Mining Indole Alkaloid Synthesis Gene Clusters from Genomes of 53 Claviceps Strains Revealed Redundant Gene Copies and an Approximate Evolutionary Hourglass Model

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Genome Assemblies
2.2. Presence of Four Classes of Alkaloid Genes in 53 Genomes
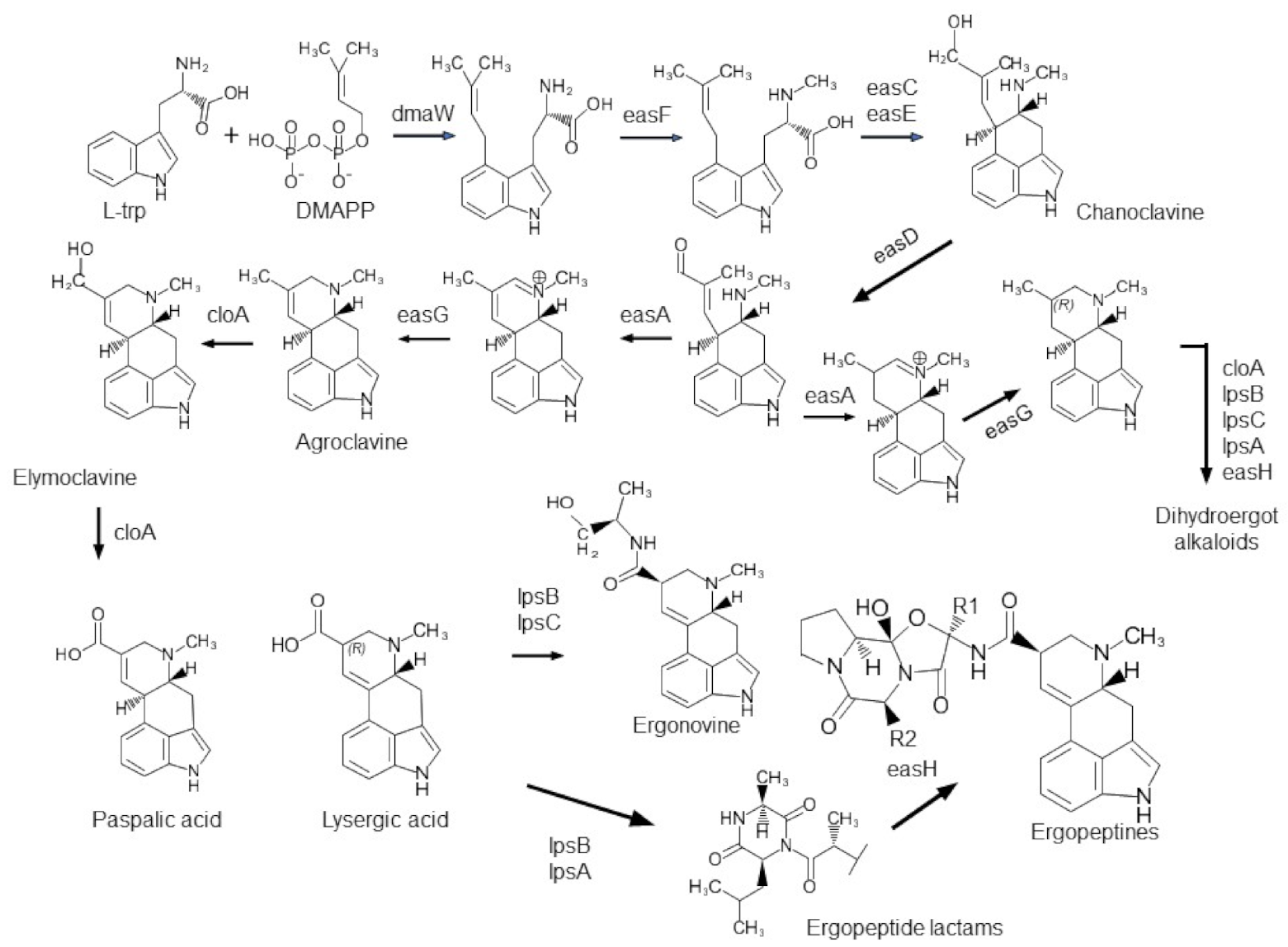
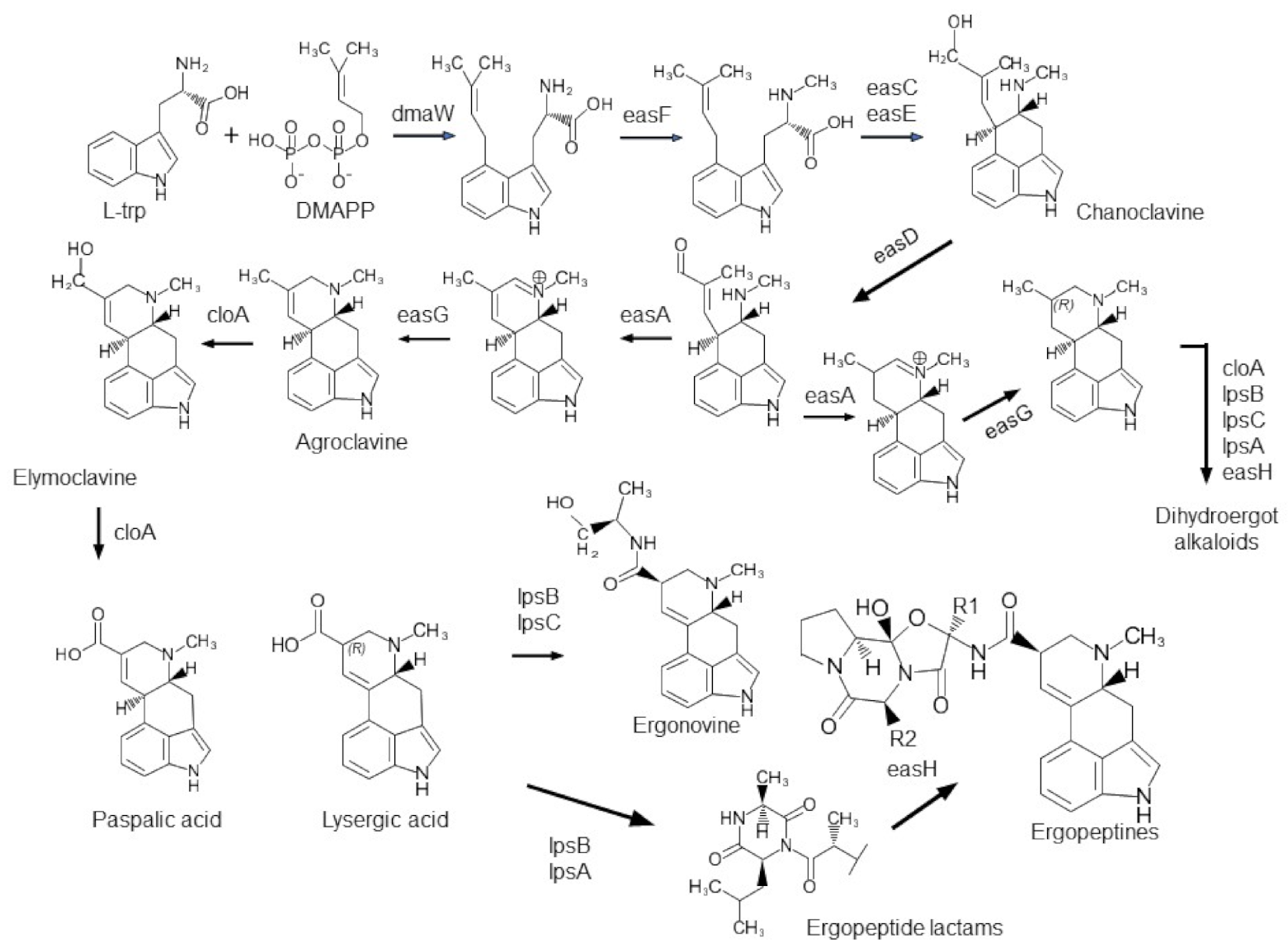
2.2.1. Ergot Alkaloid Genes (eas)
2.2.2. Indole-Diterpene/Lolitrem (idt/ltm) Genes
2.2.3. Loline Alkaloid (lol) and Peramine (per) Genes
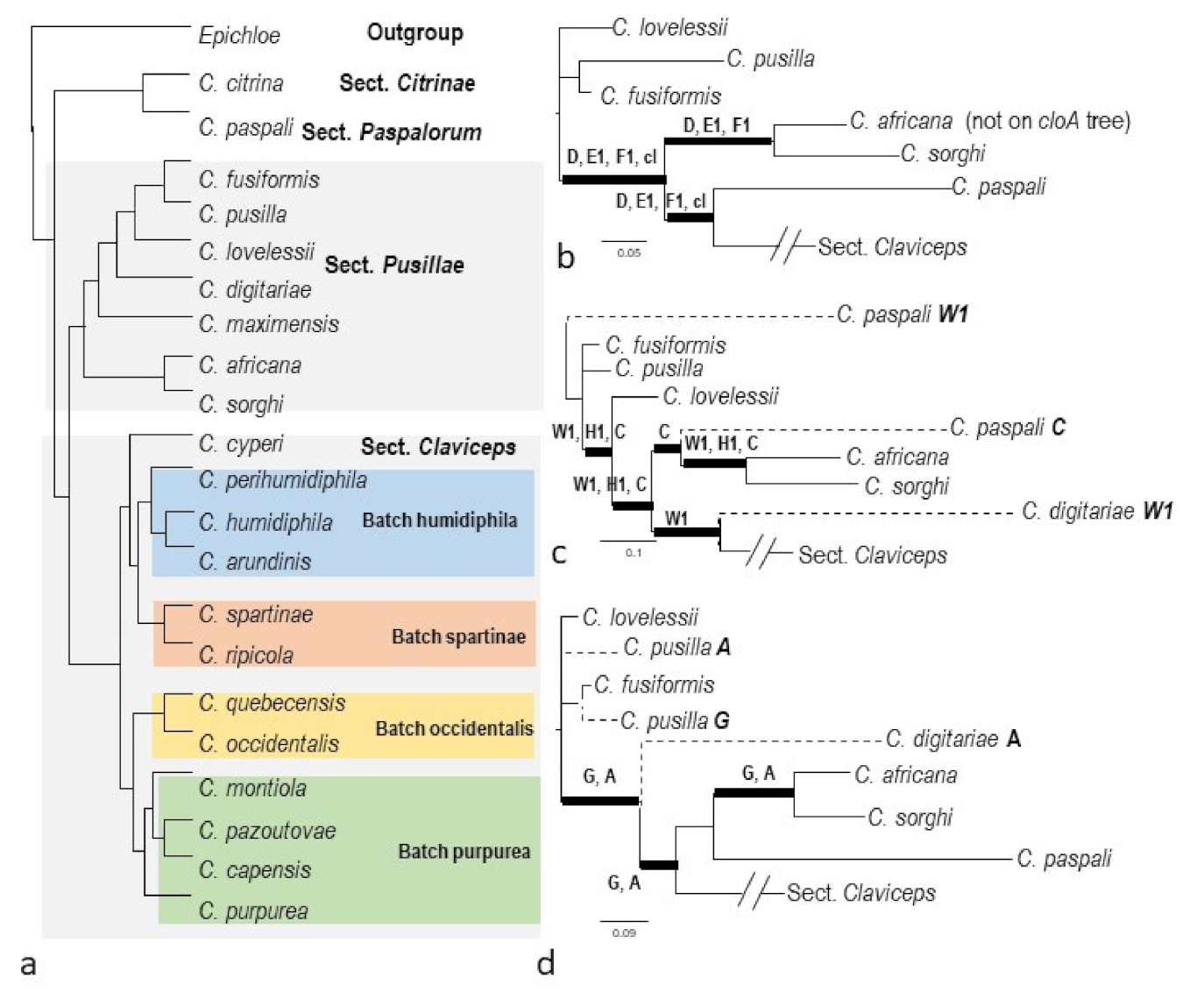
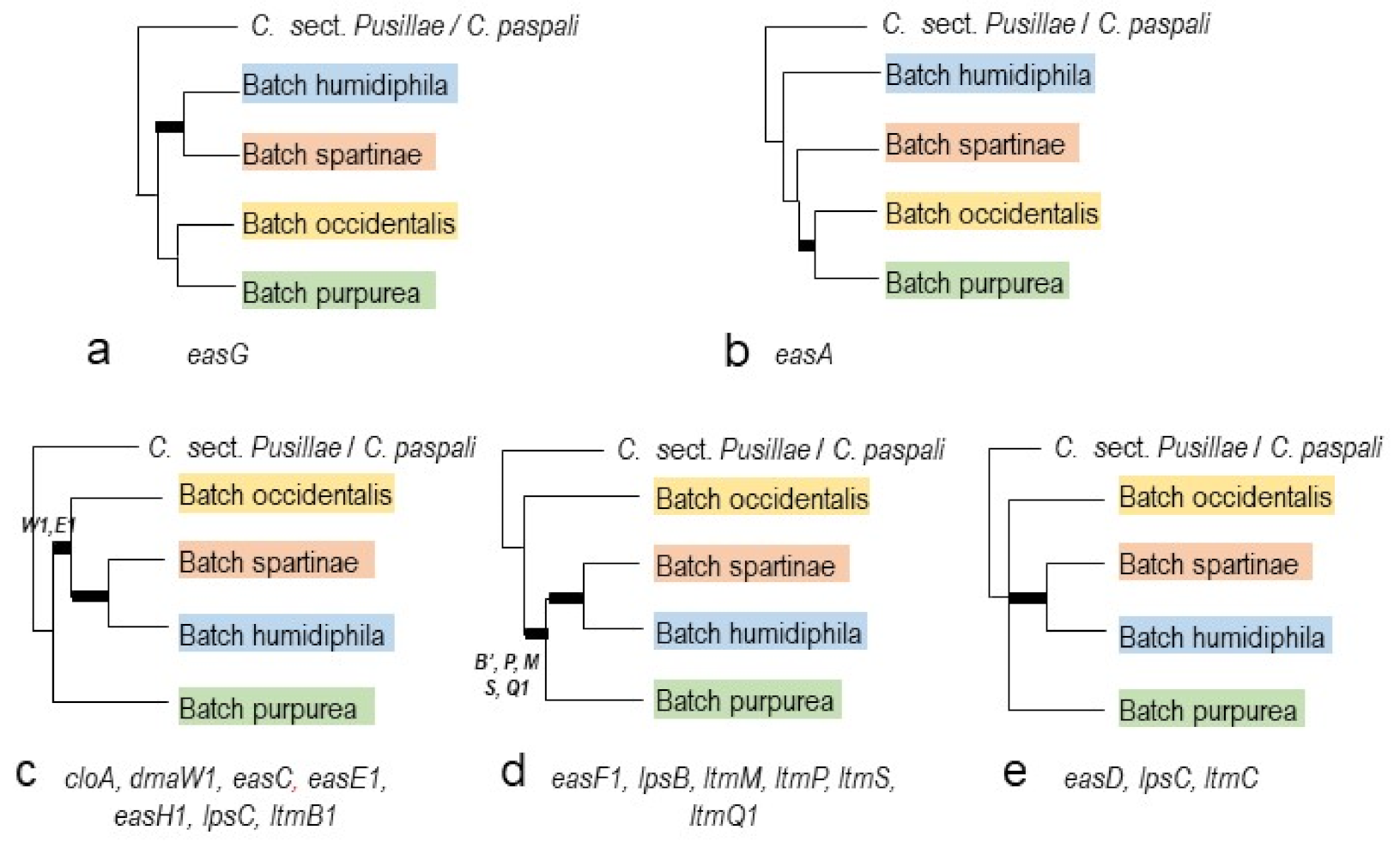
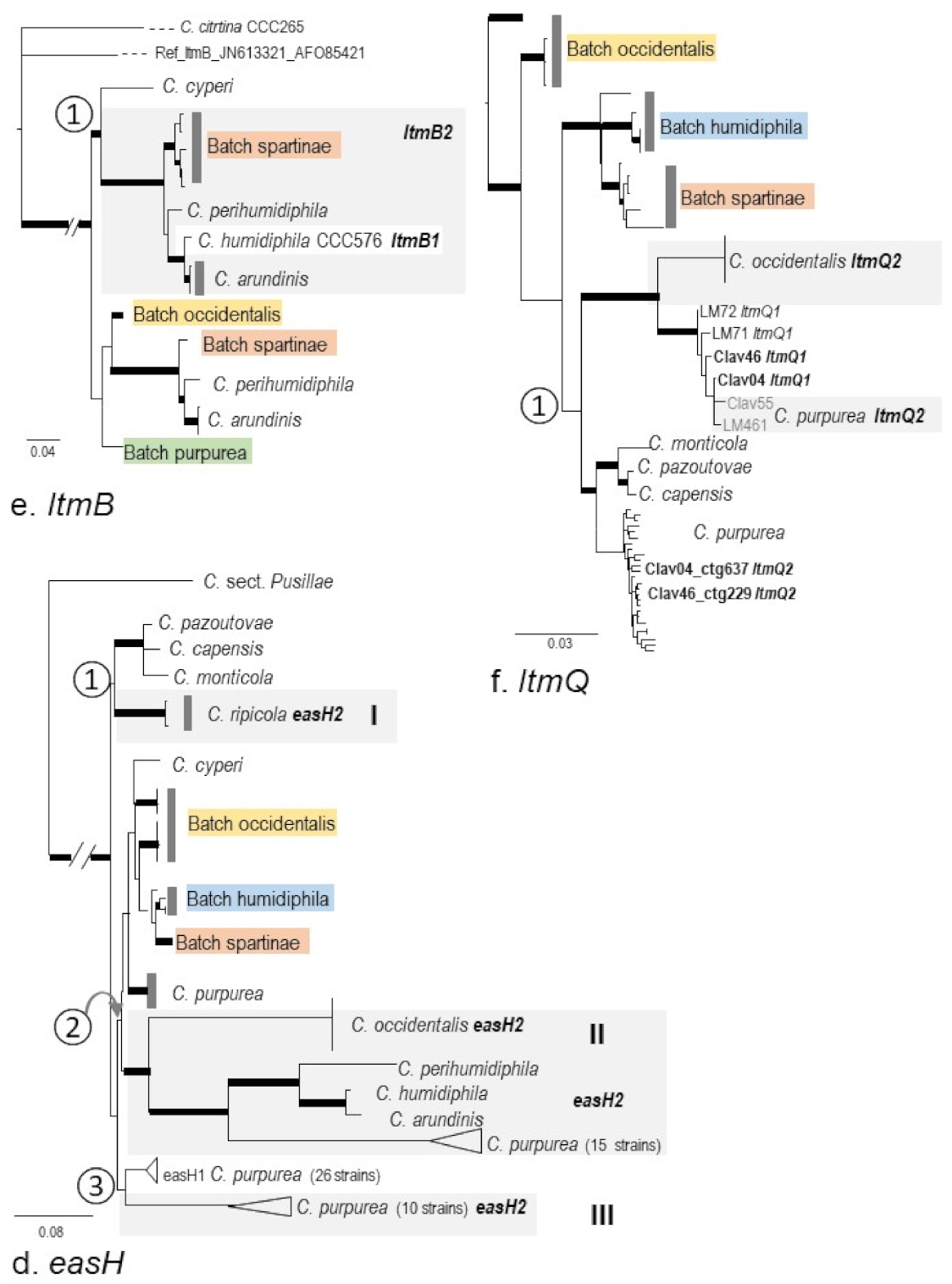
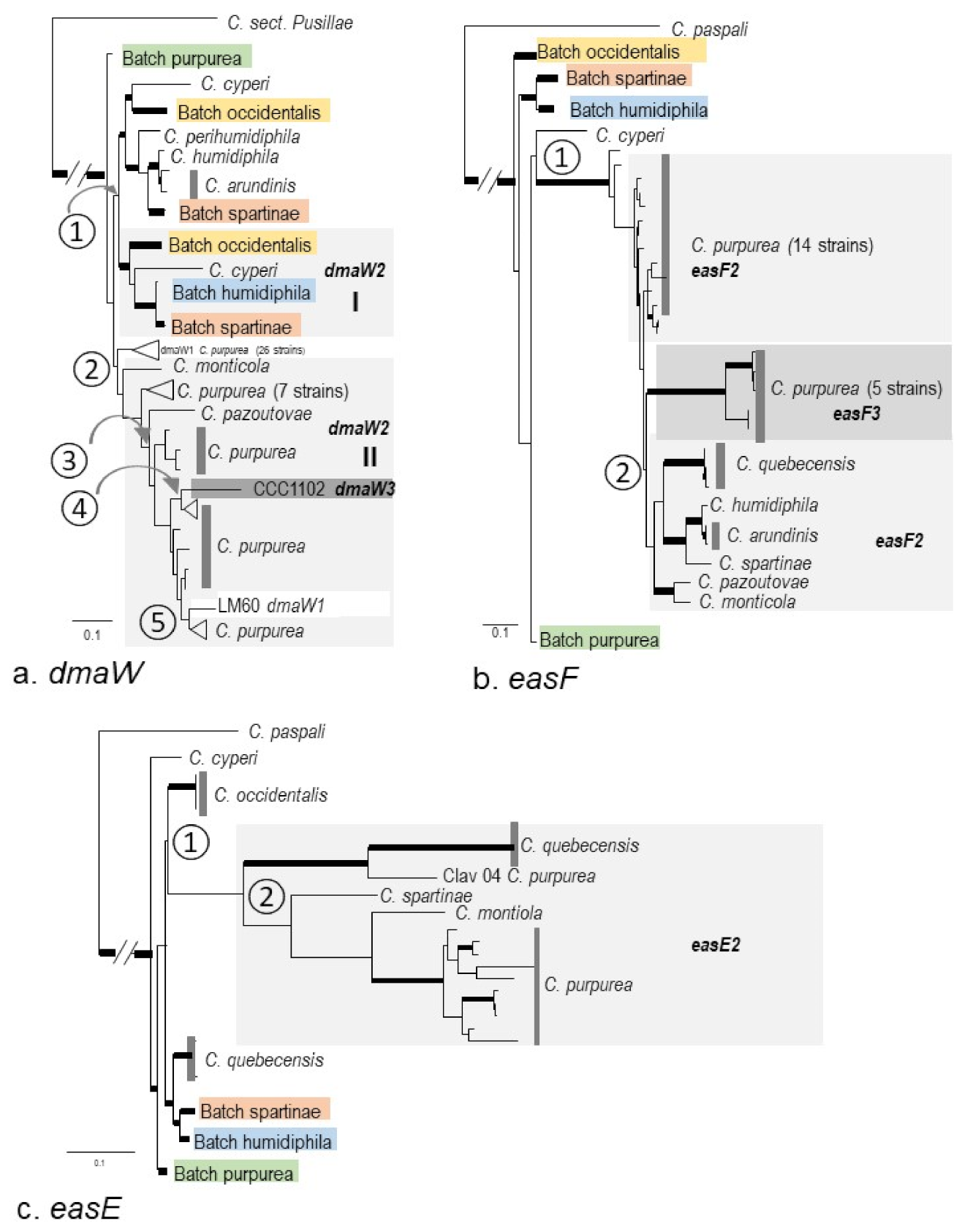
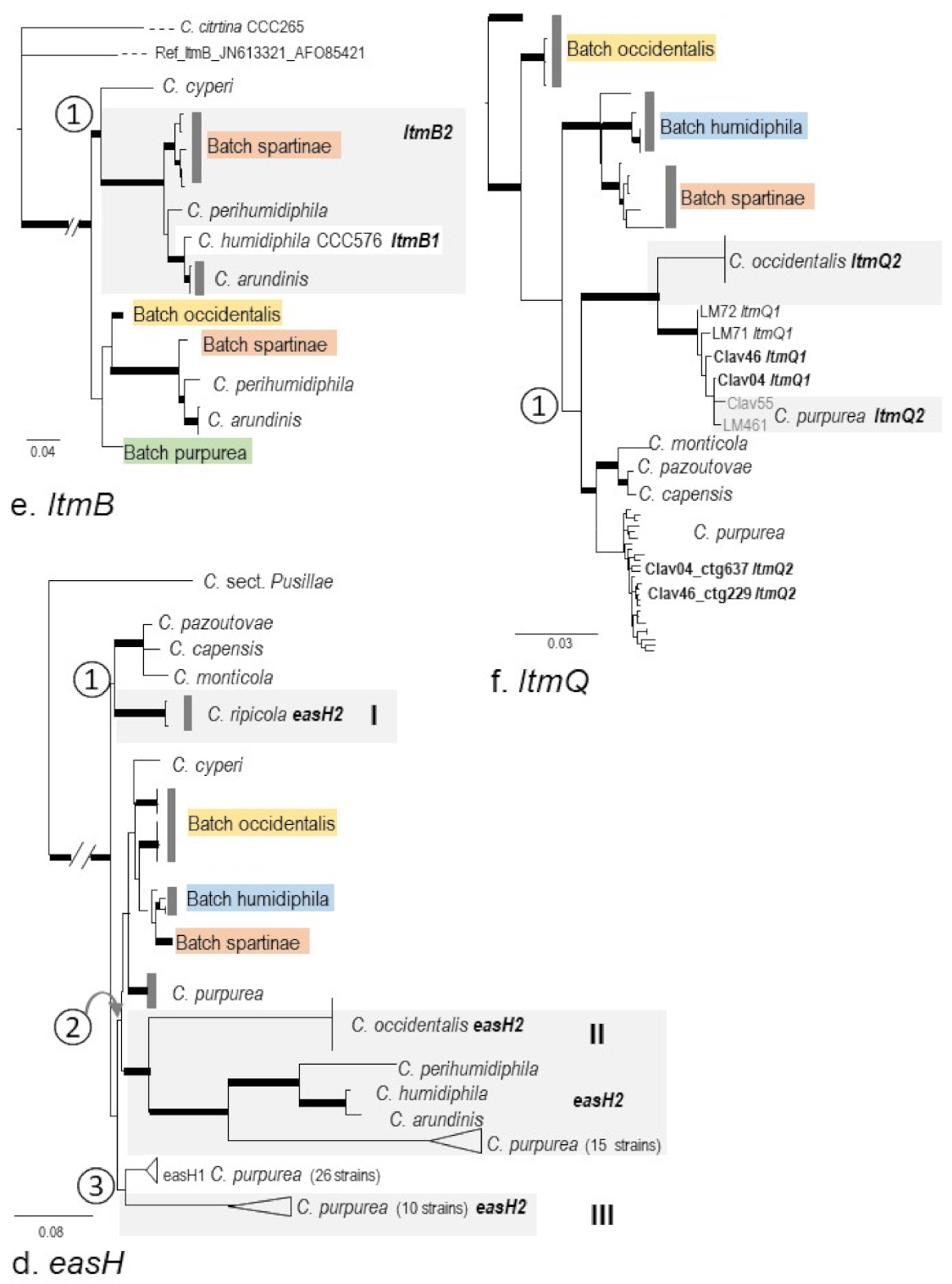
2.3. Phylogenies of eas and idt/ltm Genes
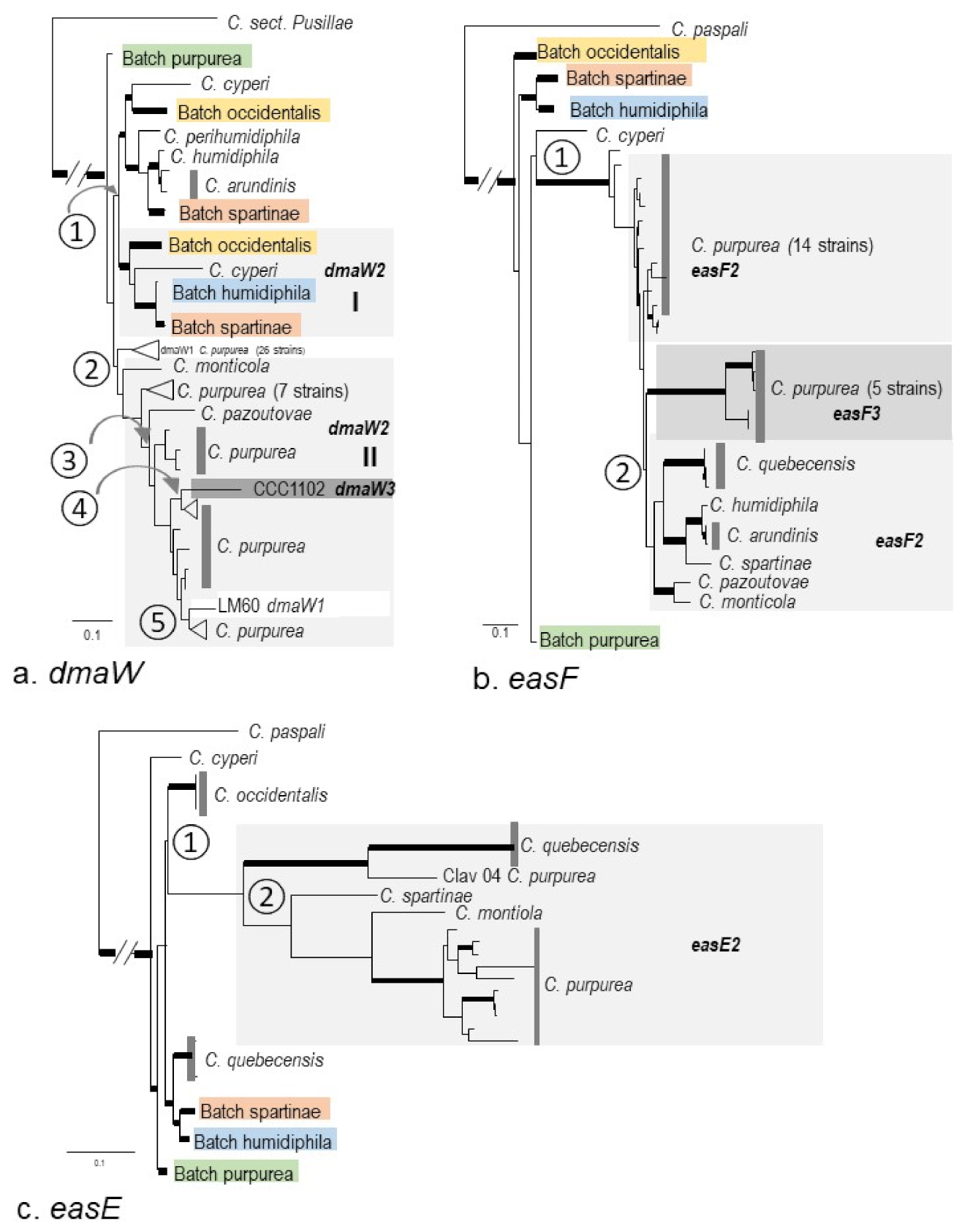
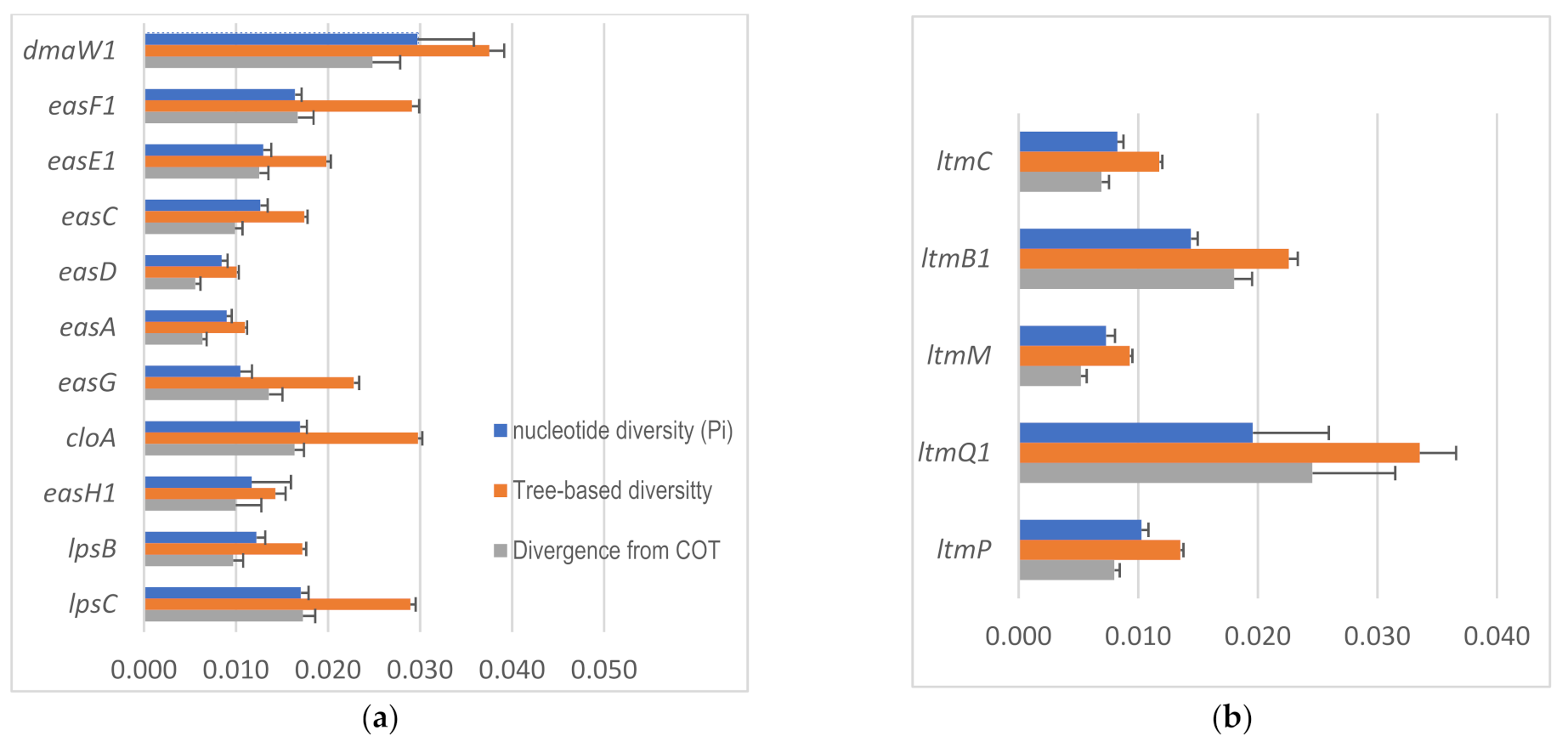
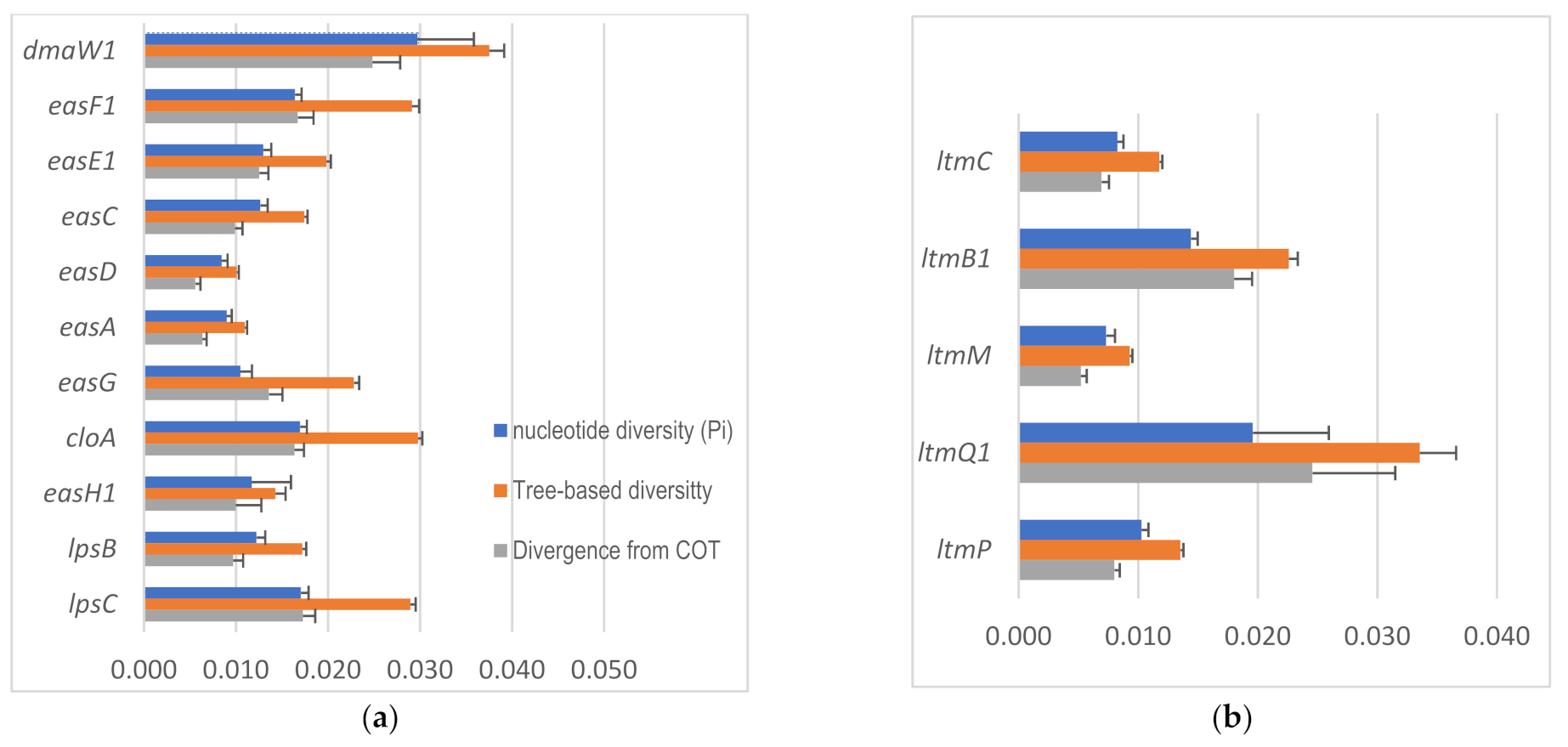
2.4. Intraspecific Genetic Variation within C. purpurea
3. Discussion
3.1. Correlations between the Presence/Absence of Alkaloid Genes and Alkaloid Production
3.2. Macro-Evolution of the Gene Clusters—Frequent Gene Duplications and Losses
3.3. Micro-Evolution of eas Genes within C. purpurea—An Approximate Hourglass Model
4. Materials and Methods
4.1. Genome Aquisition
4.2. Alkaloid Gene Screening and Extraction
4.3. Phylogenetic Analyses
4.4. Intraspecific Gene Diversity and Divergence Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tenberge, K.B. Biology and life strategy of the ergot fungi. In Ergot: The Genus Claviceps; Kren, V., Cvak, L., Eds.; Hardwood Academic Publishers (republished 2006 by Taylor and Francise e-Library): Amsterdam, The Netherlands, 1999; pp. 25–56. [Google Scholar]
- Luttrell, E.S. Host-parasite relationships and development of the ergot sclerotium in Claviceps purpurea. Can. J. Bot. 1980, 58, 942–958. [Google Scholar] [CrossRef]
- Píchová, K.; Pažoutová, S.; Kostovčík, M.; Chudíčková, M.; Stodůlková, E.; Novák, P.; Flieger, M.; van der Linde, E.; Kolařík, M. Evolutionary history of ergot with a new infrageneric classification (Hypocreales: Clavicipitaceae: Claviceps). Mol. Phylogenetics Evol. 2018, 123, 73–87. [Google Scholar] [CrossRef]
- Liu, M.; Overy, D.P.; Cayouette, J.; Shoukouhi, P.; Hicks, C.; Bisson, K.; Sproule, A.; Wyka, S.A.; Broders, K.; Popovic, Z.; et al. Four phylogenetic species of ergot from Canada and their characteristics in morphology, alkaloid production, and pathogenicity. Mycologia 2020, 112, 974–988. [Google Scholar] [CrossRef]
- Campbell, W.P.; Freisen, H.A. The control of ergot in cereal crops. Plant Dis. Rep. 1959, 43, 1266–1267. [Google Scholar]
- European Food Safety Authority. Scientific opinion on ergot allkaloids in food and feed. EFSA J. 2012, 10, 2798. [Google Scholar]
- Tfelt-Hansen, P.; Saxena, P.R.; Dahlöf, C.; Pascual, J.; Láinez, M.; Henry, P.; Diener, H.-C.; Schoenen, J.; Ferrari, M.D.; Goadsby, P.J. Ergotamine in the acute treatment of migraine: A review and European consensus. Brain 2000, 123, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.L., Jr. Ergot and its alkaloids. Am. J. Pharm. Educ. 2006, 70, 98. [Google Scholar] [CrossRef]
- Barger, G. Ergot and Ergotism: A Monograph Based on the Dohme Lecture Delivered in Johns Hopkins University, Baltimore; Gurney and Jackson: London, UK, 1931. [Google Scholar]
- Belser-Ehrlich, S.; Harper, A.; Hussey, J.; Hallock, R. Human and cattle ergotism since 1900: Symptoms, outbreaks, and regulations. Toxicol. Ind. Health 2013, 29, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Schardl, C.L.; Panaccione, D.G.; Tudzynski, P.; Geoffrey, A.C. Ergot alkaloids—Biology and molecular biology. In The Alkaloids: Chemistry and Biology; Academic Press: Cambridge, MA, USA, 2006; Volume 63, pp. 45–86. [Google Scholar]
- Tudzynski, P.; Holter, K.; Correia, T.; Arntz, C.; Grammel, N.; Keller, U. Evidence for an ergot alkaloid gene cluster in Claviceps purpurea. Mol. Gen. Genet. 1999, 261, 133–141. [Google Scholar] [CrossRef]
- Young, C.A.; Schardl, C.L.; Panaccione, D.G.; Florea, S.; Takach, J.E.; Charlton, N.D.; Moore, N.; Webb, J.S.; Jaromczyk, J. Genetics, genomics and evolution of ergot alkaloid diversity. Toxins 2015, 7, 1273–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.L.; Panaccione, D.G. Diversification of ergot alkaloids in natural and modified fungi. Toxins 2015, 7, 201–218. [Google Scholar] [CrossRef] [Green Version]
- Knaus, H.G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garcia-Calvo, M.; Helms, L.M.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P.; Smith, A.B., 3rd; et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef]
