
Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation

 , and
, and
Abstract
1. Introduction
2. Results
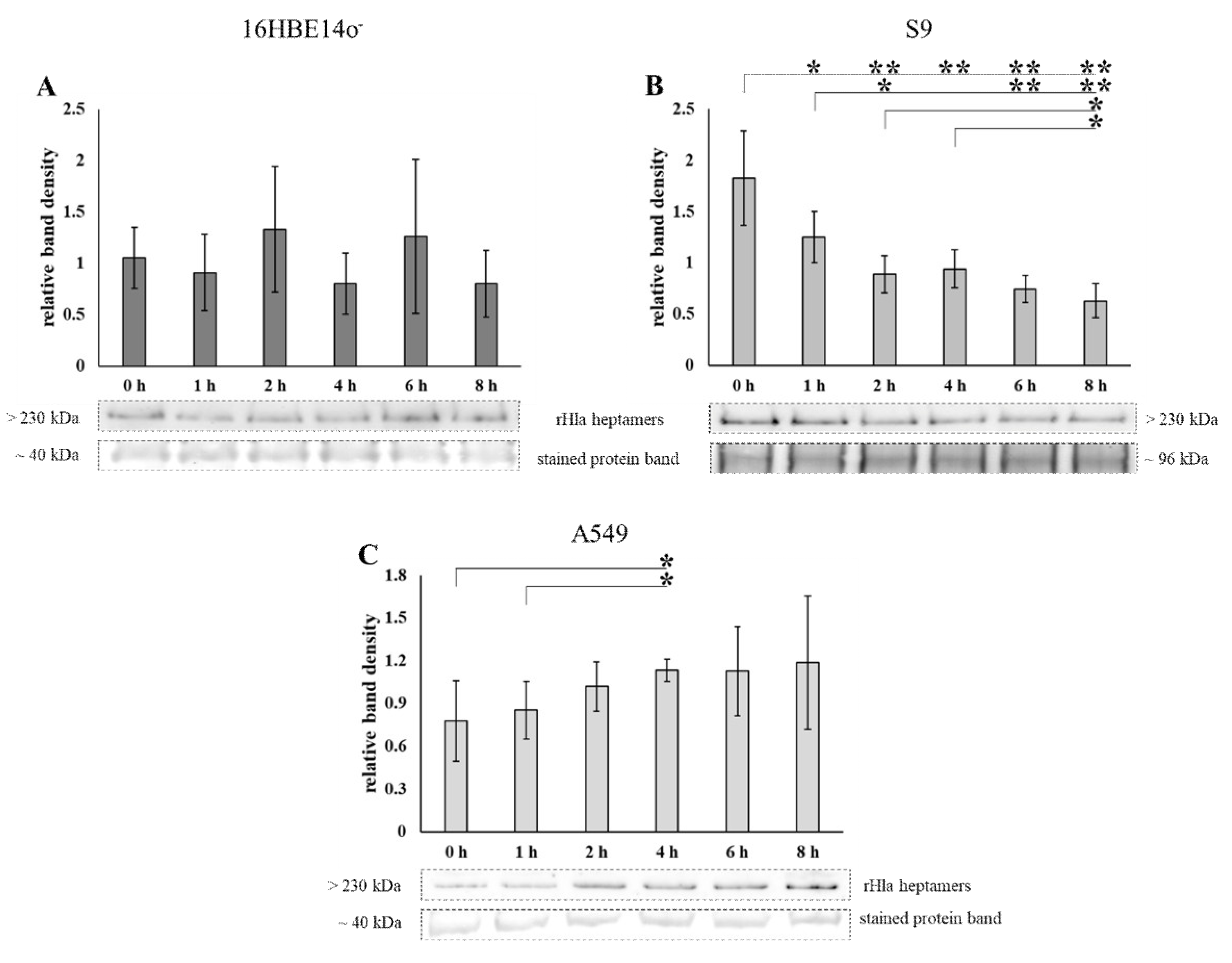
2.1. Loss of Heptamers from Airway Epithelial Cells Transiently Exposed to Alpha-Toxin
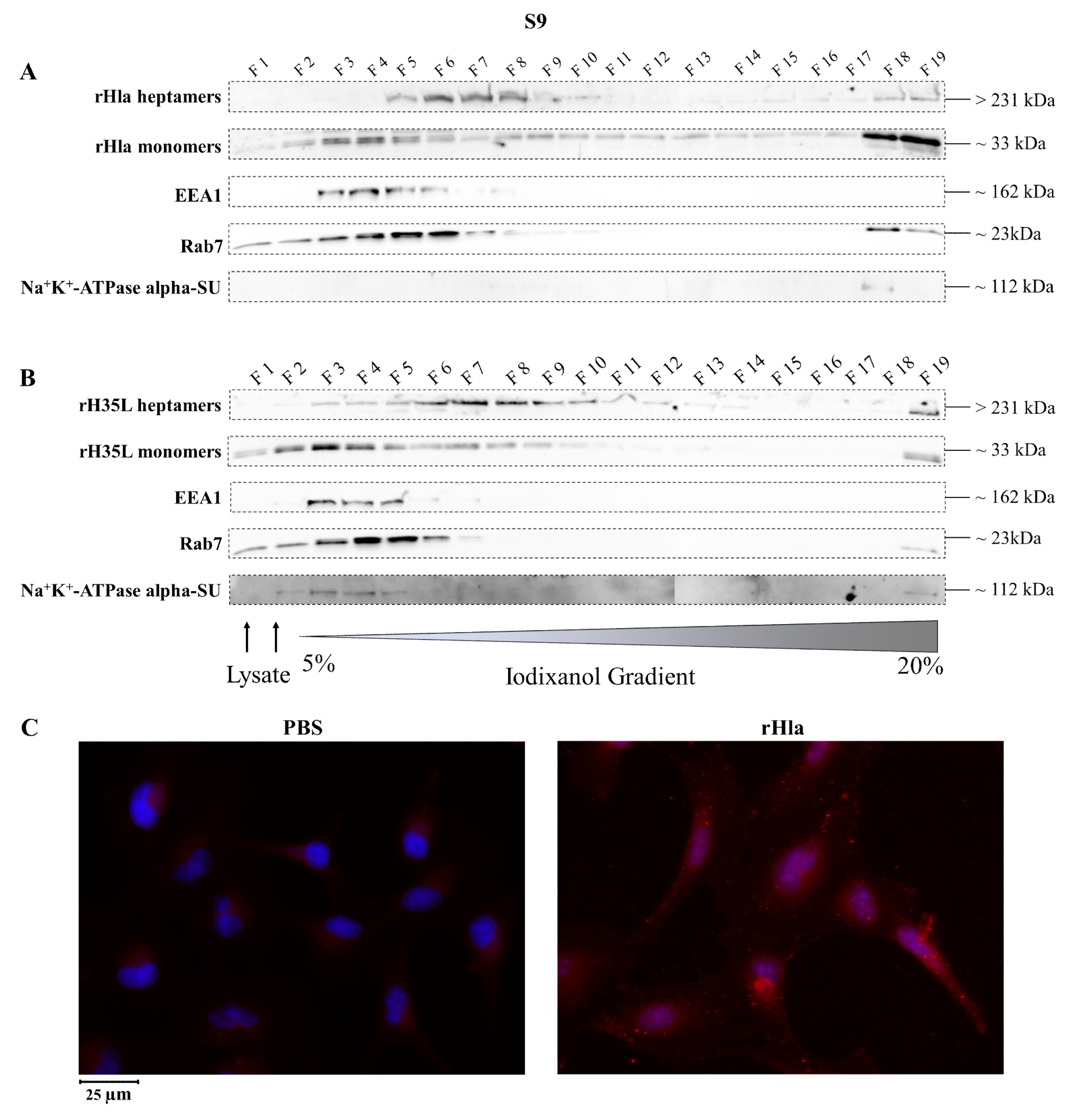
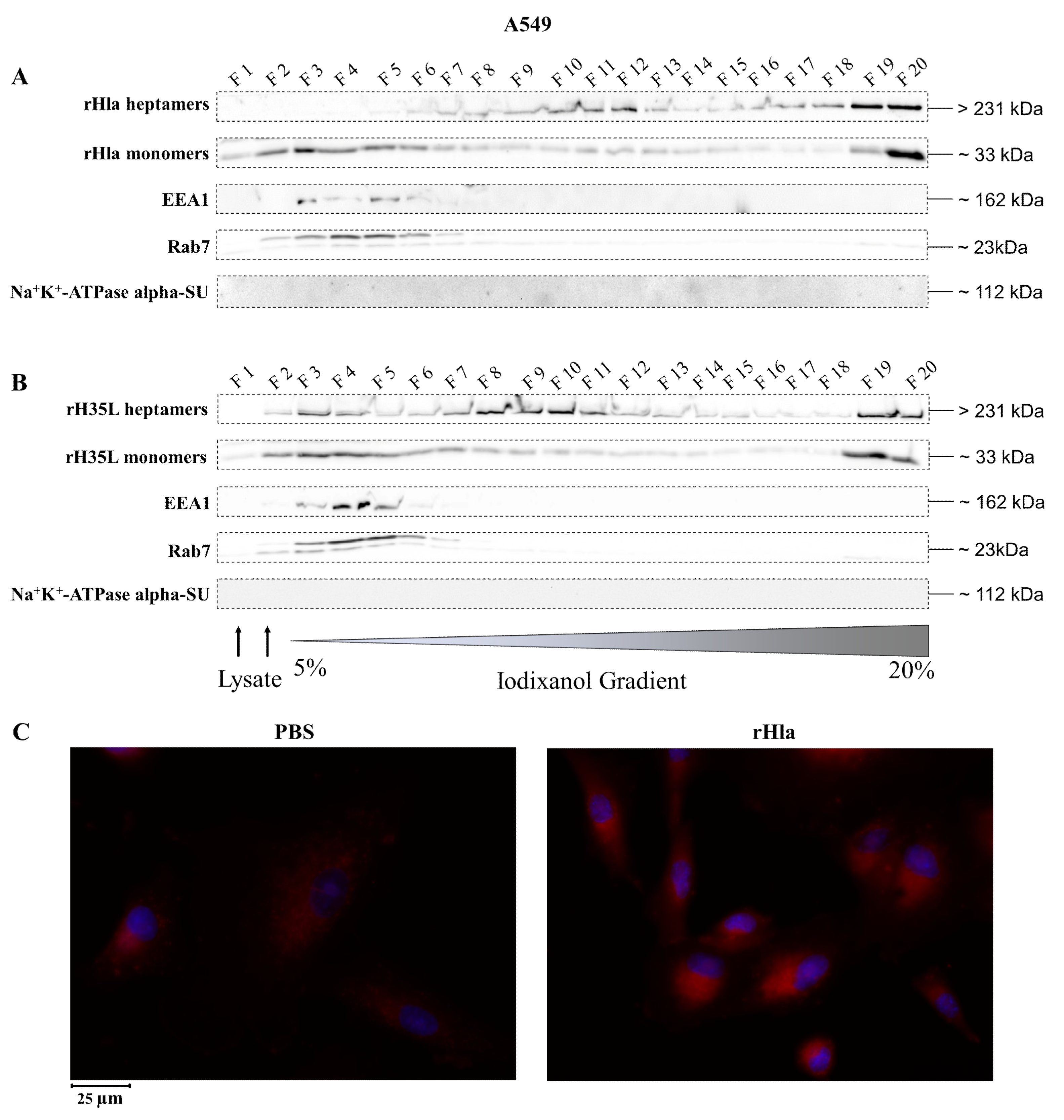
2.2. Presence of Alpha-Toxin Monomers and Heptamers in Subcellular Fractions of Toxin-Exposed Airway Epithelial Cells
2.3. Disposal of Alpha-Toxin Heptamers from Airway Epithelial Cells
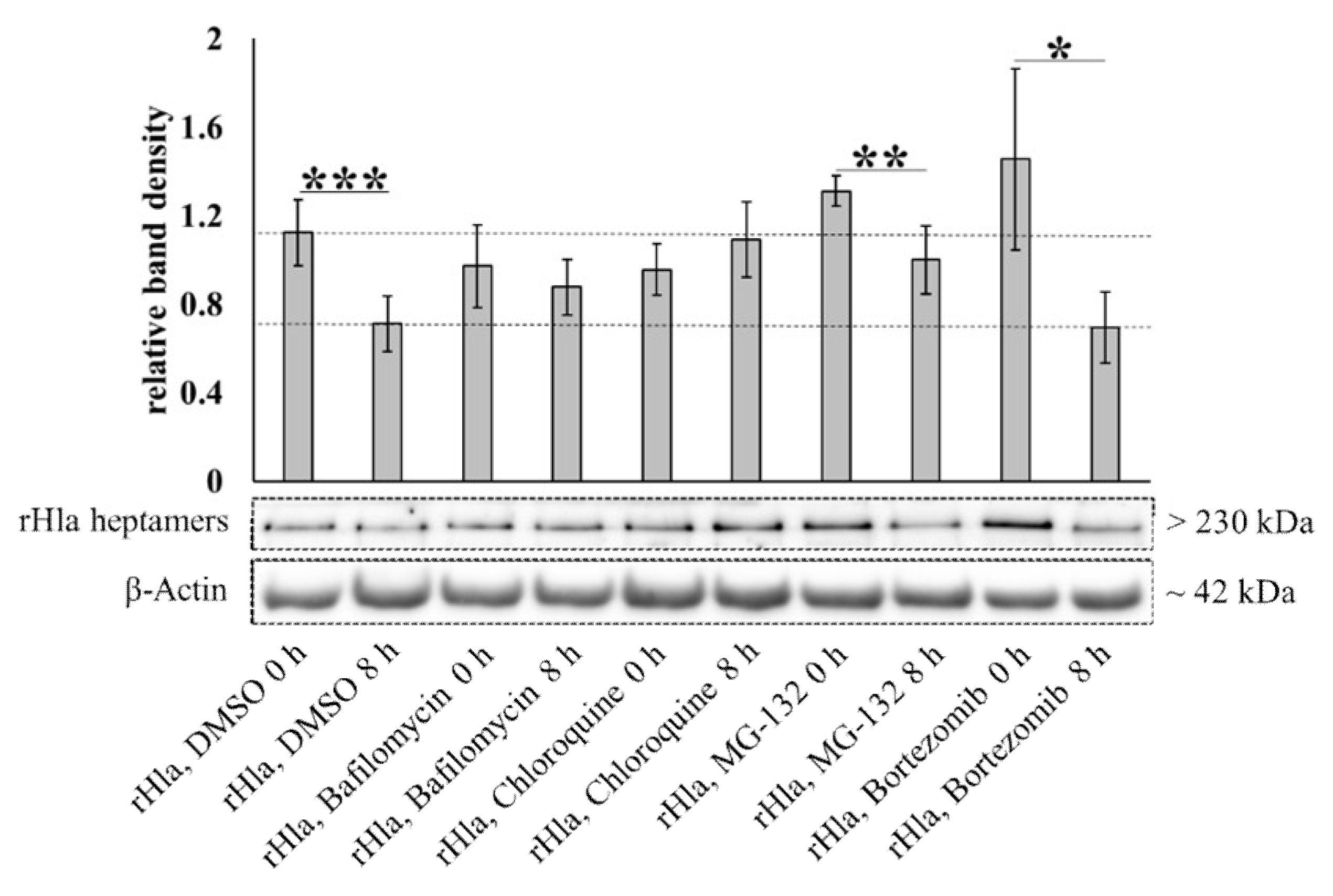
2.4. Degradation of Alpha-Toxin Heptamers through Lysosomal or Proteasomal Pathways?
2.5. Disposal of Alpha-Toxin Heptamers to The Extracellular Space: Exosome/Microsome Formation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Expression and Purification of Recombinant S. aureus Alpha-Toxin (rHla) and rH35L
4.3. Human Airway Model Epithelial Cell Cultures and Culture Conditions
4.4. Abundance of rHla-Heptamers in 16HBE14o-, S9 or A549 Cells upon Pulse-Treatment with Monomeric rHla
4.5. Continuous Iodixanol Density Gradients
4.6. Fluorescence Microscopy
4.7. Potential Proteasomal or Lysosomal rHla Heptamer Degradation in S9 Cells
4.8. Stability of rHla Heptamers at an Acidic pH
4.9. Mass Spectrometric Analysis of Proteins in Pelleted Extracellular Vesicles in rHla-Treated or in Serum-Starved S9 Cells
4.10. Data Presentation and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef]
- Hildebrandt, J.-P. Pore-forming virulence factors of Staphylococcus aureus destabilize epithelial barriers-effects of alpha-toxin in the early phases of airway infection. AIMS Microbiol. 2015, 1, 11–36. [Google Scholar] [CrossRef]
- Ulrich, M.; Herbert, S.; Berger, J.; Bellon, G.; Louis, D.; Münker, G.; Döring, G. Localization of Staphylococcus aureus in infected airways of patients with cystic fibrosis and in a cell culture model of S. aureus adherence. Am. J. Respir. Cell Mol. Biol. 1998, 19, 83–91. [Google Scholar] [CrossRef]
