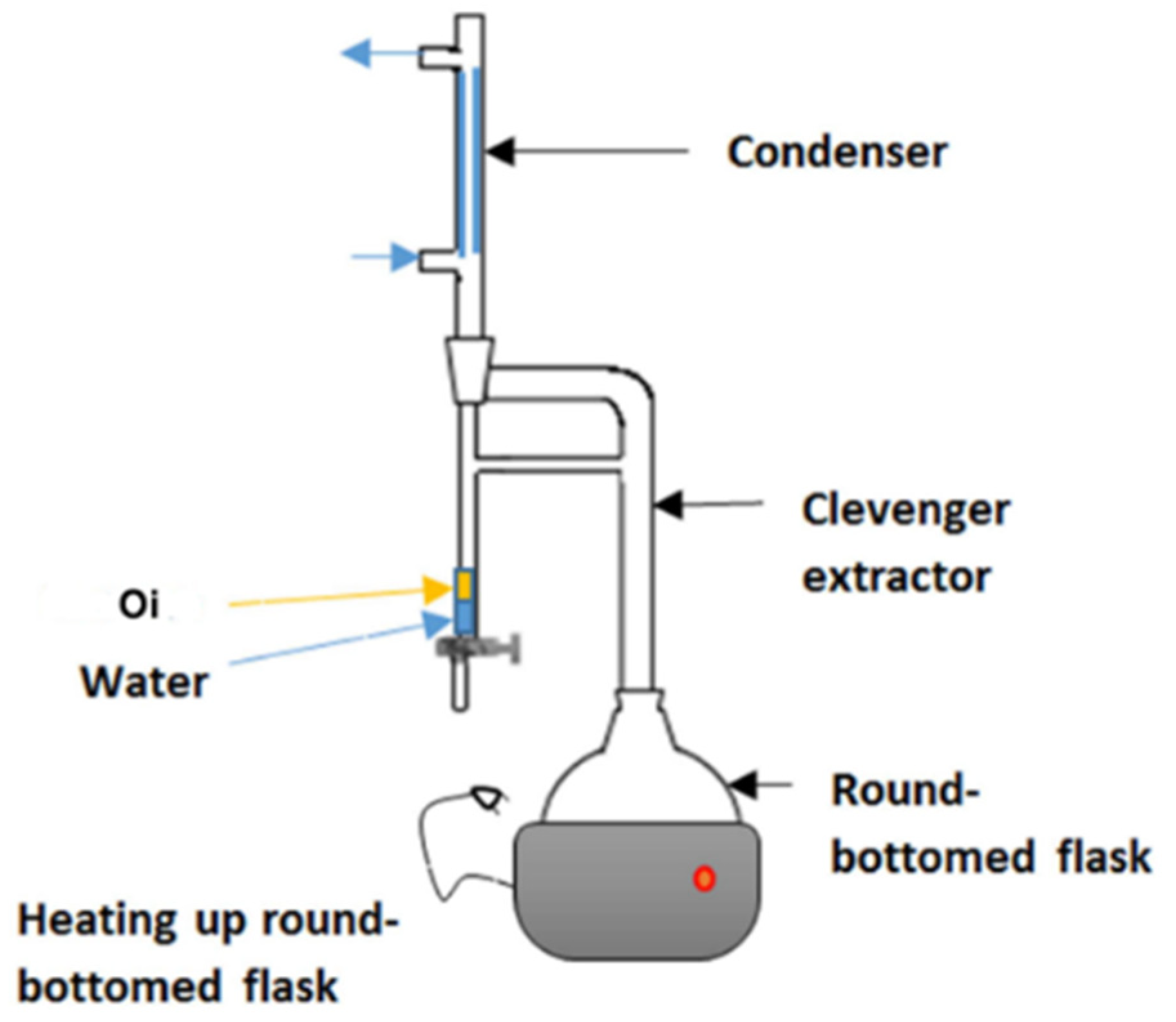{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkenylbenzenes | Sample/s | Sample Preparation | Amount of Alkenylbenzenes Found in Sample/s | GC Conditions | Detector | Ref. |
|---|---|---|---|---|---|---|
| myristicin elemicine cis-isosafrole borneol caryophyllene | Chinese Ainsliacea fragrans Champ ex Benth | hydrodistillation | myristicin (41.3%) elemicine (11.9%) cis-isosafrole (11.5%) borneol (9.1%) caryophyllene (8.8%) - % values were the % found in the extracted essential oil - the essential oil was 0.06% v/w of sample | column:HP-5, 5% diphenyl and 95% dimethylpolysiloxane, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 60 °C hold for 1 min, ramp to 180 °C (10 °C/min), hold for 1 min, ramp to 280 °C (20 °C/min), hold for 15 min. carrier gas:helium, 1 mL/min injection:split, 1:10, 1 µL, 270 °C run time: 34.0 min LOD: not reported LOQ: not reported | FID | [22] |
| safrole apiol myristicin | essential oils from Vietnam | hydrodistillation | safrole (38.1%) apiol (10.8%) myristicin (8.0%) - % values were the % found in the extracted essential oil - the essential oil was 0.2% v/w of sample | column: SE-52 capillary column, 50 m × 0.25 mm × 1.0 µm program: 60 °C for 1 min, heating to 230 °C (3 °C/min), hold for 12.3 min carrier gas: helium, 1.5 mL/min injection:split/splitless, 1:100, 0.1 µL, 230 °C run time: 70.0 min LOD:not reported LOQ:not reported | FID | [23] |
| eugenol methyleugenol | holy basil essential oils | hydrodistillation | eugenol (37–45%) methyleugenol (65%) - % values were the % found in the extracted essential oil - the essential oil was 7.9 ± 3.2 mg/g of sample | column:HP-5 fused silica capillary, 5% phenyl methylpolysiloxane, 30 m × 0.32 mm, 0.25 µm program:initial temperature 50 °C held for 5 min, increase to 120 °C (3 °C/min), to 250 °C (5 °C/min), to 300 °C (15 °C/min) and hold for 5 min. carrier gas:helium, 25 mL/min injection: split, 50:1, 250 °C run time: 62.6 min LOD: 0.21 µg/mL LOQ: 0.54 µg/mL | FID | [18] |
| MS for identification only | column:HP-5 MS fused-silica capillary column, 5% phenyl methylpolysiloxane, 30 m × 0.25 mm i.d × 0.25 µm program:50 °C for 5 min, increase to 120 °C (3 °C/min), to 250 °C (5 °C/min), hold for 0.67 min. carrier gas:helium, 25 mL/min injection:split, 50:1, 1 µL, 250 °C run time:55.0 min LOD:not reported LOQ:not reported | MS | ||||
| eugenol estragole | Ocimum species | hydrodistillation | eugenol (566.8 ± 98.0 mg/g–859.3 ± 151.3 mg/g) estragole (448.8 ± 126.8 mg/g–640.2 ± 44.8 mg/g) | column: RTX-5 column, methylpolysiloxane, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 70 °C, heating ramp up to 180 °C (4 °C/min), ramp at 250 °C (10 °C/min) carrier gas:nitrogen, 1 mL/min injection:split, 1:30, 1 µL, 250 °C run time:34.5 min LOD:1.2 µg/mL LOQ:not reported | FID | [17] |
| MS was used for identification only | column:HP-5MS column, methylpolysiloxane, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 70 °C, heating ramp up to 180 °C (4 °C/min), ramp at 250 °C (10 °C/min) carrier gas: helium, 1 mL/min injection: split, 1:100, 1 µL, 250 °C run time: 34.5 min LOD: not reported LOQ: not reported | MS | ||||
| 16 alkenylbenzenes | essential oils | not described | different % values of alkenylbenzenes in 23 essential oils | column: capillary column 30 m × 0.32 mm × 0.25 µm program:hold at 40 °C for 2 min, increase from 40 °C to 200 °C (10 °C/min), to 300 °C (20 °C/min). carrier gas: nitrogen, 2.1 mL/min injection:split, 1:50, 300 °C run time: 31.0 min LOD:not reported LOQ:not reported | FID | [19] |
| human serum | not described | eugenol (222 ± 34 ng/mL), geraniol (6.18 ± 0.67 ng/mL), methyleugenol (0.74 ± 0.08 ng/mL), cis-isoeugenol (1.87 ± 0.69 ng/mL), acetyl eugenol (30.2 ± 11 ng/mL), myristicin (12.8 ± 1.6 ng/mL) after administration of clove essential oil cream | ||||