- Smith, M.M.; Warren, V.A.; Thomas, B.S.; Brochu, R.M.; Ertel, E.A.; Rohrer, S.; Schaeffer, J.; Schmatz, D.; Petuch, B.R.; Tang, Y.S.; et al. Nodulisporic acid opens insect glutamate-gated chloride channels: Identification of a new high affinity modulator. Biochemistry 2000, 39, 5543–5554. [Google Scholar] [CrossRef] [PubMed]
- Turland, N.J.; Wiersema, J.H.; Barrie, F.R.; Greuter, W.; Hawksworth, D.L.; Herendeen, P.S.; Knapp, S.; Kusber, W.-H.; Li, D.-Z.; Marhold, K. International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) Adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017. Regnum Vegetabile 159; Koeltz Botanical Books: Glashütten, Germany, 2018; Volume 159. [Google Scholar]
- Botha, C.J.; Kellerman, T.S.; Fourie, N. A tremorgenic mycotoxicosis in cattle caused by Paspalum distichum (L.) infected by Claviceps paspali. J. S. Afr. Vet. Assoc. 1996, 67, 36–37. [Google Scholar] [PubMed]
- Prestidge, R.A. Causes and control of perennial ryegrass staggers in New Zealand. Agric. Ecosyst. Environ. 1993, 44, 283–300. [Google Scholar] [CrossRef]
- Uhlig, S.; Botha, C.J.; Vrålstad, T.; Rolén, E.; Miles, C.O. Indole−diterpenes and ergot alkaloids in Cynodon dactylon (bermuda grass) infected with Claviceps cynodontis from an outbreak of tremors in cattle. J. Agric. Food Chem. 2009, 57, 11112–11119. [Google Scholar] [CrossRef] [PubMed]
- Kozák, L.; Szilágyi, Z.; Tóth, L.; Pócsi, I.; Molnár, I. Functional characterization of the idtF and idtP genes in the Claviceps paspali indole diterpene biosynthetic gene cluster. Folia Microbiol. 2020, 65, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Saikia, S.; Takemoto, D.; Tapper, B.A.; Lane, G.A.; Fraser, K.; Scott, B. Functional analysis of an indole-diterpene gene cluster for lolitrem B biosynthesis in the grass endosymbiont Epichloë festucae. FEBS Lett. 2012, 586, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Young, C.A.; Bryant, M.K.; Christensen, M.J.; Tapper, B.A.; Bryan, G.T.; Scott, B. Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol. Genet. Genom. MGG 2005, 274, 13–29. [Google Scholar] [CrossRef]
- Young, C.A.; Felitti, S.; Shields, K.; Spangenberg, G.; Johnson, R.D.; Bryan, G.T.; Saikia, S.; Scott, B. A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet. Biol. 2006, 43, 679–693. [Google Scholar] [CrossRef]
- Young, C.A.; Tapper, B.A.; May, K.; Moon, C.D.; Schardl, C.L.; Scott, B. Indole-diterpene biosynthetic capability of Epichloë endophytes as predicted by ltm gene analysis. Appl. Environ. Microbiol. 2009, 75, 2200–2211. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Ozaki, T.; Harada, M.; Miyasaka, T.; Sato, H.; Miyamoto, K.; Kanazawa, J.; Liu, C.; Maruyama, J.-I.; Adachi, M.; et al. Biosynthesis of indole diterpene lolitrems: Radical-induced cyclization of an epoxyalcohol affording a characteristic lolitremane skeleton. Angew. Chem. Int. Ed. 2020, 59, 17996–18002. [Google Scholar] [CrossRef]
- Uhlig, S.; Egge-Jacobsen, W.; Vrålstad, T.; Miles, C.O. Indole-diterpenoid profiles of Claviceps paspali and Claviceps purpurea from high-resolution Fourier transform Orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2014, 28, 1621–1634. [Google Scholar] [CrossRef]
- Negård, M.; Uhlig, S.; Kauserud, H.; Andersen, T.; Høiland, K.; Vrålstad, T. Links between genetic groups, indole alkaloid profiles and ecology within the grass-parasitic Claviceps purpurea species complex. Toxins 2015, 7, 1431–1456. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, S.; Rangel-Huerta, O.D.; Divon, H.H.; Rolén, E.; Pauchon, K.; Sumarah, M.W.; Vrålstad, T.; Renaud, J.B. Unraveling the ergot alkaloid and indole diterpenoid metabolome in the Claviceps purpurea species complex using lc–hrms/ms diagnostic fragmentation filtering. J. Agric. Food Chem. 2021, 69, 7137–7148. [Google Scholar] [CrossRef] [PubMed]
- Schardl, C.L.; Grossman, R.B.; Nagabhyru, P.; Faulkner, J.R.; Mallik, U.P. Loline alkaloids: Currencies of mutualism. Phytochemistry 2007, 68, 980–996. [Google Scholar] [CrossRef]
- Tanaka, A.; Tapper, B.A.; Popay, A.; Parker, E.J.; Scott, B. A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol. Microbiol. 2005, 57, 1036–1050. [Google Scholar] [CrossRef] [PubMed]
- Duboule, D. Temporal colinearity and the phylotypic progression: A basis for the stability of a vertebrate Bauplan and the evolution of morphologies through heterochrony. Development 1994, 1994, 135–142. [Google Scholar] [CrossRef]