- Boucher, R.C. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur. Resp. J. 2004, 23, 146–158. [Google Scholar] [CrossRef]
- Sibbald, M.J.; Ziebandt, A.K.; Engelmann, S.; Hecker, M.; de Jong, A.; Harmsen, H.J.; Raangs, G.C.; Stokroos, I.; Arends, J.P.; Dubois, J.Y.; et al. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol. Mol. Biol. Rev. 2006, 70, 755–788. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Wang, R.; Khan, B.A.; Sturdevant, D.E.; Otto, M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun. 2011, 79, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- BubeckWardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; BubeckWardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Richter, E.; Harms, M.; Ventz, K.; Gierok, P.; Chilukoti, R.K.; Hildebrandt, J.-P.; Mostertz, J.; Hochgräfe, F. A multi-omics approach identifies key hubs associated with cell type-specific responses of airway epithelial cells to staphylococcal alpha-toxin. PLoS ONE 2015, 10, e0122089. [Google Scholar] [CrossRef]
- Möller, N.; Ziesemer, S.; Hildebrandt, P.; Assenheimer, N.; Völker, U.; Hildebrandt, J.-P.S. S. aureus alpha-toxin monomer binding and heptamer formation in host cell membranes—Do they determine sensitivity of airway epithelial cells toward the toxin? PLoS ONE 2020, 15, e0233854. [Google Scholar] [CrossRef]
- Schwiering, M.; Brack, A.; Stork, R.; Hellmann, N. Lipid and phase specificity of alpha-toxin from S. aureus. Biochim. Biophys. Acta 2013, 1828, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Ziesemer, S.; Möller, N.; Nitsch, A.; Müller, C.; Beule, A.G.; Hildebrandt, J.-P. Sphingomyelin depletion from plasma membranes of human airway epithelial cells completely abrogates the deleterious actions of S. aureus alpha-toxin. Toxins 2019, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, L.; Miles, G.; Bayley, H. Role of the amino latch of staphylococcal alpha-hemolysin in pore formation: A co-operative interaction between the N terminus and position 217. J. Biol. Chem. 2006, 281, 2195–2204. [Google Scholar] [CrossRef]
