Structural Modeling of Cell Wall Peptidase CwpFM (EntFM) Reveals Distinct Intrinsically Disordered Extensions Specific to Pathogenic Bacillus cereus Strains

 ,
, 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. CwpFM is Present and Highly Conserved within the Members of the B. cereus Group
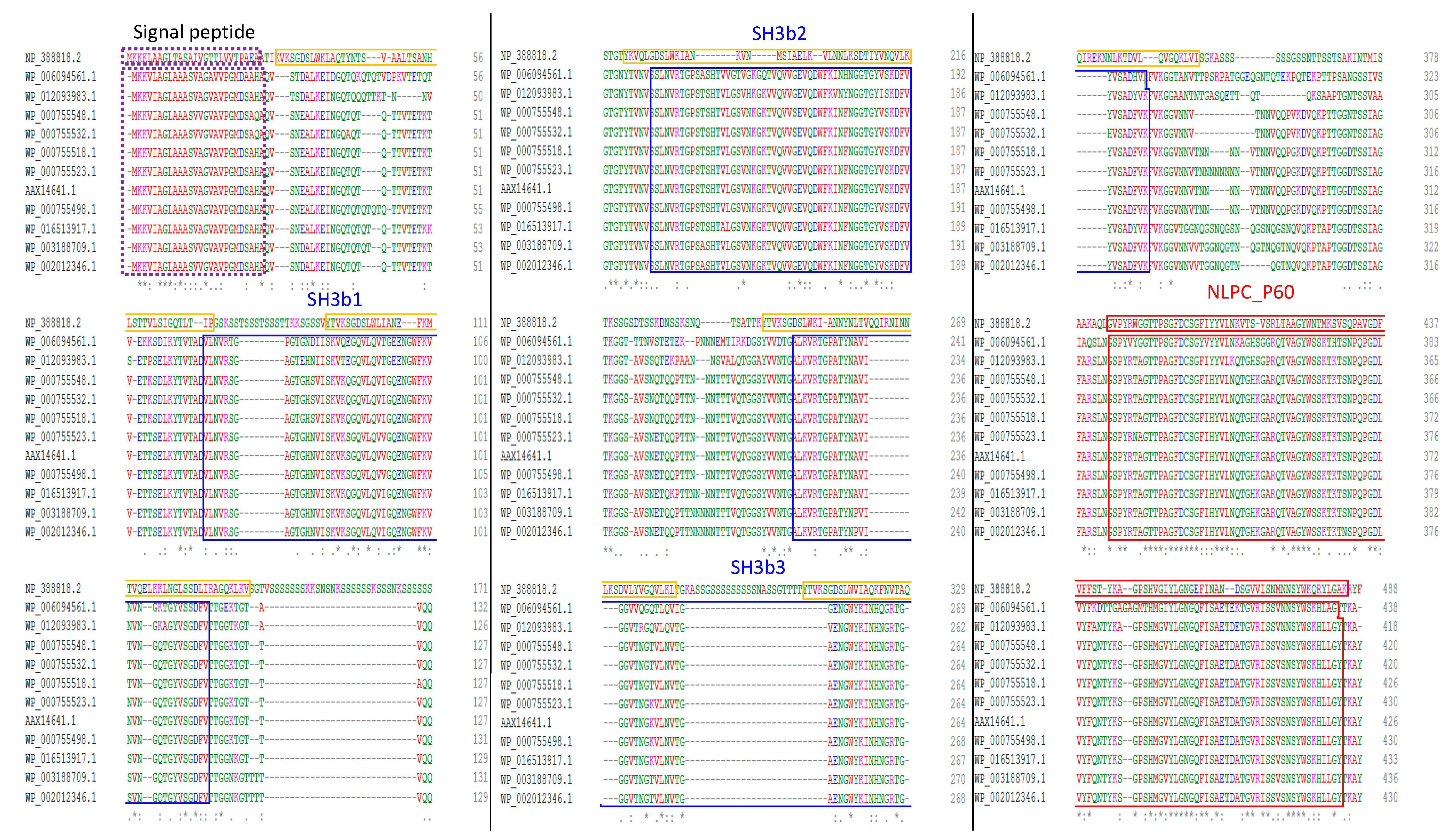
2.2. Domain Organization of the CwpFM Homologues from the B. cereus Group Indicates the Presence of Cell Wall Degradation Domains
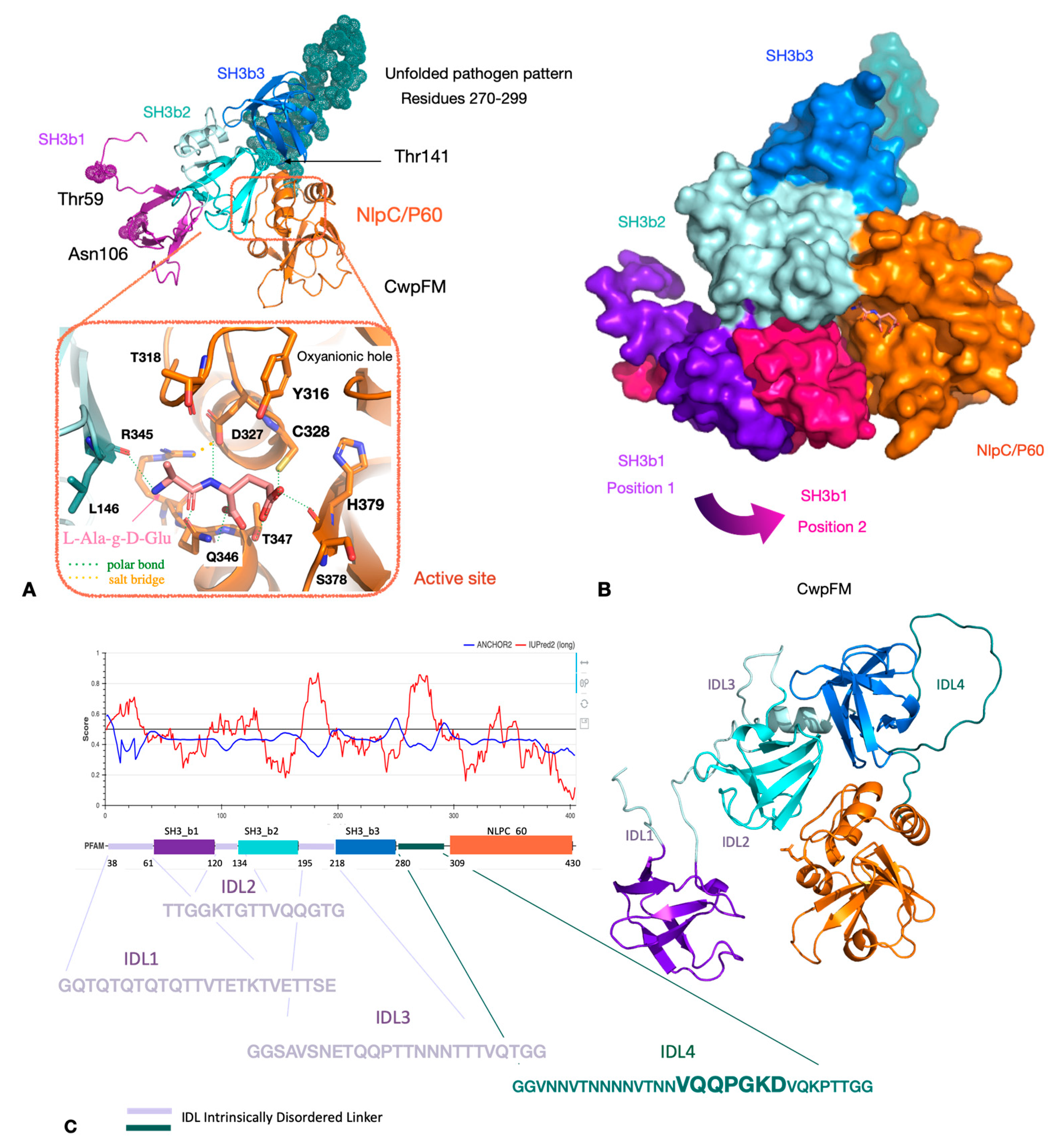
2.3. Homology Modeling of CwpFM 3D Structure Highlights a Conserved Modular Topology Composed of Three SH3b Domains Connected to an NLPC_P60 Cysteine Peptidase Domain, and Emphasizes the Presence of Disordered Connecting Domains that Could Play a Role in Specificity and Affinity towards PG and Proteins
2.4. CwpFM Structure Is Able to Accommodate a PG Fragment
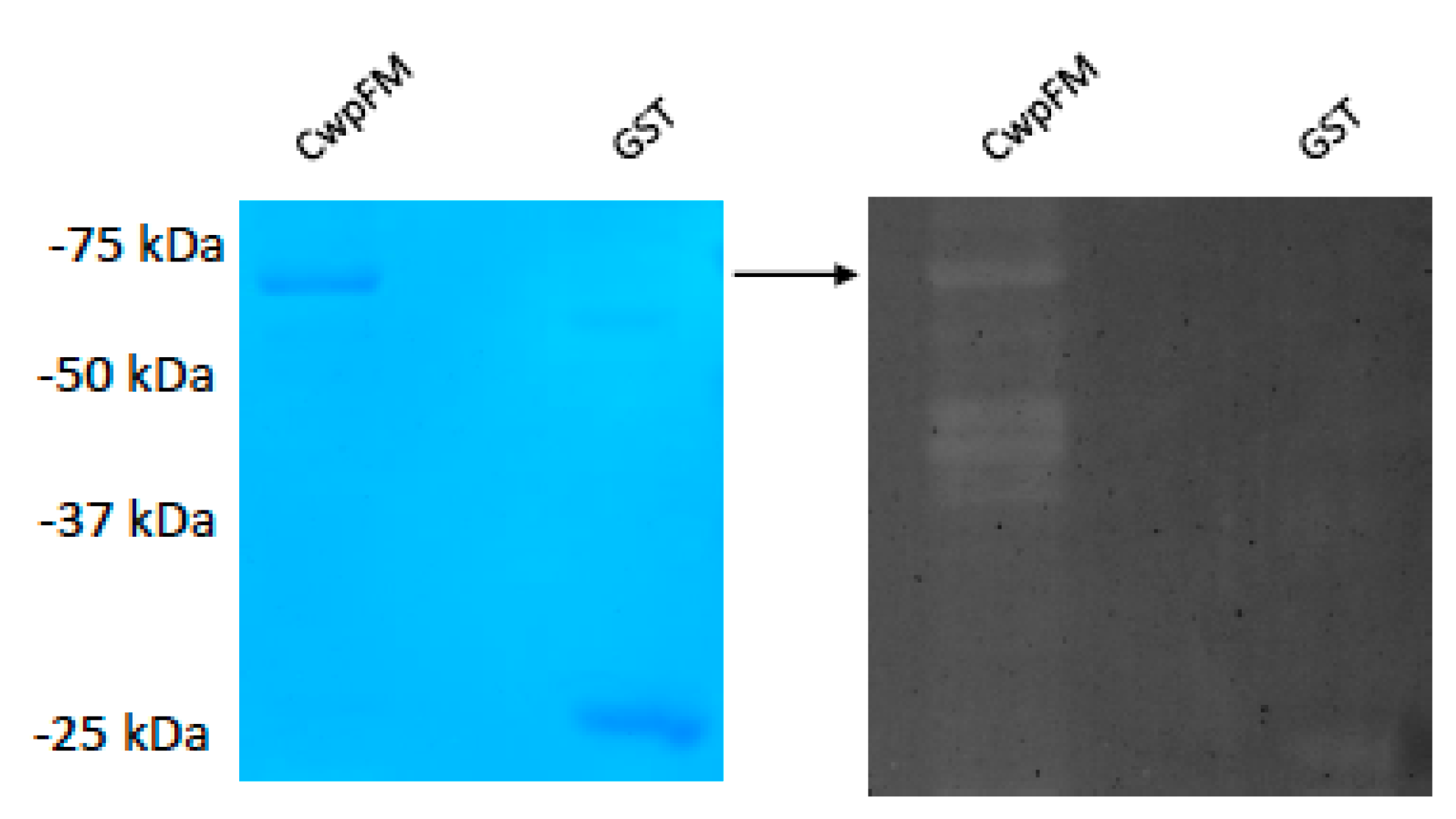
2.5. B. cereus CwpFM Shows a Weak PG Hydrolase Activity
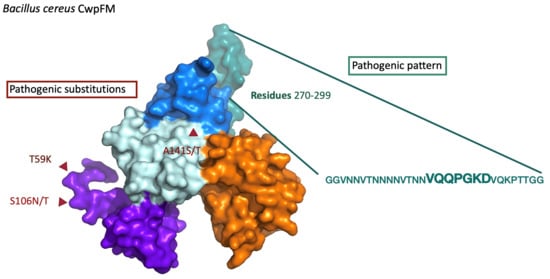
2.6. CwpFM Distribution in B. cereus Strains of Various Pathogenic Potentials
2.7. Analysis of the Differences at the 2D and 3D Levels
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain Sequences
4.2. Sequence Alignment, Conserved Motifs and Domain Analysis of the Proteins