- Slack, J.M.W.; Holland, P.W.H.; Graham, C.F. The zootype and the phylotypic stage. Nature 1993, 361, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Von Baer, K.E. Über Entwickelungsgeschichte der Thiere: Beobachtung und Reflexion; Bei den gebrüdern Bornträger: Königsberg, Russia, 1828; Volume 1. [Google Scholar]
- Richardson, M.K. Vertebrate evolution: The developmental origins of adult variation. Bioessays 1999, 21, 604–613. [Google Scholar] [CrossRef]
- Poe, S.; Wake, M.H. Quantitative tests of general models for the evolution of development. Am. Nat. 2004, 164, 415–422. [Google Scholar] [CrossRef]
- Cheng, X.; Hui, J.H.; Lee, Y.Y.; Wan Law, P.T.; Kwan, H.S. A “developmental hourglass in fungi”. Mol. Biol. Evol. 2015, 32, 1556–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prud‘homme, B.; Gompel, N. Genomic hourglass. Nature 2010, 468, 768–769. [Google Scholar] [CrossRef]
- Quint, M.; Drost, H.-G.; Gabel, A.; Ullrich, K.K.; Bönn, M.; Grosse, I. A transcriptomic hourglass in plant embryogenesis. Nature 2012, 490, 98–101. [Google Scholar] [CrossRef]
- Haeckel, E. Generelle Morphologie der Organismen. Allgemeine Grundzüge der Organischen Formen-Wissenschaft, Mechanisch Begründet Durch Die von Charles Darwin Reformirte Descendenztheorie; G. Reimer: Berlin, Germany, 1866; Volume 1. [Google Scholar]
- Gould, S.J. Ontogeny and Phylogeny; Harvard University Press: Cambridge, MA, USA, 1985. [Google Scholar]
- Cruickshank, T.; Wade, M.J. Microevolutionary support for a developmental hourglass: Gene expression patterns shape sequence variation and divergence in Drosophila. Evol. Dev. 2008, 10, 583–590. [Google Scholar] [CrossRef]
- Wyka, S.A.; Mondo, S.J.; Liu, M.; Dettman, J.; Nalam, V.; Broders, K.D. Whole-genome comparisons of ergot fungi reveals the divergence and evolution of species within the genus Claviceps are the result of varying mechanisms driving genome evolution and host range expansion. Genome Biol. Evol. 2021, 13, evaa267. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, B.D.; Liu, M.; Nguyen, H.D.T.; Lane, F.A.; Morgan, S.W.; De Vos, L.; Wilken, P.M.; Duong, T.A.; Aylward, J.; Coetzee, M.P.A.; et al. Nine draft genome sequences of Claviceps purpurea s.lat., including C. arundinis, C. humidiphila, and C. cf. spartinae, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria eucalypti, and Teratosphaeria destructans. IMA Fungus 2018, 9, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Flieger, M.; Wurst, M.; Shelby, R. Ergot alkaloids--sources, structures and analytical methods. Folia Microbiol. 1997, 42, 3–29. [Google Scholar] [CrossRef]
- Tanda, S. Mycological studies on ergot in Japan (Part 9). Distinct variety of Claviceps purpurea Tul. on Phalaris arundinacea L. and P. arundinacea var. picta L. J. Agric. Sci. Tokyo Nogyo Daigaku 1979, 24, 67–95. [Google Scholar]
- Pažoutová, S.; Parbery, D.P. The taxonomy and phylogeny of Claviceps. In Ergot: The Genus Claviceps; Kren, V., Cvak, L., Eds.; Hardwood Academic Publishers (republished 2006 by Taylor and Francise e-Library): Amsterdam, The Netherlands, 1999; pp. 57–77. [Google Scholar]
- Pažoutová, S.; Olsovská, J.; Linka, M.; Kolínská, R.; Flieger, M. Chemoraces and habitat specialization of Claviceps purpurea populations. Appl. Envron. Microbiol. 2000, 66, 5419–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudzynski, P.; Correia, T.; Keller, U. Biotechnology and genetics of ergot alkaloids. Appl. Microbiol. Biotechnol. 2001, 57, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Panaccione, D.G.; Schardl, C.L. Phylogenetic analyses reveal monophyletic origin of the ergot alkaloid gene dmaW in fungi. Evol. Bioinform. 2009, 5, EBO–S2633. [Google Scholar] [CrossRef]
- Rokas, A.; Wisecaver, J.H.; Lind, A.L. The birth, evolution and death of metabolic gene clusters in fungi. Nat. Rev. Microbiol. 2018, 16, 731–744. [Google Scholar] [CrossRef]
- Lorenz, N.; Haarmann, T.; Pazoutová, S.; Jung, M.; Tudzynski, P. The ergot alkaloid gene cluster: Functional analyses and evolutionary aspects. Phytochemistry 2009, 70, 1822–1832. [Google Scholar] [CrossRef]
- Raff, R.A. The Shape of Life: Genes, Development, and the Evolution of Animal Form; University of Chicago Press: Chicago, IL, USA, 2012. [Google Scholar]
- Galis, F.; van Dooren, T.J.; Metz, J.A. Conservation of the segmented germband stage: Robustness or pleiotropy? Trends Genet. TIG 2002, 18, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Schlosser, G.; Wagner, G.P. Modularity in Development and Evolution; University of Chicago Press: Chicago, IL, USA, 2004. [Google Scholar]