- Valeva, A.; Palmer, M.; Bhakdi, S. Staphylococcal alpha-toxin: Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages. Biochemistry 1997, 36, 13298–13304. [Google Scholar] [CrossRef]
- Walev, I.; Martin, E.; Jonas, D.; Mohamadzadeh, M.; Müller-Klieser, W.; Kunz, L.; Bhakdi, S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun. 1993, 61, 4972–4979. [Google Scholar] [CrossRef]
- Eichstaedt, S.; Gäbler, K.; Below, S.; Müller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, P.; Völker, U.; Hecker, M.; Hildebrandt, J.-P. Effects of Staphylococcus aureus-hemolysin A on calcium signalling in immortalized human airway epithelial cells. Cell Calcium 2009, 45, 165–176. [Google Scholar] [CrossRef]
- Eiffler, I.; Behnke, J.; Ziesemer, S.; Müller, C.; Hildebrandt, J.-P. Staphylococcus aureus alpha-toxin-mediated cation entry depolarizes membrane potential and activates p38 MAP kinase in airway epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2016, 311, L676–L685. [Google Scholar] [CrossRef]
- Baaske, R.; Richter, M.; Möller, N.; Ziesemer, S.; Eiffler, I.; Müller, C.; Hildebrandt, J.-P. ATP release from human airway epithelial cells exposed to Staphylococcus aureus alpha-toxin. Toxins 2016, 8, 365. [Google Scholar] [CrossRef]
- Räth, S.; Ziesemer, S.; Witte, A.; Konkel, A.; Müller, C.; Hildebrandt, P.; Völker, U.; Hildebrandt, J.-P.S. S. aureus haemolysin A-induced IL-8 and IL-6 release from human airway epithelial cells is mediated by activation of p38- and Erk-MAP kinases and additional, cell type-specific signalling mechanisms. Cell Microbiol. 2013, 15, 1253–1265. [Google Scholar] [CrossRef]
- Hermann, I.; Räth, S.; Ziesemer, S.; Volksdorf, T.; Dress, R.J.; Gutjahr, M.; Müller, C.; Beule, A.G.; Hildebrandt, J.-P. Staphylococcus aureus hemolysin A disrupts cell-matrix adhesions in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 14–24. [Google Scholar] [CrossRef]