4.3. D Alignments and Clustering of the Strains
4.4. Molecular Modeling
4.5. Docking
4.6. Expression and Purification of the CwpFM-GST-Tagged Protein
4.7. Zymogram
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcorlo, M.; Martinez-Caballero, S.; Molina, R.; Hermoso, J.A. Carbohydrate recognition and lysis by bacterial peptidoglycan hydrolases. Curr. Opin. Struct. Biol. 2017, 44, 87–100. [Google Scholar] [CrossRef]
- Do, T.; Page, J.E.; Walker, S. Uncovering the activities, biological roles, and regulation of bacterial cell wall hydrolases and tailoring enzymes. J. Biol. Chem. 2020, 295, 3347–3361. [Google Scholar] [CrossRef]
- Dorr, T.; Moynihan, P.J.; Mayer, C. Editorial: Bacterial Cell Wall Structure and Dynamics. Front. Microbiol. 2019, 10, 2051. [Google Scholar] [CrossRef]
- He, J.; Fu, W.; Zhao, S.; Zhang, C.; Sun, T.; Jiang, T. Lack of MSMEG_6281, a peptidoglycan amidase, affects cell wall integrity and virulence of Mycobacterium smegmatis. Microb. Pathog. 2019, 128, 405–413. [Google Scholar] [CrossRef]
- Healy, C.; Gouzy, A.; Ehrt, S. Peptidoglycan Hydrolases RipA and Ami1 Are Critical for Replication and Persistence of Mycobacterium tuberculosis in the Host. MBio 2020, 11, e03315-19. [Google Scholar] [CrossRef]