- Panaccione, D.G. Origins and significance of ergot alkaloid diversity in fungi. FEMS Microbiol. Lett. 2005, 251, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shoukouhi, P.; Bisson, K.R.; Wyka, S.A.; Broders, K.D.; Menzies, J.G. Sympatric divergence of the ergot fungus, Claviceps purpurea, populations infecting agricultural and nonagricultural grasses in North America. Ecol. Evol. 2021, 11, 273–293. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Maust, B.S.; Nickle, D.C.; Learn, G.H.; Liu, Y.; Heath, L.; Kosakovsky Pond, S.L.; Mullins, J.I. DIVEIN: A web server to analyze phylogenies, sequence divergence, diversity, and informative sites. Biotechniques 2010, 48, 405–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | BioSample | WGS # | Contigs | Total Length (bp) | Largest Contig (bp) | N50 (bp) | L50 | GC (%) | Coverage (x) | Complete BUSCO’s (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. arundinis | CCC1102 | SAMN11159893 | JAIUSP000000000 | 1406 | 29,878,863 | 375,533 | 55,909 | 156 | 51.42 | 61x | 98.1 | |
| C. capensis | CCC1504 | SAMN11159898 | JAIUSL000000000 | 1497 | 27,462,555 | 202,637 | 39,758 | 198 | 51.69 | 66x | 98.8 | |
| C. cyperi | CCC1219 | SAMN11159895 | JAIUSO000000000 | 2467 | 26,149,012 | 130,032 | 19,946 | 386 | 51.72 | 56x | 98.3 | |
| C. monticola | CCC1483 | SAMN11159896 | JAIUSN000000000 | 1787 | 27,131,110 | 129,905 | 29,639 | 279 | 51.6 | 58x | 98.8 | |
| C. occidentalis | LM77 | SAMN11159879 | JAIURJ000000000 | 2285 | 28,557,246 | 118,746 | 21,556 | 410 | 51.37 | 58x | 97.8 | |
| C. occidentalis | LM84 | SAMN11159876 | JAIURI000000000 | 2119 | 28,639,296 | 133,797 | 23,641 | 389 | 51.39 | 164x | 97.0 | |
| C. pazoutovae | CCC1485 | SAMN11159897 | JAIUSM000000000 | 1619 | 27,544,752 | 151,196 | 35,477 | 229 | 51.7 | 61x | 98.3 | |
| C. purpurea s.s. | Clav04 | SAMN11159846 | JAIUSJ000000000 | 2581 | 30,594,081 | 349,533 | 29,378 | 296 | 51.69 | 46x | 98.8 | |
| C. purpurea s.s. | Clav26 | SAMN11159847 | JAIUSI000000000 | 1821 | 30,253,558 | 299,368 | 36,369 | 242 | 51.48 | 59x | 98.8 | |
| C. purpurea s.s. | Clav46 | SAMN11159848 | JAIUSG000000000 | 1887 | 30,292,940 | 231,314 | 36,582 | 246 | 51.08 | 58x | 99.1 | |
| C. purpurea s.s. | Clav52 | SAMN11159849 | JAIUSE000000000 | 1714 | 29,291,845 | 175,165 | 35,956 | 250 | 51.42 | 60x | 98.9 | |
| C. purpurea s.s. | Clav55 | SAMN11159850 | JAIUSD000000000 | 2023 | 30,195,775 | 203,523 | 33,461 | 261 | 51.55 | 59x | 98.4 | |
| C. purpurea s.s. | LM14 | SAMN11159853 | JAIUSC000000000 | 1888 | 30,259,282 | 163,532 | 32,812 | 268 | 51.74 | 49x | 97.9 | |
| C. purpurea s.s. | LM207 | SAMN11159861 | JAIUSB000000000 | 1910 | 30,165,540 | 260,847 | 31,428 | 273 | 51.74 | 53x | 98.7 | |
| C. purpurea s.s. | LM223 | SAMN11159862 | JAIURY000000000 | 1894 | 30,223,423 | 195,661 | 31,693 | 291 | 51.73 | 74x | 98.4 | |
| C. purpurea s.s. | LM232 | SAMN11159863 | JAIURX000000000 | 1911 | 30,304,653 | 216,996 | 33,376 | 265 | 51.73 | 53x | 98.8 | |
| C. purpurea s.s. | LM233 | SAMN11159864 | JAIURW000000000 | 1928 | 30,249,987 | 183,378 | 33,023 | 273 | 51.74 | 49x | 98.3 | |
| C. purpurea s.s. | LM30 | SAMN11159855 | JAIURV000000000 | 1816 | 30,203,936 | 160,353 | 35,005 | 265 | 51.75 | 64x | 98.4 | |
| C. purpurea s.s. | LM33 | SAMN11159856 | JAIURU000000000 | 2011 | 30,162,301 | 157,176 | 28,954 | 306 | 51.75 | 45x | 97.9 | |
| C. purpurea s.s. | LM39 | SAMN11159857 | JAIURT000000000 | 1797 | 30,183,718 | 168,047 | 34,902 | 258 | 51.75 | 81x | 98.3 | |
| C. purpurea s.s. | LM4 | SAMN11159851 | JAIURS000000000 | 1866 | 30,197,808 | 200,831 | 31,054 | 281 | 51.74 | 64x | 98.3 | |
| C. purpurea s.s. | LM46 | SAMN11159858 | JAIURR000000000 | 1842 | 30,109,785 | 205,399 | 32,503 | 270 | 51.76 | 79x | 98.6 | |
| C. purpurea s.s. | LM461 | SAMN11159865 | JAIURQ000000000 | 2041 | 30,157,824 | 190,247 | 28,928 | 307 | 51.74 | 37x | 97.7 | |
| C. purpurea s.s. | LM469 | SAMN11159866 | JAIURP000000000 | 1836 | 30,218,091 | 199,880 | 34,408 | 269 | 51.74 | 75x | 98.3 | |
| C. purpurea s.s. | LM470 | SAMN11159867 | JAIURO000000000 | 2482 | 30,086,038 | 123,014 | 23,231 | 384 | 51.75 | 26x | 97.9 | |
| C. purpurea s.s. | LM474 | SAMN11159868 | JAIURN000000000 | 1917 | 30,149,711 | 232,504 | 30,855 | 283 | 51.75 | 64x | 98 | |
| C. purpurea s.s. | LM5 | SAMN11159852 | JAIURM000000000 | 1817 | 30,171,863 | 188,144 | 34,174 | 271 | 51.74 | 67x | 98.4 | |
| C. purpurea s.s. | LM60 | SAMN11159859 | JAIURL000000000 | 1871 | 30,274,458 | 180,242 | 31,977 | 275 | 51.73 | 81x | 98.6 | |