| essential oils and human serum | not described | MS was used for identification only | column:DB-5 fused silica capillary column, 5% phenyl methylpolysiloxane, 30 m × 0.25 mm i.d × 0.25 µm program:increase from 45 °C to 250 °C (5 °C/min) carrier gas:helium, 0.82 mL/min injection: not reported run time:41 min LOD: not reported LOQ: not reported | MS | ||
| methyleugenol | Cymbo-pogon khasia-nus Hack. | hydrodistillation | methyleugenol (73.2%) - % values were the % found in the extracted essential oil - the essential oil was 0.73% v/w of sample | column: HP-5 fused silica capillary, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 40 °C, hold for 2 min, increase to 250 °C (5 °C/min), to 300 °C (30 °C/min), hold for 10 min carrier gas: helium, 1 mL/min injection:split, 1:20, 1 µL, 250 °C run time:55.7 min LOD:not reported LOQ:not reported | MS | [24] |
| methyleugenol | Melaleuca alternifolia oils | solvent dilution | methyleugenol (160.0 µg/mL–552.0 µg/mL) | column: Varian Factor Four VF-5, 30 m × 0.25 mm × 0.25 µm program: 130 °C to 180 °C (15 °C/min), increase to 230 °C (30 °C/min), hold for 4 min carrier gas:helium, 1.2 mL/min injection:split, 7:1, 1 µL, 240 °C run time:9.0 min LOD: 150 ppb LOQ: 500 ppb | MS | [25] |
| anethole estragole eugenol methyleugenol safrole myristicin | aroma-therapy massage oil products | dispersive liquid-liquid microextraction (DLLME), dual DLLME | anethole (up to 862.1 µg/g) estragole (up to 0.7 µg/g) eugenol (0.5 µg/g–851.5 µg/g) methyleugenol (0.1 µg/g–0.5 µg/g) safrole (up to 0.2 µg/g) myristicin (up to 0.7 µg/g) | column:VF-5MS fused silica capillary column, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 90 °C for 1 min, ramp to 130 °C (40 °C/min), ramp to 137 °C (3.5 °C/min), ramp to 139.4 °C (0.3 °C/min), ramp to 280 °C (70 °C/min), hold for the remaining time carrier gas: helium, 2 mL/min injection: split, 10:1, 1 µL, 260 °C run time: 16.0 min LOD: 1.0–3.0 ng/mL LOQ: 2.5–10.0 ng/mL | MS | [26] |
| eugenol methyleugenol | Ocimum micran-thum | hydrodistillation | eugenol (12%) methyleugenol (14%) of the total area in the chromatogram of the distillate | column: ZB-5HT INFERNO, 5% phenyl 95% polydimethylsiloxane, 30 m × 0.25 mm i.d × 0.25 µm program: 60 °C to 68 °C (0.7 °C/min), hold for 7 min, increase to 100 °C (10 °C/min), to 130 °C (5 °C/min), hold for 7 min, to 135 °C (1 °C/min) and hold for 6 min carrier gas:helium, 1 mL/min injection:split, 1:100, 1 µL, 280 °C run time: 45.6 min LOD: not reported LOQ:not reported | MS | [21] |
| estragole eugenol | basil species and pot cultures | steam distillation | estragole (2.3 mg/mL in Lettuce Leaf) eugenol (1.2 mg/mL in Mammolo Genovese, 0.4 mg/mL in Manes) | column: DB-WAX, 30 m × 0.25 mm × 0.25 µm program: 40 °C for 3 min, increase to 60 °C (8 °C/min), to 70 °C (5 °C/min), to 230 °C (4 °C/min), keep for constant for 1 min. carrier gas: helium, 0.5 mL/min injection:split, 1:100, 1 µL, 240 °C run time:48.5 min LOD:0.0085 mg/mL (estragole), 0.0063 mg/mL (eugenol) LOQ:0.0118 mg/mL (estragole), 0.0066 mg/mL (eugenol) | MS | [27] |
| estragole methyleugenol safrole | food and beverage samples | QuEChERS | estragole (0.7 mg/kg–5.2 mg/kg in fish samples) methyleugenol (0.6 mg/kg–3.3 mg/kg in bakery, meat, dairy and vegetable samples) safrole (up to 2.4 mg/kg in butter with spices sample) | column: REStek Rtx®- CLPesticides, 30 m × 0.25 mm × 0.25 µm program: 60 °C for 1 min, increase to 80 °C (50 °C/min), to 125 °C (3 °C/min), to 300 °C (10 °C/min), hold for 5 min carrier gas: helium, 1 mL/min injection: splitless, 1 µL, 250 °C run time: 38.9 min LOD:not reported LOQ: 0.05 mg/kg (non-alcoholic beverages), 0.5 mg/kg (other food matrices) | MS | [28] |
| methyleugenol estragole | Anthriscus cerefolium L. Hoffm | hydrodistillation | methyleugenol (47.2% in El-Sharkia essential oil) El-Sharkia essential oil is 0.075–0.083 mL/plant estragole (18.0% in El-Fayoum essential oil) El-Fayoum essential oil is 0.12–0.16 mL/plant | column: TG-WAX, 30 m × 0.25 mm × 0.25 µm program: 40 °C for 1 min, increase to 160 °C (4 °C/min), hold for 6 min, increase to 210 °C (6 °C/min), hold for 1 min. carrier gas: helium, 1 mL/min injection: split, 1:10, 0.2 µL, 210 °C run time: 46.3 min LOD: not reported LOQ: not reported | MS | [29] |