- Mahasenan, K.V.; Batuecas, M.T.; De Benedetti, S.; Kim, C.; Rana, N.; Lee, M.; Hesek, D.; Fisher, J.F.; Sanz-Aparicio, J.; Hermoso, J.A.; et al. Catalytic Cycle of Glycoside Hydrolase BglX from Pseudomonas aeruginosa and Its Implications for Biofilm Formation. ACS Chem. Biol. 2020, 15, 189–196. [Google Scholar] [CrossRef]
- Lonergan, Z.R.; Nairn, B.L.; Wang, J.; Hsu, Y.P.; Hesse, L.E.; Beavers, W.N.; Chazin, W.J.; Trinidad, J.C.; VanNieuwenhze, M.S.; Giedroc, D.P.; et al. An Acinetobacter baumannii, Zinc-Regulated Peptidase Maintains Cell Wall Integrity during Immune-Mediated Nutrient Sequestration. Cell Rep. 2019, 26, 2009–2018.e6. [Google Scholar] [CrossRef]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front. Microbiol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Ramarao, N.; Lereclus, D.; Sorokin, A. The Bacillus cereus group. In Molecular Medical Microbiology, 2nd ed.; Academic Press: Cambrige, MA, USA, 2015; Volume III, pp. 1041–1078. [Google Scholar]
- Kolsto, A.B.; Tourasse, N.J.; Okstad, O.A. What sets Bacillus anthracis apart from other Bacillus species? Annu. Rev. Microbiol. 2009, 63, 451–476. [Google Scholar] [CrossRef]