| C. purpurea s.s. | LM63 | SAMN20436330 | JAIUSS000000000 | 1674 | 30,276,205 | 210,630 | 40,954 | 218 | 51.79 | 68x | 98.4 | |
| C. purpurea s.s. | LM65 | SAMN20436331 | JAIUSR000000000 | 1822 | 30,277,382 | 206,609 | 37,976 | 241 | 51.78 | 71x | 98.4 | |
| C. purpurea s.s. | LM71 | SAMN11159860 | JAIURK000000000 | 1919 | 30,241,564 | 172,997 | 32,324 | 282 | 51.76 | 168x | 98 | |
| C. purpurea s.s. | LM72 | SAMN20436332 | JAIUSQ000000000 | 1986 | 30,160,156 | 282,506 | 36,805 | 249 | 51.81 | 63x | 98.4 | |
| C. quebecensis | Clav32 | SAMN11159882 | JAIUSH000000000 | 1362 | 28,435,427 | 248,888 | 42,252 | 192 | 51.61 | 64x | 99.0 | |
| C. quebecensis | Clav50 | SAMN11159881 | JAIUSF000000000 | 1404 | 28,499,699 | 294,425 | 47,797 | 178 | 51.6 | 59x | 98.8 | |
| C. ripicola | LM219 | SAMN11159874 | JAIUSA000000000 | 1847 | 30,428,256 | 154,690 | 34,898 | 254 | 51.39 | 55x | 97.1 | |
| C. ripicola c.f. | LM220 | SAMN11159873 | JAIURZ000000000 | 1662 | 30,409,961 | 205,881 | 43,971 | 211 | 51.43 | 91x | 97.7 | |
| C. spartinae | CCC535 | SAMN11159888 | JAIUSK000000000 | 2017 | 28,974,645 | 142,723 | 28,332 | 300 | 51.38 | 60x | 98.1 | |
| Assemblies from previous studies | ||||||||||||
| C. arundinis | CCC1102 | SAMN11159893 | SRPS01 | 1406 | 29,878,863 | 375,533 | 55,909 | 156 | 51.42 | 61x | 97.7 * | |
| C. africana | CCC489 | SAMN11159887 | SRPY01 | 5329 | 31,933,801 | 98,049 | 12,225 | 752 | 44.68 | 56x | 95 * | |
| C. arundinis | LM583 | SAMN08798359 | QEQZ01 | 1613 | 30,055,381 | 164,904 | 39,306 | 223 | 51.42 | 69x | 96.9 | |
| C. citrina | CCC265 | SAMN11159885 | SRQA01 | 4830 | 25,056,896 | 81,802 | 8747 | 871 | 47.57 | 64x | 92.2* | |
| C. digitariae | CCC659 | SAMN11159892 | SRPT01 | 3821 | 31,170,596 | 116,859 | 16,077 | 572 | 45.5 | 57x | 95.9 * | |
| C. humidiphila | LM576 | SAMN08798355 | QERB01 | 1831 | 30,488,243 | 190,085 | 34,787 | 261 | 51.51 | 77x | 97.9 | |
| C. lovelessii | CCC647 | SAMN11159891 | SRPU01 | 8201 | 34,575,813 | 65,439 | 5747 | 1781 | 43.61 | 53x | 91.6 * | |
| C. maximensis | CCC398 | SAMN11159886 | SRPZ01 | 2317 | 29,114,417 | 192,851 | 37,101 | 230 | 46.66 | 58x | 98.3 * | |
| C. occidentalis | LM78 | SAMN08800200 | QEQY01 | 2321 | 28,571,683 | 125,459 | 21,416 | 422 | 51.37 | 64x | 97.3 | |
| C. perihumidiphila | LM81 | SAMN08800226 | QEQX01 | 1423 | 30,694,913 | 232,029 | 46,526 | 192 | 51.5 | 140x | 96.9 | |
| C. purpurea s.s. | LM28 | SAMN08797627 | QERD01 | 1930 | 30,251,797 | 260,842 | 31,815 | 274 | 51.74 | 49x | 97.9 | |
| C. purpurea s.s. | LM582 | SAMN08798357 | QERA01 | 2207 | 30,199,509 | 132,072 | 27,199 | 334 | 51.74 | 89x | 98.6 | |
| C. pusilla | CCC602 | SAMN11159889 | SRPW01 | 9171 | 37,319,484 | 83,555 | 5659 | 1917 | 41.84 | 52x | 90.9 * | |
| C. quebecensis | LM458 | SAMN08851611 | QEQW01 | 1700 | 35,882,593 | 1,850,351 | 41,784 | 191 | 51.87 | 78x | 98.0 | |
| C. ripicola | LM454 | SAMN08798353 | QERC01 | 2108 | 30,692,668 | 189,162 | 28,587 | 314 | 51.37 | 156x | 97.9 | |
| C. ripicola | LM218 | SAMN08798202 | QERE01 | 1630 | 30,598,250 | 206,723 | 39,763 | 232 | 51.4 | 146x | 97.6 | |
| C. sorghi | CCC632 | SAMN11159890 | SRPV01 | 7206 | 31,897,900 | 112,296 | 6643 | 1389 | 45.24 | 60x | 89.9 * | |
| Section | Organism | Asbl * | Sample | lpsC | easA | lpsB | cloA | easC | easD | easE | easF | easG | dmaW | easH | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E1 | E2 | F1 | F2 | F3 | W1 | W2 | W3 | H1 | H2 | |||||||||||||||||||||
| Claviceps | C. arundinis | WF | CCC1102 | 418 | /411 | 411 | 411 | 411 | 411 | 411 | 411 | 411 | 273 | 411 | 411 | /583 | 273 | 707 | 583 | |||||||||||
| SW | CCC1102 | 157 | 157 | 157 | 157 | 157 | 157 | 157 | 73 | 157 | 157 | 73 | 305 | 157 | 458 | |||||||||||||||
| BW | LM583 | 455 | 455 | 455 | 455 | 455 | 455 | 455 | 187 | 455 | /805 | 805 | 187 | 805 | 822 | |||||||||||||||
| C. capensis | WF | CCC1504 | 173 | 173 | 173 | 173 | 173 | 173 | 173 | 173 | 173 | 1354 | 347 | |||||||||||||||||
| C. cyperi | WF | CCC1219 | 277 | 277 | 277 | 277 | 277 | 277 | 277 | 277 | 277 | 277 | /2037 | 2094 | /696 | 525 | ||||||||||||||
| C. humidiphila | BW | LM576 | 599 | 259 | 259 | 259 | 259 | 259 | 259 | 259 | 390 | 259 | 259 | 390 | 259 | 701 | ||||||||||||||
| C. monticola | WF | CCC1483 | 367 | 367 | 367 | 367 | 367 | 367 | 367 | 986 | 367 | 986 | 367 | 367 | /1745 | 986 | /966 | 494 | ||||||||||||
| C. occidentalis | WF | LM77 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 1262 | 702 | 1887 | 1693 | |||||||||||||||
| BW | LM78 | 192 | 192 | 192 | 192 | 192 | 192 | 192 | 192 | 192 | 1273 | 722 | 1871 | 1675 | ||||||||||||||||