- Ziesemer, S.; Eiffler, I.; Schönberg, A.; Müller, C.; Hochgräfe, F.; Beule, A.G.; Hildebrandt, J.-P. Staphylococcus aureusα-toxin induces actin filament remodeling in human airway epithelial model cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; BubeckWardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef]
- Gierok, P.; Harms, M.; Richter, E.; Hildebrandt, J.-P.; Lalk, M.; Mostertz, J.; Hochgräfe, F. Staphylococcus aureus alpha-toxin mediates general and cell type-specific changes in metabolite concentrations of immortalized human airway epithelial cells. PLoS ONE 2014, 9, e94818. [Google Scholar] [CrossRef]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef]
- Sheff, D.R.; Daro, E.A.; Hull, M.; Mellman, I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J. Cell Biol. 1999, 145, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.T.; Callaghan, J.M.; Steele-Mortimer, O.; Stenmark, H.; Parton, R.G.; Campbell, P.L.; McCluskey, J.; Yeo, J.P.; Tock, E.P.; Toh, B.H. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 1995, 270, 13503–13511. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Hundal, H.S.; Maxwell, D.L.; Ahmed, A.; Darakhshan, F.; Mitsumoto, Y.; Klip, A. Subcellular distribution and immunocytochemical localization of Na,K-ATPase subunit isoforms in human skeletal muscle. Mol. Membr. Biol. 1994, 11, 255–262. [Google Scholar] [CrossRef]
- Mobasheri, A.; Oukrif, D.; Dawodu, S.P.; Sinha, M.; Greenwell, P.; Stewart, D.; Djamgoz, M.B.; Foster, C.S.; Martín-Vasallo, P.; Mobasheri, R. Isoforms of Na+, K+-ATPase in human prostate; specificity of expression and apical membrane polarization. Histol. Histopathol. 2001, 16, 141–154. [Google Scholar] [CrossRef]
- Menzies, B.E.; Kernodle, D.S. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: Role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 1994, 62, 1843–1847. [Google Scholar] [CrossRef]