| eugenol isoeugenol methyleugenol | fish fillet | solvent extraction and solid phase extraction (SPE) | eugenol (259.0 µg/kg–2329.0 µg/kg) isoeugenol (86.2 µg/kg–1032.0 µg/kg) methyleugenol (not found) | column:DB-17 capillary column, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 80 °C, hold for 2 min, increase to 220 °C (25 °C/min) and hold for 1 min, increase to 280 °C (30 °C/min), hold for 1 min carrier gas: helium, 2 mL/min injection:split, 1 µL, 260 °C run time:11.6 min LOD: 0.4 µg/kg (eugenol), 1.2 µg/kg (isoeugenol), 0.2 µg/kg (methyleugenol) LOQ: 1.2 µg/kg (eugenol), 4 µg/kg (isoeugenol), 0.7 µg/kg (methyleugenol) | MS | [30] |
| methyleugenol | food samples | QuEChERS | methyleugenol (6.1 ± 0.4 mg/kg) | column: DB-1 capillary column, 30 m × 0.25 mm × 0.25 µm program: 70 °C for 1 min, increase to 120 °C (40 °C/min), to 180 °C (8 °C/min), hold for 1 min, to 280 °C (40 °C/min) and hold for 1 min carrier gas: not reported injection:splitless, 2 µL, 280 °C run time: 14.3 min LOD:20 µg/kg (solid/semi-solid food samples), 0.4 µg/kg (beverages) LOQ: 50 µg/kg (solid/semi-solid food samples), 1 µg/kg (beverages) | MS | [31] |
| methyleugenol estragole | food samples | liquid-liquid extraction | methyleugenol (4288.0 mg/kg for allspice pimento, 1351.0 mg/kg for nutmeg and n.d–1184.0 mg/kg for basil) estragole (not reported) | column: HP-Innowax, fused silica capillary column, 41 m × 0.25 mm × 0.25 µm program: 40 °C for 1 min, increase to 200 °C (8 °C/min), hold for 5 min carrier gas: helium, 1 mL/min injection:split, 1:5, 1 µL, 240 °C run time: 26.0 min LOD: 2.1 mg/L (methyleugenol), 1.3 mg/L (estragole) LOQ: 5.3 mg/L (methyleugenol), 4.7 mg/L (estragole) | MS | [32] |
| estragole tr-anethole safrole eugenol tr-iso-eugenol acetyl eugenol methyleugenol myristicin | pepper and its varieties | ultrasound-assisted extraction | estragole (2.2 mg/kg–45.7 mg/kg) tr-anethole (10.7 mg/kg–42.7 mg/kg) safrole (0.2 mg/kg–3.0 mg/kg) eugenol (10.5 mg/kg–120.0 mg/kg) tr-iso-eugenol (0.7 mg/kg–3.6 mg/kg) acetyl eugenol (45.8 mg/kg in red pepper) methyleugenol (0.5 mg/kg–20.1 mg/kg) myristicin (0.2 mg/kg–6.1 mg/kg) | column: BP5MS capillary analytical, 30 m × 0.25 mm × 0.25 µm program:initial temperature at 70 °C (3 min), increase from 70 °C to 250 °C (10 °C/min) then increase to 280 °C (50 °C/min) and maintain for 3 min carrier gas: helium, 1 mL/min injection:splitless, 2 µL, 280 °C run time: 24.6 min LOD:0.02 mg/kg (estragole), 0.02 mg/kg (trans-anethole), 0.01 mg/kg (safrole), 0.01 mg/kg (eugenol), 0.01 (trans-iso-eugenol), 0.01 mg/kg (acetyl eugenol), 0.01 mg/kg (methyleugenol), 0.01 mg/kg (myristicin) LOQ:0.2 mg/kg (estragole), 0.2 mg/kg (trans-anethole), 0.2 mg/kg (safrole), 0.2 mg/kg (eugenol), 0.2 (trans-iso-eugenol), 0.2 mg/kg (acetyl eugenol), 0.2 mg/kg (methyleugenol), 0.2 mg/kg (myristicin) | HRMS-Q-Orbitrap | [20] |
2.2. Sample Preparation
2.2.1. Solvent Dilution/Extraction
2.2.2. LLE
2.2.3. LPME
2.2.4. QuEChERS
2.2.5. SPE
2.2.6. Distillation
3. HPLC
3.1. Analysis
3.2. Sample Preparation
Solvent Extraction
4. Capillary Electrophoresis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, D.; Pimentel, M. Sustainability of meat-based and plant-based diets and the environment. Am. J. Clin. Nutr. 2003, 78, 660S–663S. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, I.M.C.M.; Martena, M.J.; Boersma, M.G.; Spiegelenberg, W.; Alink, G.M. Molecular mechanisms of toxicity of important food-borne phytotoxins. Mol. Nutr. Food Res. 2005, 49, 131–158. [Google Scholar] [CrossRef] [PubMed]