| WF | LM84 | 290 | 290 | 290 | 290 | 290 | 290 | 290 | 290 | /1715 | 1715 | 1340 | 721 | 1779 | 1618 | |||||||||||||||
| C. pazoutovae | WF | CCC1485 | 307 | 307 | 307 | 307 | 307 | 307 | 307 | 307 | 767 | 307 | 307 | /1479 | 767 | /622 | 430 | |||||||||||||
| C. perihumidiphila | BW | LM81 | 114 | 114 | 114 | 114 | 114 | 114 | 114 | 114 | /604 | 604 | 359 | 604 | 710 | |||||||||||||||
| C. purpruea | WF | Clav52 | 131 | 131 | 131 | 131 | 131 | 131 | 131 | 739 | 131 | /1395 | 739 | 1395 | 1395 | /923 | 879 | 923 | 835 | |||||||||||
| WF | Clav04 | 71 | 71 | 71 | 71 | 71 | 71 | 71 | 1407 | 71 | 1514 | /2293 | 71 | 71 | /993 | 2459 | /594 | 993 | 874 | |||||||||||
| WF | Clav26 | 105 | 105 | 105 | 105 | 105 | 105 | 105 | 105 | 816 | /1596 | 105 | 105 | /1453 | 1596 | 759 | 836 | |||||||||||||
| WF | Clav46 | 201 | 201 | 201 | 201 | 201 | 201 | 201 | 1255 | 201 | /1358 | 1255 | /1668 | 1358 | 1358 | /928 | 1481 | /1294 | 928 | 1043 | ||||||||||
| WF | Clav55 | 419 | 419 | 419 | 419 | 419 | 419 | 419 | 1226 | 419 | /1416 | 1226 | /1797 | 1416 | 1416 | /1316 | 1797 | /1399 | 1316 | 1168 | ||||||||||
| WF | LM14 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | 85 | /699 | 1391 | /539 | 699 | 716 | ||||||||||||||
| WF | LM207 | 374 | 374 | 374 | 374 | 374 | 374 | 374 | 374 | 374 | 374 | /1169 | 1625 | /1506 | 1169 | 1183 | ||||||||||||||
| WF | LM223 | 444 | 444 | 444 | 444 | 444 | 444 | 444 | 444 | /1556 | 1027 | /1718 | 1556 | 1556 | /783 | 1460 | 783 | 762 | ||||||||||||
| WF | LM232 | 112 | 112 | 112 | 112 | 112 | 112 | 112 | 112 | 112 | 112 | 1843 | 1148 | 908 | ||||||||||||||||
| WF | LM233 | 89 | 89 | 89 | 89 | 89 | 89 | 89 | 89 | 89 | /658 | 658 | 533 | 658 | 692 | |||||||||||||||
| BW | LM28 | 126 | 126 | 126 | 126 | 126 | 126 | 126 | 126 | 126 | 126 | 1874 | /563 | 126 | 1208 | |||||||||||||||
| WF | LM30 | 88 | 88 | 88 | 88 | 88 | 88 | 88 | 88 | 88 | /1022 | 1022 | 610 | 1022 | 1180 | |||||||||||||||
| WF | LM33 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | 106 | /1343 | 1781 | /649 | 1343 | 1210 | ||||||||||||||
| WF | LM39 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 855 | 100 | /1391 | 855 | /1766 | 1340 | 1391 | 1391 | /753 | 1340 | /546 | 753 | 703 | |||||||||
| WF | LM4 | 152 | 152 | 152 | 152 | 152 | 152 | 152 | 152 | 152 | 152 | /1570 | 1797 | 857 | 726 | |||||||||||||||
| WF | LM46 | 209 | 209 | 209 | 209 | 209 | 209 | 209 | 209 | 209 | 209 | 1386 | 209 | 1025 | ||||||||||||||||
| WF | LM461 | 121 | 121 | 121 | 121 | 121 | 121 | /1443 | 1443 | 1006 | 1443 | 1006 | 1643 | 1006 | 1006 | /1505 | 1846 | /1490 | 1505 | 1402 | ||||||||||
| WF | LM469 | 91 | 91 | 91 | 91 | 91 | 91 | 91 | 980 | 91 | /1419 | 980 | /1609 | 1727 | 1419 | 1419 | /1018 | 1727 | /419 | 1018 | 680 | |||||||||
| WF | LM470 | 294 | 294 | 294 | 294 | 294 | 294 | 294 | 2080 | 294 | /1428 | 2080 | /2165 | 1428 | 1428 | 2358 | 949 | 736 | ||||||||||||
| WF | LM474 | 158 | 158 | 158 | 158 | 158 | 158 | 158 | 829 | 158 | /1493 | 829 | /1365 | 1493 | 1493 | /788 | 1782 | /41 | 788 | 719 | ||||||||||
| WF | LM5 | 121 | 121 | 121 | 121 | 121 | 121 | 121 | 121 | 121 | 121 | /1217 | 1546 | /679 | 1217 | 1198 | ||||||||||||||
| BW | LM582 | 98 | 98 | 98 | 98 | 98 | 98 | 98 | 98 | 98 | 98 | 1036 | 800 | |||||||||||||||||
| WF | LM60 | 333 | 333 | 333 | 333 | 333 | 333 | 333 | 333 | 333 | /1791 | 1790 | /1266 | 1480 | /676 | 1266 | 1242 | |||||||||||||
| WF | LM71 | 159 | 159 | 159 | 159 | 159 | 159 | 159 | 159 | 655 | 655 | 931 | 655 | 727 | ||||||||||||||||
| WF | LM63 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 1178 | 160 | 1178 | 985 | 621 | /160 | 621 | 985 | 621 | 918 | ||||||||||||
| WF | LM65 | 21 | 21 | 21 | 21 | 21 | 21 | 21 | 21 | 1018 | 254 | 21 | 21 | 254 | 21 | |||||||||||||||
| WF | LM72 | 190 | 190 | 190 | 190 | 190 | 190 | 190 | 1156 | 190 | 1156 | 5 | 5 | 912 | 5 | |||||||||||||||
| C. quebecensis | WF | Clav32 | 308 | 308 | 308 | 308 | 308 | 308 | 308 | 753 | 308 | /201 | 753 | /730 | 201 | 201 | 730 | 201 | ||||||||||||
| WF | Clav50 | 227 | 227 | 227 | 227 | 227 | 227 | 227 | 731 | 227 | /231 | 731 | /679 | 231 | 231 | 679 | 231 | |||||||||||||
| BW | LM458 | 165 | 165 | 165 | 165 | 165 | 165 | 165 | 1446 | 165 | 1446 | /1061 | 498 | 498 | 1061 | 498 | ||||||||||||||
| C. ripicola | BW | LM218 | 81 | 81 | 81 | 81 | 81 | 81 | 81 | 81 | 81 | 81 | 527 | 81 | 820 | |||||||||||||||
| WF | LM219 | 78 | 78 | 78 | 78 | 78 | 78 | 78 | 78 | 78 | 78 | 533 | 78 | |||||||||||||||||