- Jessberger, N.; Krey, V.M.; Rademacher, C.; Bohm, M.E.; Mohr, A.K.; Ehling-Schulz, M.; Scherer, S.; Martlbauer, E. From genome to toxicity: A combinatory approach highlights the complexity of enterotoxin production in Bacillus cereus. Front. Microbiol. 2015, 6, 560. [Google Scholar]
- Celandroni, F.; Salveti, S.; Senesi, S.; Ghelardi, E. Bacillus thuringiensis membrane-damaging toxins acting on mammalian cells. FEMS 2014, 361, 95–103. [Google Scholar]
- Carlson, C.R.; Caugant, D.A.; Kolstø, A.-B. Genotypic diversity among Bacillus cereus and Bacillus thuringiensis strains. Appl. Environ. Microbiol. 1994, 60, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef]
- Stevens, M.J.A.; Tasara, T.; Klumpp, J.; Stephan, R.; Ehling-Schulz, M.; Johler, S. Whole-genome-based phylogeny of Bacillus cytotoxicus reveals different clades within the species and provides clues on ecology and evolution. Sci. Rep. 2019, 9, 1984. [Google Scholar] [CrossRef]
- Glasset, B.; Herbin, S.; Guillier, L.; Cadel-Six, S.; Vignaud, M.L.; Grout, J.; Pairaud, S.; Michel, V.; Hennekinne, J.A.; Ramarao, N.; et al. Bacillus cereus-induced food-borne outbreaks in France, 2007 to 2014: Epidemiology and genetic characterisation. Eurosurveillance 2016, 21, 30413. [Google Scholar] [CrossRef]