- Monien, B.H.; Sachse, B.; Niederwieser, B.; Abraham, K. Detection of N-Acetyl-S-[3′-(4-methoxyphenyl)allyl]-l-Cys (AMPAC) in human urine samples after controlled exposure to fennel tea: A new metabolite of estragole and trans-anethole. Chem. Res. Toxicol. 2019, 32, 2260–2267. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, R.; Herrmann, K.; Engst, W.; Meinl, W.; Klein, K.; Glatt, H.; Zanger, U.M. Methyleugenol DNA adducts in human liver are associated with SULT1A1 copy number variations and expression levels. Arch. Toxicol. 2017, 91, 3329–3339. [Google Scholar] [CrossRef] [PubMed]
- Sangster, S.A.; Caldwell, J.; Hutt, A.J.; Anthony, A.; Smith, R.L. The metabolic disposition of [methoxy- 14 C]-labelled trans -anethole, estragole and p -propylanisole in human volunteers. Xenobiotica 1987, 17, 1223–1232. [Google Scholar] [CrossRef]
- Choong, Y.M.; Lin, H.J. A rapid and simple gas chromatographic method for direct determination of safrole in soft drinks. J. Food Drug Anal. 2001, 9, 27–32. [Google Scholar] [CrossRef]
- Raffo, A.; D’Aloise, A.; Magrì, A.L.; Leclercq, C. Quantitation of tr-cinnamaldehyde, safrole and myristicin in cola-flavoured soft drinks to improve the assessment of their dietary exposure. Food Chem. Toxicol. 2013, 59, 626–635. [Google Scholar] [CrossRef]
- European Commission Health & Consumer Protection Directorate-General. Opinion of the Scientific Committee on Food on Estragole (1-Allyl-4-methoxybenzene); Scientific Committee on Food: Brussel, Belgium, 2001. [Google Scholar]
- European Commission Health & Consumer Protection Directorate-General. Opinion of the Scientific Committee on Food on Methyleugenol 4-Allyl-1, 2-Dimethoxybenzene; Scientific Committee on Food: Brussel, Belgium, 2001. [Google Scholar]
- European Commission Health & Consumer Protection Directorate-General. Opinion of the Scientific Committee on Food on the Safety of the Presence of Safrole (1-allyl-3, 4-Methylene Dioxy Benzene) in Flavourings and other Food Ingredients with Flavouring Properties; Scientific Committee on Food: Brussel, Belgium, 2002. [Google Scholar]
- Auerbach, S.S.; Shah, R.R.; Mav, D.; Smith, C.S.; Walker, N.J.; Vallant, M.K.; Boorman, G.A.; Irwin, R.D. Predicting the hepatocarcinogenic potential of alkenylbenzene flavoring agents using toxicogenomics and machine learning. Toxicol. Appl. Pharmacol. 2010, 243, 300–314. [Google Scholar] [CrossRef]
- Suparmi, S.; Widiastuti, D.; Wesseling, S.; Rietjens, I.M.C.M. Natural occurrence of genotoxic and carcinogenic alkenylbenzenes in Indonesian jamu and evaluation of consumer risks. Food Chem. Toxicol. 2018, 118, 53–67. [Google Scholar] [CrossRef]
- McNair, H.M.; Miller, J.M.; Snow, N.H. Basic Gas Chromatography; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Stauffer, E.; Dolan, J.A.; Newman, R. Chapter 8—Gas chromatography and gas chromatography—Mass spectrometry. In Fire Debris Analysis; Academic Press: Burlington, VT, USA, 2008; pp. 235–293. [Google Scholar]
- Zhao, J.; Lv, G.-P.; Chen, Y.-W.; Li, S.-P. Advanced development in analysis of phytochemicals from medicine and food dual purposes plants used in China. J. Chromatogr. A 2011, 1218, 7453–7475. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.V.B.; Alves Filho, E.G.; Silva, L.M.A.; Zocolo, G.J.; de Brito, E.S.; Gramosa, N.V. Chemometric analysis of NMR and GC datasets for chemotype characterization of essential oils from different species of Ocimum. Talanta 2018, 180, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Joy, N.; Berle, D.; Affolter, J.; Pegg, R.B. Employing predicted response factors and a validated chromatographic method for the relative quantitation of holy basil essential oils. J. Essent. Oil Res. 2020, 32, 407–418. [Google Scholar] [CrossRef]
- Wang, L.H.; Wang, C.C.; Chuang, S.K. Simultaneous determination of alkenyl benzenes in essential oils and human serum by gas chromatrography and GC-MS. Asian J. Chem. 2010, 22, 3835–3842. [Google Scholar]
- Rivera-Pérez, A.; López-Ruiz, R.; Romero-González, R.; Garrido Frenich, A. A new strategy based on gas chromatography–high resolution mass spectrometry (GC–HRMS-Q-Orbitrap) for the determination of alkenylbenzenes in pepper and its varieties. Food Chem. 2020, 321, 126727. [Google Scholar] [CrossRef]