- Greening, D.W.; Simpson, R.J. Understanding extracellular vesicle diversity—Current status. Expert. Rev. Proteomics 2018, 15, 887–910. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in exosome isolation and analysis in health and disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Hildebrand, A.; Pohl, M.; Bhakdi, S. Staphylococcus aureus alpha-toxin. Dual mechanism of binding to target cells. J. Biol. Chem. 1991, 266, 17195–17200. [Google Scholar] [CrossRef]
- Popov, L.M.; Marceau, C.D.; Starkl, P.M.; Lumb, J.H.; Shah, J.; Guerrera, D.; Cooper, R.L.; Merakou, C.; Bouley, D.M.; Meng, W.; et al. The adherens junctions control susceptibility to Staphylococcus aureus α-toxin. Proc. Natl. Acad. Sci. USA 2015, 112, 14337–14342. [Google Scholar] [CrossRef]
- von Hoven, G.; Rivas, A.J.; Neukirch, C.; Klein, S.; Hamm, C.; Qin, Q.; Meyenburg, M.; Füser, S.; Saftig, P.; Hellmann, N.; et al. Dissecting the role of ADAM10 as a mediator of Staphylococcus aureus α-toxin action. Biochem. J. 2016, 473, 1929–1940. [Google Scholar] [CrossRef]
- Virreira Winter, S.; Zychlinsky, A.; Bardoel, B.W. Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus α-hemolysin-mediated toxicity. Sci. Rep. 2016, 6, 24242. [Google Scholar] [CrossRef]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef]
- Ursos, L.M.B.; Dzekunov, S.M.; Roepe, P.D. The effects of chloroquine and verapamil on digestive vacuolar pH of P. falciparum either sensitive or resistant to chloroquine. Mol. Biochem. Parasitol. 2000, 110, 125–134. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Below, S.; Konkel, A.; Zeeck, C.; Müller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, J.-P. Virulence factors of Staphylococcus aureus induce Erk-MAP kinase activation and c-Fos expression in S9 and 16HBE14o- human airway epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 296, L470–L479. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Blankenburg, S.; Hentschker, C.; Nagel, A.; Hildebrandt, P.; Michalik, S.; Dittmar, D.; Surmann, K.; Völker, U. Improving proteome coverage for small sample amounts: An advanced method for proteomics approaches with low bacterial cell numbers. Proteomics 2019, 19, e1900192. [Google Scholar] [CrossRef]
- Palma Medina, L.M.; Becker, A.K.; Michalik, S.; Yedavally, H.; Raineri, E.; Hildebrandt, P.; Gesell Salazar, M.; Surmann, K.; Pförtner, H.; Mekonnen, S.A.; et al. Metabolic cross-talk between human bronchial epithelial cells and internalized Staphylococcus aureus as a driver for infection. Mol. Cell Proteomics. 2019, 18, 892–908. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | SwissProt # | Protein Description | Treated with rHla | 8 h in Starvation Medium | 48 h in Starvation Medium |
|---|---|---|---|---|---|
| ESCRT | Q99816 | Tumor susceptibility gene 101 protein, TSG101 | 8 | 7 | 10 |
| Q9H9H4 | Vacuolar protein sorting-associated protein 37B, VPS37B | 4 | 4 | 4 | |
| A5D8V6 | Vacuolar protein sorting-associated protein 37C, VPS37C | 0 | 0 | 2 | |
| O43633 | Charged multivesicular body protein 2a, CHMP2A | 4 | 4 | 5 | |
| ESCRT-associated | Q8WUM4 | Programmed cell death 6 interacting protein, PDCD6IP/Alix | 34 | 31 | 35 |
| Tetraspanins | P08962 | CD63 antigen, CD63 | 0 | 3 | 3 |
| P21926 | CD9 antigen, CD9 | 5 | 6 | 6 | |
| P27701 | CD82 antigen, CD82 | 2 | 4 | 4 | |
| P48509 | CD151 antigen, CD151 | 2 | 3 | 4 | |
| O43657 | Tetraspanin-6, TSPNA6 | 0 | 0 | 2 | |
| RNA binding protein, ribosomal protein | P26373 | 60S ribosomal protein L13, RPL13 | 9 | 10 | 10 |
| P61254 | 60S ribosomal protein L26, RPL26 | 0 | 0 | 2 | |
| P62851 | 40S ribosomal protein S25, RPS25 | 6 | 6 | 7 | |
| Cargo selection | P46934 | E3 ubiquitin-protein ligase NEDD4, NEDD4 | 0 | 0 | 2 |
| Q96PU5 | E3 ubiquitin-protein ligase NEDD4-like, NEDD4L | 5 | 5 | 7 | |