| WF | LM220 | 77 | 77 | 77 | 588 | 588 | 588 | 588 | 588 | 588 | 588 | 95 | 588 | 285 | ||||||||||||||||
| BW | LM454 | 120 | 120 | 120 | 623 | 623 | 623 | 623 | 623 | 623 | 107 | 623 | ||||||||||||||||||
| C. spartinae | WF | CCC535 | 1156 | /1375 | 853 | 853 | 853 | 680 | 680 | 680 | 27 | 680 | 27 | 680 | 680 | 27 | 680 | |||||||||||||
| Pusillae | C. africana | SW | CCC489 | 424 | 424 | 424 | 424 | 424 | 424 | 424 | 424 | /1076 | ||||||||||||||||||
| C. digitariae | SW | CCC659 | 403 | 403 | ||||||||||||||||||||||||||
| C. lovelessii | SW | CCC647 | 1632 | 1632 | 2885 | 2885 | /4933 | 556 | /4933 | 556 | 556 | 556 | 556 | 143 | ||||||||||||||||
| C. maximensis | WF | CCC398 | ||||||||||||||||||||||||||||
| C. pusilla | SW | CCC602 | 3688 | 1435 | 3688 | 3968 | /3891 | 3968 | 3968 | /3180 | 3180 | 1918 | 1809 | |||||||||||||||||
| C. sorghi | SW | CCC632 | 2692 | 2475 | 2475 | 186 | 186 | 186 | 186 | 186 | 3370 | |||||||||||||||||||
| Section | Organism | Assembly * | ltmQ1 | ltmQ2 | ltmP | ltmB1 | ltmB2 | ltmC | ltmS | ltmM | ltmG | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Citrinae | C. citrina | WF | CCC265 | 1947 | 1947 | 582 | 2211 | |||||
| Claviceps | C. arundinis | WF | CCC1102 | 50 | 50 | 50 | 332 | 50 | 50 | 50 | ||
| BW | LM583 | 158 | 158 | 158 | 124 | 158 | 158 | 158 | ||||
| C. capensis | WF | CCC1504 | 29 | 29 | 29 | 29 | 29 | 29 | ||||
| C. cyperi | WF | CCC1219 | 25 | 25 | 25 | 25 | ||||||
| C. humidiphila | BW | LM576 | 945 | 945 | 478 | 745 | 745 | 745 | ||||
| C. monticola | WF | CCC1483 | 568 | 568 | 591 | 591 | 591 | 591 | ||||
| C. occidentalis | WF | LM77 | 1456 | 1898 | 1456 | 1538 | 1538 | 1538 | 657 | |||
| BW | LM78 | 985 | 1877 | 985 | 985 | 985 | 985 | 691 | ||||
| WF | LM84 | 376 | 1789 | 376 | 376 | 376 | 376 | 376 | ||||
| C. pazoutovae | WF | CCC1485 | 225 | 225 | 185 | 185 | 185 | 185 | ||||
| C. perihumidiphila | BW | LM81 | 27 | 27 | 27 | 7 | 27 | 27 | 27 | |||
| C. purpruea | WF | Clav52 | 174 | 174 | 174 | 174 | 174 | 1230 | ||||
| WF | Clav04 | 130 | 637 | 130 | 130 | 130 | 130 | 130 | ||||
| WF | Clav26 | 116 | 116 | 116 | 116 | 116 | 116 | |||||
| WF | Clav46 | 43 | 229 | 43 | 43 | 43 | 43 | 43 | ||||
| WF | Clav55 | 1358/1838/1286 | 1444 | 1286 | 557 | 557 | 557 | 557 | ||||
| WF | LM14 | 243 | 243 | 243 | 243 | 243 | 243 | |||||
| WF | LM207 | 255 | 255 | 255 | 255 | 255 | 255 | |||||
| WF | LM223 | 327 | 327 | 327 | 327 | 327 | 327 | |||||
| WF | LM232 | 315 | 315 | 315 | 315 | 315 | 315 | |||||
| WF | LM233 | 7 | 7 | 7 | 7 | 7 | 7 | |||||
| BW | LM28 | 258 | 258 | 258 | 258 | 258 | 258 | |||||
| WF | LM30 | 87 | 87 | 87 | 87 | 87 | 87 | |||||
| WF | LM33 | 51 | 51 | 51 | 51 | 51 | 51 | |||||
| WF | LM39 | 192 | 192 | 192 | 192 | 192 | 192 | |||||
| WF | LM4 | 361 | 361 | 361 | 361 | 1220 | 1220 | |||||
| WF | LM46 | 29 | 29 | 29 | 29 | 29 | 29 | |||||
| WF | LM461 | 529/65 | 1592 | 965 | 965 | 965 | 965 | 1500 | ||||
| WF | LM469 | 37 | 37 | 37 | 37 | 37 | 37 | |||||
| WF | LM470 | 646 | 787 | 787 | 787 | 787 | 787 | |||||
| WF | LM474 | 243 | 243 | 243 | 243 | 243 | 243 | |||||
| WF | LM5 | 17 | 17 | 17 | 17 | 17 | 17 | |||||
| BW | LM582 | 112 | 112 | 112 | 112 | 112 | 112 | |||||
| WF | LM60 | 765 | 765 | 765 | 765 | 765 | 440 | |||||
| WF | LM71 | 393 | 1283 | 977 | 977 | 977 | 977 | |||||
| WF | LM63 | 433 | 433 | 433 | 433 | 433 | 433 | |||||
| WF | LM65 | 406 | 406 | 406 | 406 | 406 | 406 | |||||
| WF | LM72 | 549/1151 | 1151 | 361 | 361 | 361 | 361 | |||||
| C. quebecensis | WF | Clav32 | 56 | 56 | 56 | 56 | 56 | 56 | ||||
| WF | Clav50 | 91 | 91 | 91 | 91 | 91 | 91 | |||||
| BW | LM458 | 536 | 536/1563 | 475 | 475 | 475 | 475 | |||||
| C. ripicola | BW | LM218 | 191 | 191 | 191 | 136 | 191 | 191 | 191 | |||
| WF | LM219 | 395 | 395 | 638 | 589 | 638 | 638 | 638 | ||||
| WF | LM220 | 368 | 368 | 368 | 591 | 368 | 368 | 368 | ||||
| BW | LM454 | 138 | 138 | 764 | 949 | 764 | 764 | 764 | ||||
| C. spartinae | WF | CCC535 | 225 | 225 | 225 | 47 | 225 | 1212 | 1212 |
| Biosynthesis Genes | # of Sequences 1 | Total # of Sites | # of Sites (Excluding Indel) | Segregating Sites | Ratio | # of Haplotypes | Haplotype (Gene) Diversity | Nucleotide Diversity | Average Number of Nucleotide Differences | Tree-Based Divergence from COT 2 | Tree-Based Diversity | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | n° | n | s | s/n | h | Hd | Pi | Std dev | K | Mean | Std Error | Mean | Std. Error | ||
| Ergot Alkaloid (eas) Genes | |||||||||||||||
| dmaW | 35 | 1516 | 921 | 196 | 0.213 | 32 | 0.995 | 0.055 | 0.004 | 50.523 | 0.062 | 0.004 | 0.086 | 0.002 | |