- Caamal-Herrera, I.O.; Muñoz-Rodríguez, D.; Madera-Santana, T.; Azamar-Barrios, J.A. Identification of volatile compounds in essential oil and extracts of ocimum micranthum willd leaves using GC/MS. Int. J. Appl. Res. Nat. Prod. 2016, 9, 31–40. [Google Scholar]
- Zhao, M.P.; Liu, X.C.; Liu, Q.Z.; Liu, Z.L. Gas chromaotography-mass spectrometry analysis of insecticidal essential oil derived from Chinese Ainsliaea fragrans Champ ex Benth (Compositae). Trop. J. Pharm. Res. 2015, 14, 1685–1689. [Google Scholar] [CrossRef]
- Schmidt, E.; Huong, L.T.; Dai, D.N.; Thang, T.D.; Wanner, J.; Jirovetz, L. Analysis and olfactory description of four essential oils from Vietnam. Nat. Prod. Commun. 2016, 11, 1551–1554. [Google Scholar] [CrossRef]
- Gogoi, R.; Loying, R.; Sarma, N.; Begum, T.; Pandey, S.K.; Lal, M. Comparative analysis of in-vitro biological activities of methyl eugenol rich Cymbopogon khasianus hack., leaf essential oil with pure methyl eugenol compound. Curr. Pharm. Biotechnol. 2020, 21, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.A.; Davies, N.W.; Larkman, T. GC-MS method validation and levels of methyl eugenol in a diverse range of tea tree (Melaleuca alternifolia) oils. Anal. Bioanal. Chem. 2017, 409, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Li, J.H.; Feng, C.H. Dual dispersive liquid-liquid microextraction for determination of phenylpropenes in oils by gas chromatography-mass spectrometry. J. Chromatogr. A 2015, 1410, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Muráriková, A.; Ťažký, A.; Neugebauerová, J.; Planková, A.; Jampílek, J.; Mučaji, P.; Mikuš, P. Characterization of essential oil composition in different basil species and pot cultures by a GC-MS Method. Molecules 2017, 22, 1221. [Google Scholar] [CrossRef]
- Lopez, P.; Van Sisseren, M.; De Marco, S.; Jekel, A.; De Nijs, M.; Mol, H.G.J. A straightforward method to determine flavouring substances in food by GC-MS. Food Chem. 2015, 174, 407–416. [Google Scholar] [CrossRef]
- Hendawy, S.F.; Hussein, M.S.; El-Gohary, A.E.; Soliman, W.S. Chemical constituents of essential oil in chervil (Anthriscus cerefolium L. Hoffm.) cultivated in different locations. J. Essent. Oil-Bear. Plants 2019, 22, 264–272. [Google Scholar] [CrossRef]
- Ke, C.; Liu, Q.; Li, L.; Chen, J.; Wang, X.; Huang, K. Simultaneous determination of eugenol, isoeugenol and methyleugenol in fish fillet using gas chromatography coupled to tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1031, 189–194. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Wang, C.; Yang, J.; Han, G. Stable isotope labeling-assisted GC/MS/MS method for determination of methyleugenol in food samples. J. Sci. Food Agric. 2018, 98, 3485–3491. [Google Scholar] [CrossRef]
- Grosch, S.; Monakhova, Y.B.; Kuballa, T.; Ruge, W.; Kimmich, R.; Lachenmeier, D.W. Comparison of GC/MS and NMR for quantification of methyleugenol in food. Eur. Food Res. Technol. 2013, 236, 267–275. [Google Scholar] [CrossRef]
- Falaki, F. Sample Preparation Techniques for Gas Chromatography; IntechOpen: London, UK, 2019. [Google Scholar]
- Chemat, F.; Rombaut, N.; Sicaire, A.-G.; Meullemiestre, A.; Fabiano-Tixier, A.-S.; Abert-Vian, M. Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols and applications. A review. Ultrason. Sonochem. 2017, 34, 540–560. [Google Scholar] [CrossRef]
- Mussatto, S.I. Chapter 11—Generating biomedical polyphenolic compounds from spent coffee or silverskin. In Coffee in Health and Disease Prevention; Preedy, V.R., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 93–106. [Google Scholar]
- Kyle, P.B. Toxicology: GCMS. In Mass Spectrometry for the Clinical Laboratory; Nair, H., Clarke, W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 131–163. [Google Scholar]