| Protein trafficking, protein sorting | O00560 | Syntenin-1, Syndecan-binding protein 1, SDCBP | 10 | 10 | 10 |
| Q8N5I2 | Arrestin domain-containing protein 1, ARRDC1 | 2 | 3 | 4 | |
| Q9H0U4 | Ras-related protein Rab-1B, Rab1B | 12 | 11 | 11 | |
| P20339 | Ras-related protein Rab-5A, Rab5A | 8 | 8 | 9 | |
| P61020 | Ras-related protein Rab-5B, Rab5B | 6 | 6 | 6 | |
| P61006 | Ras-related protein Rab-8A, Rab8A | 6 | 6 | 6 | |
| Q15907 | Ras-related protein Rab-11B, Rab11B | 12 | 10 | 12 | |
| Integral membrane protein | P06756 | Integrin alpha-V, ITGAV | 12 | 14 | 16 |
| O14672 | A disintegrin and metalloproteinase domain-containing protein 10, ADAM10 | 7 | 5 | 7 | |
| Q969P0 | Immunoglobulin superfamily member 8, IGSF8 | 10 | 10 | 10 | |
| GPI-anchor | P35052 | Glypican-1, GPC1 | 9 | 8 | 9 |
| Scaffolding protein | O75955 | Flotillin-1, FLOT1 | 19 | 17 | 20 |
| P11142 | Heat shock 70 kDa protein 8, HSPA8 | 33 | 32 | 35 |
| Category | Swiss Prot # | Protein Description | Treated with rHla | 8 h in Starvation Medium | 48 h in Starvation Medium |
|---|---|---|---|---|---|
| Ribonuclear protein | P31943 | Heterogeneous nuclear ribonucleoprotein H, HNRNPH1 | 10 | 7 | 10 |
| P14866 | Heterogeneous nuclear ribonucleoprotein L, HNRNPl | 14 | 11 | 15 | |
| Calcium-bindingchaperone | P27797 | Calreticulin, CALR | 8 | 9 | 9 |
| Nuclear export receptor | P55060 | Exportin-2, CSE1L | 28 | 21 | 29 |
| Calcium-dependent phospholipid binding protein | O75131 | Copine-3, CPNE3 protein, CPNE3 | 7 | 6 | 7 |
| Mitochondrial outer membrane protein | P21796 | Voltage-dependent anion-selective channel protein 1, VDAC1 | 7 | 4 | 8 |
| P45880 | Voltage-dependent anion-selective channel protein 2, VDAC2 | 6 | 6 | 6 | |
| Mitochondrial andnuclear protein | Q99623 | Prohibitin-2, PHB2 | 6 | 6 | 8 |
| Mitochondrial inner membrane protein | P48047 | ATP synthase subunit O, mitochondrial, ATP5O | 0 | 0 | 5 |
| Q00325 | Solute carrier family 25 member 3, SLC25A3 | 3 | 4 | 4 | |