| dmaW1 | 21 | 1480 | 938 | 154 | 0.164 | 19 | 0.99 | 0.030 | 0.006 | 27.886 | 0.025 | 0.003 | 0.038 | 0.002 | |
| dmaW2 | 14 | 1516 | 923 | 102 | 0.111 | 13 | 0.989 | 0.042 | 0.004 | 39.11 | 0.042 | 0.008 | 0.065 | 0.004 | |
| easF | 32 | 1232 | 630 | 121 | 0.192 | 24 | 0.972 | 0.058 | 0.015 | 36.548 | 0.049 | 0.011 | 0.081 | 0.003 | |
| easF1 | 25 | 1232 | 642 | 40 | 0.062 | 17 | 0.953 | 0.016 | 0.001 | 10.54 | 0.017 | 0.002 | 0.029 | 0.001 | |
| easF2 | 8 | 1232 | 634 | 101 | 0.159 | 8 | 1 | 0.048 | 0.020 | 30.464 | 0.031 | 0.015 | 0.055 | 0.010 | |
| easE | 34 | 2283 | 1607 | 577 | 0.359 | 28 | 0.979 | 0.085 | 0.021 | 135.938 | 0.085 | 0.028 | 0.147 | 0.008 | |
| easE1 | 28 | 1921 | 1891 | 112 | 0.059 | 22 | 0.968 | 0.013 | 0.001 | 24.526 | 0.013 | 0.001 | 0.020 | 0.000 | |
| easE2 | 7 | 2283 | 1614 | 536 | 0.332 | 7 | 1 | 0.132 | 0.034 | 212.238 | 0.115 | 0.042 | 0.218 | 0.032 | |
| easC | 28 | 1508 | 1503 | 89 | 0.059 | 23 | 0.979 | 0.013 | 0.001 | 10.034 | 0.010 | 0.001 | 0.017 | 0.000 | |
| easD | 28 | 851 | 846 | 37 | 0.044 | 22 | 0.976 | 0.008 | 0.001 | 7.1510 | 0.006 | 0.001 | 0.010 | 0.000 | |
| easA | 28 | 1143 | 1143 | 51 | 0.045 | 21 | 0.966 | 0.009 | 0.001 | 10.286 | 0.006 | 0.000 | 0.011 | 0.000 | |
| easG | 28 | 1151 | 923 | 57 | 0.062 | 20 | 0.974 | 0.010 | 0.001 | 9.6720 | 0.014 | 0.001 | 0.023 | 0.001 | |
| cloA | 28 | 2754 | 2110 | 173 | 0.082 | 23 | 0.979 | 0.017 | 0.001 | 35.796 | 0.016 | 0.001 | 0.030 | 0.000 | |
| easH | 51 | 1195 | 569 | 247 | 0.434 | 23 | 0.936 | 0.132 | 0.014 | 75.151 | 0.102 | 0.013 | 0.154 | 0.004 | |
| easH1 | 28 | 947 | 943 | 64 | 0.068 | 15 | 0.944 | 0.012 | 0.004 | 11.053 | 0.010 | 0.003 | 0.014 | 0.001 | |
| easH2 | 23 | 1195 | 571 | 232 | 0.406 | 15 | 0.858 | 0.168 | 0.017 | 95.85 | 0.150 | 0.033 | 0.188 | 0.010 | |
| lpsB | 28 | 4006 | 3961 | 255 | 0.064 | 23 | 0.979 | 0.012 | 0.001 | 48.484 | 0.010 | 0.001 | 0.017 | 0.000 | |
| lpsC | 27 | 5431 | 5416 | 421 | 0.078 | 25 | 0.994 | 0.017 | 0.001 | 92.379 | 0.017 | 0.001 | 0.029 | 0.001 | |
| Indole-Diterpene/Lolitrem (idt/ltm) Genes | |||||||||||||||
| ltmC | 28 | 1249 | 1246 | 60 | 0.048 | 25 | 0.992 | 0.008 | 0.000 | 10.299 | 0.007 | 0.001 | 0.012 | 0.000 | |
| ltmB1 | 28 | 871 | 868 | 38 | 0.044 | 21 | 0.971 | 0.014 | 0.001 | 12.516 | 0.066 | 0.010 | 0.024 | 0.001 | |
| ltmM | 28 | 1766 | 1731 | 74 | 0.043 | 25 | 0.992 | 0.007 | 0.001 | 12.68 | 0.005 | 0.000 | 0.009 | 0.000 | |
| ltmQ | 25 | 2180 | 2119 | 293 | 0.138 | 24 | 0.997 | 0.019 | 0.006 | 41 | 0.026 | 0.010 | 0.040 | 0.004 | |
| ltmQ1 | 24 | 2180 | 2119 | 292 | 0.138 | 23 | 0.996 | 0.020 | 0.006 | 41.486 | 0.025 | 0.007 | 0.034 | 0.003 | |
| ltmP | 28 | 1949 | 1895 | 111 | 0.059 | 24 | 0.981 | 0.010 | 0.001 | 19.479 | 0.008 | 0.000 | 0.014 | 0.000 | |
| ltmS | 28 | 955 | 924 | 34 | 0.037 | 24 | 0.989 | 0.007 | 0.001 | 6.839 | 0.008 | 0.001 | 0.015 | 0.000 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by Her Majesty the Queen in Right of Canada, as represented by Agriculture and Agri Food Canada. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Findlay, W.; Dettman, J.; Wyka, S.A.; Broders, K.; Shoukouhi, P.; Dadej, K.; Kolařík, M.; Basnyat, A.; Menzies, J.G. Mining Indole Alkaloid Synthesis Gene Clusters from Genomes of 53 Claviceps Strains Revealed Redundant Gene Copies and an Approximate Evolutionary Hourglass Model. Toxins 2021, 13, 799. https://doi.org/10.3390/toxins13110799
Liu M, Findlay W, Dettman J, Wyka SA, Broders K, Shoukouhi P, Dadej K, Kolařík M, Basnyat A, Menzies JG. Mining Indole Alkaloid Synthesis Gene Clusters from Genomes of 53 Claviceps Strains Revealed Redundant Gene Copies and an Approximate Evolutionary Hourglass Model. Toxins. 2021; 13(11):799. https://doi.org/10.3390/toxins13110799
Chicago/Turabian StyleLiu, Miao, Wendy Findlay, Jeremy Dettman, Stephen A. Wyka, Kirk Broders, Parivash Shoukouhi, Kasia Dadej, Miroslav Kolařík, Arpeace Basnyat, and Jim G. Menzies. 2021. "Mining Indole Alkaloid Synthesis Gene Clusters from Genomes of 53 Claviceps Strains Revealed Redundant Gene Copies and an Approximate Evolutionary Hourglass Model" Toxins 13, no. 11: 799. https://doi.org/10.3390/toxins13110799
APA StyleLiu, M., Findlay, W., Dettman, J., Wyka, S. A., Broders, K., Shoukouhi, P., Dadej, K., Kolařík, M., Basnyat, A., & Menzies, J. G. (2021). Mining Indole Alkaloid Synthesis Gene Clusters from Genomes of 53 Claviceps Strains Revealed Redundant Gene Copies and an Approximate Evolutionary Hourglass Model. Toxins, 13(11), 799. https://doi.org/10.3390/toxins13110799