- Bicchi, C.; Cordero, C.; Liberto, E.; Sgorbini, B.; Rubiolo, P. Headspace sampling in flavor and fragrance field. In Comprehensive Sampling and Sample Preparation; Pawliszyn, J., Ed.; Academic Press: Oxford, UK, 2012; pp. 1–25. [Google Scholar]
- Kim, J.; Choi, K.; Chung, D.S. Sample Preparation for Capillary Electrophoretic Applications. In Comprehensive Sampling and Sample Preparation; Pawliszyn, J., Ed.; Academic Press: Oxford, UK, 2012; pp. 701–721. [Google Scholar]
- Rezaee, M.; Assadi, Y.; Milani Hosseini, M.-R.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.M.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS—Fundamentals, relevant improvements, applications and future trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef]
- González-Curbelo, M.Á.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Evolution and applications of the QuEChERS method. Trac. Trends Anal. Chem. 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Joshi, D.R.; Adhikari, N. An overview on common organic solvents and their toxicity. J. Pharm. Res. Int. 2019, 28, 1–18. [Google Scholar] [CrossRef]
- Mottram, D.S.; Elmore, J.S. Sensory evaluation—Aroma. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Oxford, UK, 2003; pp. 5174–5180. [Google Scholar]
- Pushpangadan, P.; George, V. Basil. In Handbook of Herbs and Spices, 2nd ed.; Peter, K.V., Ed.; Woodhead Publishing: Cambridge, UK, 2012; pp. 55–72. [Google Scholar]
- Prado, J.M.; Vardanega, R.; Debien, I.C.N.; Meireles, M.A.d.A.; Gerschenson, L.N.; Sowbhagya, H.B.; Chemat, S. Conventional extraction. In Food Waste Recovery; Galanakis, C.M., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 127–148. [Google Scholar]
- Oreopoulou, A.; Tsimogiannis, D.; Oreopoulou, V. Chapter 5—Extraction of polyphenols from aromatic and medicinal plants: An overview of the methods and the effect of extraction parameters. In Polyphenols in Plants, 2nd ed.; Watson, R.R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 243–259. [Google Scholar]
- Tahina Rabeharitsara, A. Determination of water rate in gas oil and fuel oil by extraction with betacarotenes molecules using a heavy oil clevenger extractor, process validation by SPC. Am. J. Appl. Chem. 2016, 4, 111. [Google Scholar] [CrossRef][Green Version]
- El Hassan, G.M. Effect of drying method on spearmint (Mentha spicata var. Viridis L.) oil content and physicochemical properties. Am. J. Phytomed. Clin. Ther. 2018, 10, 151–159. [Google Scholar]
- Niculau, E.D.S.; Ribeiro, L.D.P.; Ansante, T.F.; Fernandes, J.B.; Forim, M.R.; Vieira, P.C.; Vendramim, J.D.; da Silva, M.F.D.G.F. Isolation of chavibetol and methyleugenol from essential oil of Pimenta pseudocaryophyllus by high performance liquid chromatography. Molecules 2018, 23, 2909. [Google Scholar] [CrossRef]
- Kazakevich, Y.V.; Lobrutto, R. HPLC for Pharmaceutical Scientists; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Gursale, A.; Dighe, V.; Parekh, G. Simultaneous quantitative determination of cinnamaldehyde and methyl eugenol from stem bark of cinnamomum zeylanicum blume using RP-HPLC. J. Chromatogr. Sci. 2010, 48, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.A.; Rajput, S.J.; Raval, R.R. Development and validation of RP-HPLC method for determination of boswellic acid and myristicin in commercial herbal formulation. Int. J. Pharm. Pharm. Sci. 2013, 5, 379–383. [Google Scholar]
- Lung, I.; Stan, M.; Opriş, O.; Soran, M.L. Determination of myristicin and linalool in plants exposed to microwave radiation by high-performance liquid chromatography. Anal. Lett. 2015, 48, 567–574. [Google Scholar] [CrossRef]
- Al-Malahmeh, A.J.; Al-Ajlouni, A.; Wesseling, S.; Soffers, A.E.M.F.; Al-Subeihi, A.; Kiwamoto, R.; Vervoort, J.; Rietjens, I.M.C.M. Physiologically based kinetic modeling of the bioactivation of myristicin. Arch. Toxicol. 2017, 91, 713–734. [Google Scholar] [CrossRef] [PubMed]