| P25705 | ATP synthase subunit alpha, mitochondrial, ATP5A1 | 26 | 16 | 24 | |
| ABC transporter | P61221 | ATP-binding cassette sub-family E member 1, ABCE1 | 11 | 6 | 11 |
| Integral membraneprotein | Q07065 | Cytoskeleton-associated protein 4, CKAP4 | 6 | 12 | 16 |
| Cytoskeleton/microtubule regulation, cell motility | Q9UM54 | Unconventional myosin-VI, MYO6 | 8 | 6 | 12 |
| O75369 | Filamin-B, FLNB | 61 | 65 | 80 | |
| Q9H0H5 | Rac GTPase-activating protein 1, RACGAP1 | 8 | 9 | 12 | |
| Q02241 | Kinesin-like protein, KIF23 | 12 | 16 | 19 | |
| P06753 | Tropomyosin alpha-3 chain, TPM3 | 6 | 5 | 6 | |
| Cytosolic enzymes | P49588 | Alanine-tRNA ligase, cytoplasmic, AARS | 24 | 18 | 25 |
| P31939 | Bifunctional purine biosynthesis protein, ATIC | 21 | 17 | 25 | |
| Enzyme, organizer of 3D structure of proteins | Q96HE7 | ERO1-like protein alpha, ERO1L | 5 | 5 | 7 |
| P13667 | Protein disulfide-isomerase A4, PDIA4 | 11 | 15 | 17 | |
| Beta-subunit of heterotrimeric protein | P62879 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2, GNB2 | 7 | 5 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, N.; Ziesemer, S.; Hentschker, C.; Völker, U.; Hildebrandt, J.-P. Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation. Toxins 2021, 13, 173. https://doi.org/10.3390/toxins13030173
Möller N, Ziesemer S, Hentschker C, Völker U, Hildebrandt J-P. Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation. Toxins. 2021; 13(3):173. https://doi.org/10.3390/toxins13030173
Chicago/Turabian StyleMöller, Nils, Sabine Ziesemer, Christian Hentschker, Uwe Völker, and Jan-Peter Hildebrandt. 2021. "Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation" Toxins 13, no. 3: 173. https://doi.org/10.3390/toxins13030173
APA StyleMöller, N., Ziesemer, S., Hentschker, C., Völker, U., & Hildebrandt, J.-P. (2021). Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation. Toxins, 13(3), 173. https://doi.org/10.3390/toxins13030173