- Al-Malahmeh, A.J.; Al-ajlouni, A.M.; Wesseling, S.; Vervoort, J.; Rietjens, I.M.C.M. Determination and risk assessment of naturally occurring genotoxic and carcinogenic alkenylbenzenes in basil-containing sauce of pesto. Toxicol. Rep. 2017, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M. Application of computational methods in isolation of plant secondary metabolites. In Computational Phytochemistry; Sarker, S.D., Nahar, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 107–139. [Google Scholar]
- Quirino, J.P.; Alejandro, F.M.; Bissember, A.C. Towards cleaner downstream processing of biomass waste chemical products by liquid chromatography: A review and recommendations. J. Clean. Prod. 2020, 253, 119937. [Google Scholar] [CrossRef]
- Alajlouni, A.M.; Al-Malahmeh, A.J.; Wesseling, S.; Kalli, M.; Vervoort, J.; Rietjens, I.M.C.M. Risk assessment of combined exposure to alkenylbenzenes through consumption of plant food supplements containing parsley and dill. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Altemimi, A.; Lakhssassi, N.; Baharlouei, A.; Watson, D.G.; Lightfoot, D.A. Phytochemicals: Extraction, isolation, and identification of bioactive compounds from plant extracts. Plants 2017, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Guidelines for the Validation of Chemical Methods for the FDA FVM Program. FDA Foods Program Regulatory Science Steering Committee; U.S. Food and Drug Administration: Rockville, ML, USA, 2019; Volume 3.
- Ávila, M.; Zougagh, M.; Escarpa, A.; Ríos, Á. Determination of alkenylbenzenes and related flavour compounds in food samples by on-column preconcentration-capillary liquid chromatography. J. Chromatogr. A 2009, 1216, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Muhandiramge, R.; Quirino, J.P. Sample preparation in capillary electrophoresis for the determination of small molecule drugs and metabolites in urine. Bioanalysis 2021, 13, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Quirino, J.P.; Wuethrich, A. Electrophoresis|capillary electrophoresis: Overview. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2019; pp. 377–386. [Google Scholar]
- Quirino, J.P.; Terabe, S. Electrokinetic chromatography. J. Chromatogr. A 1999, 856, 465–482. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Ionic liquids in electrokinetic chromatography. J. Chromatogr. A 2021, 1637, 461801. [Google Scholar] [CrossRef] [PubMed]
- Huhn, C.; Pütz, M.; Pyell, U. Separation of very hydrophobic analytes by micellar electrokinetic chromatography. III. Characterization and optimization of the composition of the separation electrolyte using carbon number equivalents. Electrophoresis 2008, 29, 783–795. [Google Scholar] [CrossRef]
- Quirino, J.P.; Tarongoy, F.M. Liquid chromatography with micelles in open-tube capillaries. Green Chem. 2018, 20, 2486–2493. [Google Scholar] [CrossRef]
- Kulsing, C.; Nolvachai, Y.; Marriott, P.J. Concepts, selectivity options and experimental design approaches in multidimensional and comprehensive two-dimensional gas chromatography. TrAC Trends Anal. Chem. 2020, 130, 115995. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Felisilda, B.M.B.; Quirino, J.P. Recent advancements in open-tubular liquid chromatography and capillary electrochromatography during 2014–2018. Anal. Chim. Acta 2019, 1088, 20–34. [Google Scholar] [CrossRef] [PubMed]







| Analytes | Sample/s | Sample Preparation | Amount of Alkenylbenzenes Found in Sample/s | HPLC/UPLC Conditions | Detectors | Ref. |
|---|---|---|---|---|---|---|
| methyleugenol | pimenta pseudocaryo-phyllus | hydrodistillation | methyleugenol (27.9 mg/g) | mobile phase:hexane (A), ethanol (B) column:Phenomenex Luna amino (4.6 mm × 150 mm × 10 µm) condition:isocratic condition with ratio hexane and ethanol at 92:8 flow rate: 1 mL/min injection volume:10 µL wavelength: 230 nm LOD: not reported LOQ: not reported | UV-VIS | [49] |
| methyleugenol | cinnamomum zeylanicum blume | methanol extraction | methyleugenol (0.5 mg/g) | mobile phase:water (A), ACN & methanol (B) at ratio 45:20:35 column: reversed-phase C18 Intersil ODS-3V-C18 (150 mm × 4.6 mm × 5 µm) condition:isocratic with the ratio of methanol:ACN:water is 35:20:45 flow rate:1 mL/min injection volume: not reported wavelength: 221 nm LOD: 0.10 µg/mL (methyleugenol) LOQ: 0.30 µg/mL (methyleugenol) | UV-VIS | [51] |
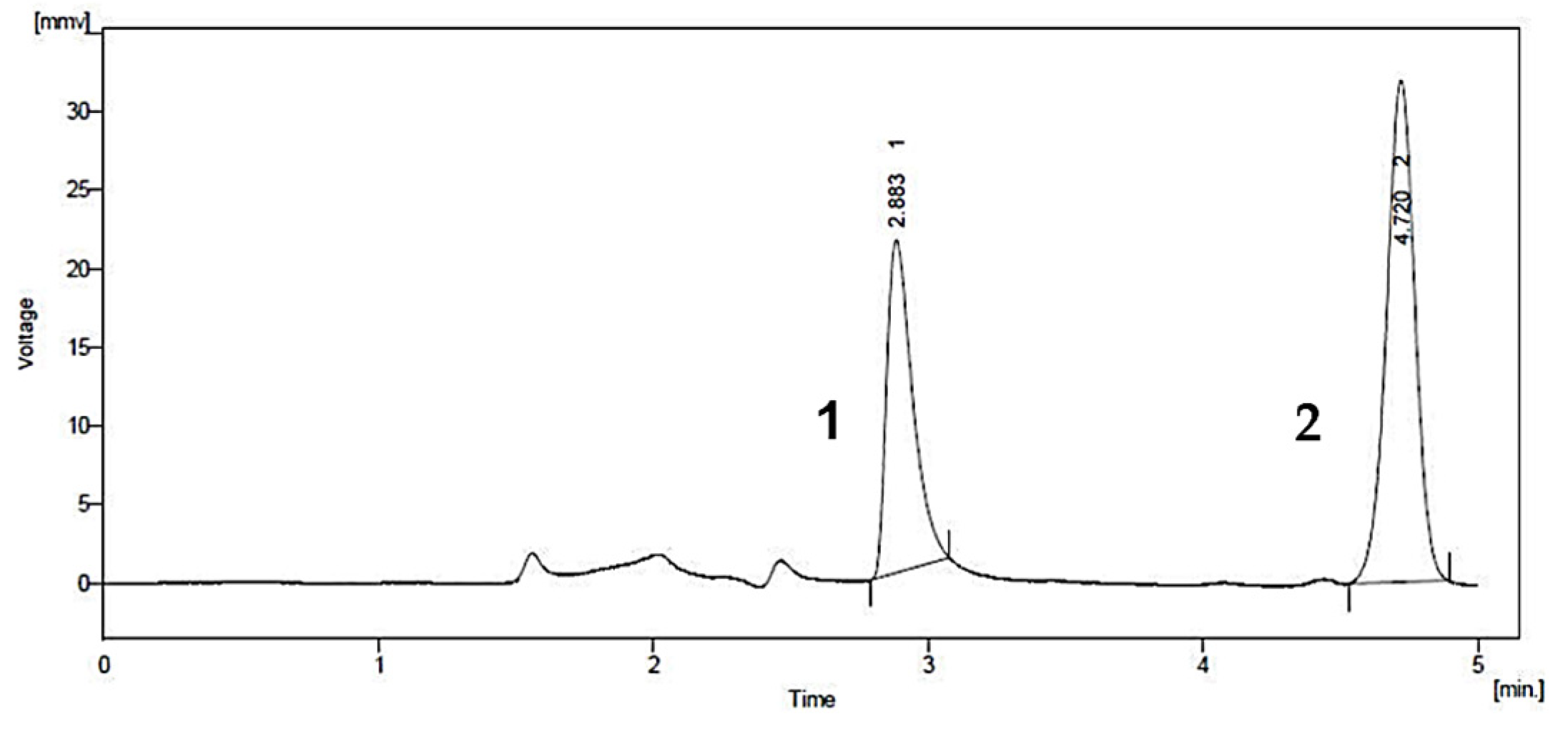
| myristicin | commercial herbal formulation | methanol extraction | myristicin (0.3 mg/g) † | mobile phase: water (A), ACN (B) column:Supelco 516 C18 (250 mm × 4.6 mm × 5 µm) condition: ACN:water is 85:15 flow rate: 1 mL/min injection volume: 20 µL wavelength: 205 nm LOD: 0.63 µg/mL (myristicin) LOQ: 1.91 µg/mL (myristicin) | UV-VIS | [52] |
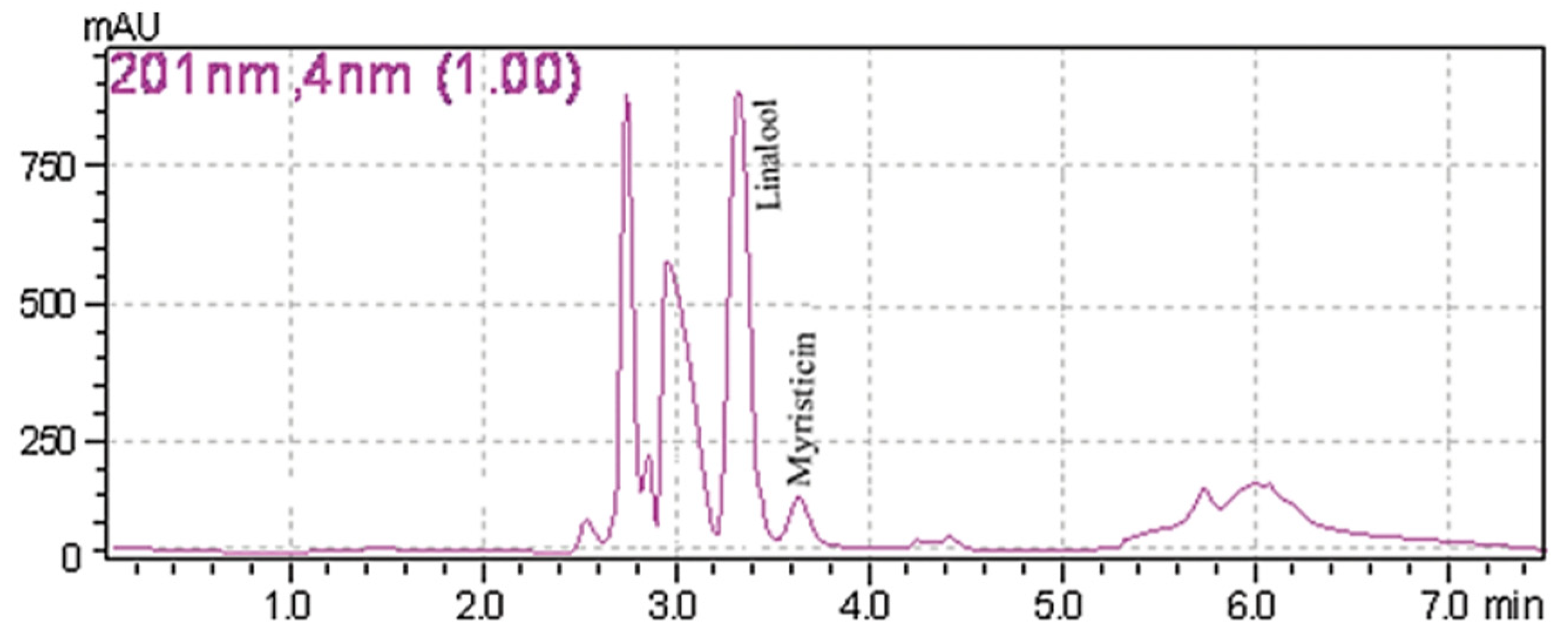
| myristicin | plants | n-hexane-diethyl ether extraction | myristicin (66.3 µg/mL) | mobile phase:water (A), ACN (B) column: LiChrosorb RP-18 (250 mm × 4 mm × 5 µm) condition:start from 0 to 1 min with 100%B, decrease to 25%B in 15 min. flow rate: 1 mL/min injection volume: 5 µL wavelength: 201 nm LOD:32.68 µg/mL (myristicin) LOQ: 64.57 µg/mL (myristicin) | DAD | [53] |
| estragole myristicin apiol | plant food supplements | methanol extraction | estragole (17.2 µg/g) myristicin (26.0 µg/g–1804.5 µg/g) apiol (93.0 µg/g–6486.6 µg/g) | mobile phase: water with 0.1%TFA (A), ACN (B) column: Waters Acquity C18 (50 mm × 2.1 mm × 1.7 µm) condition: start at 31% ACN, keep at 31%ACN for 5 min, increase to 80%ACN over 4 min and keep for 1 min, decrease to 0% over 1.5 min and keep for 1 min, increase back to 31%ACN. flow rate: 0.6 mL/min injection volume:not reported wavelength:209 nm (apiol, myristicin), 201 nm (estragole) LOD: not reported LOQ: not reported | DAD | [58] |
| elemicin methyleugenol myristicin safrole apiol estragole | Indonesian jamu | methanol extraction | elemicin (not found) methyleugenol (4.8 ± 1.6 µg/g–128.6 ± 0.9 µg/g) myristicin (33.9 ± 7.2 µg/g–440.1 ± 24.8 µg/g) safrole (3.8 ± 0.5 µg/g–18.8 ± 3.2 µg/g) apiol (not found) estragole (13.3 ± 1.3 µg/g–23.9 ± 6.3 µg/g) | mobile phase: water with 0.1%TFA (A), ACN (B) column: Waters Acquity UPLC BEH RP 18 (25 mm × 2.1 mm × 1.7 µm) condition: start at 30.5%ACN for 15 min, increase to 80%ACN over 1 min, keep at 80%ACN for 0.5 min, decrease to 0%ACN over 1.5 min, keep at 0%ACN for 1 min and back to 30.5%ACN. flow rate:0.6 mL/min injection volume: 3.5 µL wavelength: 206 nm (elemicin), 202 nm (methyleugenol, safrole), 210 nm (myristicin, apiol) and 225 nm (estragole) LOD: actual w/w values were not reported LOQ: actual w/w values were not reported | DAD | [13] |
| methyleugenol myristicin estragole apiol | pesto sauce | methanol extraction | methyleugenol (22.9 ± 3.1 µg/g–56.4 ± 7.5 µg/g) estragole (3.2 ± 1.5 µg/g–34.1 ± 2.8 µg/g) myristicin (13.2 ± 1.2 µg/g–15.8 ± 0.0 µg/g) apiol (3.4 ± 0.2 µg/g) | mobile phase: water with 0.1%TFA (A), ACN (B) column: Acquity UPLC BEH C18 (50 mm × 2.1 mm × 1.7 µm) condition:isocratic at 40%ACN for 4 min flow rate:0.6 mL/min injection volume: 3.5 µL wavelength: 201 nm (methyleugenol, estragole), 210 nm (myristicin, apiol) LOD: not reported LOQ: not reported | DAD | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, H.N.P.; Quirino, J.P. Analytical Separation of Carcinogenic and Genotoxic Alkenylbenzenes in Foods and Related Products (2010–2020). Toxins 2021, 13, 387. https://doi.org/10.3390/toxins13060387
Dang HNP, Quirino JP. Analytical Separation of Carcinogenic and Genotoxic Alkenylbenzenes in Foods and Related Products (2010–2020). Toxins. 2021; 13(6):387. https://doi.org/10.3390/toxins13060387
Chicago/Turabian StyleDang, Huynh N. P., and Joselito P. Quirino. 2021. "Analytical Separation of Carcinogenic and Genotoxic Alkenylbenzenes in Foods and Related Products (2010–2020)" Toxins 13, no. 6: 387. https://doi.org/10.3390/toxins13060387
APA StyleDang, H. N. P., & Quirino, J. P. (2021). Analytical Separation of Carcinogenic and Genotoxic Alkenylbenzenes in Foods and Related Products (2010–2020). Toxins, 13(6), 387. https://doi.org/10.3390/toxins13060387