- Agata, N.; Ohta, M.; Mori, M.; Isobe, M. A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus cereus. FEMS Microbiol. Lett. 1995, 129, 17–20. [Google Scholar]
- Mahler, H.; Pasa, A.; Kramer, J.; Schulte, P.; Scoging, A.; Bar, W.; Krahenbuhl, S. Fulminant liver failure in association with the emetic toxin of Bacillus cereus. N. Engl. J. Med. 1997, 336, 1142–1148. [Google Scholar] [CrossRef]
- Naranjo, M.; Denayer, S.; Botteldoorn, N.; Delbrassinne, L.; Veys, J.; Waegenaere, J.; Sirtaine, N.; Driesen, R.B.; Sipido, K.R.; Mahillon, J.; et al. Sudden death of a young adult associated with Bacillus cereus food poisoning. J. Clin. Microbiol. 2011, 49, 4379–4381. [Google Scholar] [CrossRef]
- Ramarao, N.; Sanchis, V. The pore-forming hemolysins of Bacillus cereus: A review. Toxins 2013, 5, 1119–1139. [Google Scholar] [CrossRef]
- Granum, E.; Lund, T. Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Lett. 2006, 157, 223–228. [Google Scholar] [CrossRef]
- Fox, D.; Mathur, A.; Xue, Y.; Liu, Y.; Tan, W.H.; Feng, S.; Pandey, A.; Ngo, C.; Hayward, J.A.; Atmosukarto, I.I.; et al. Bacillus cereus non-haemolityc enterotoxin activates the NLRP3 inflammasome. Nat. Commun. 2020, 11, 760–775. [Google Scholar] [CrossRef] [PubMed]
- Haydar, A.; Tran, S.L.; Guillemet, E.; Darrigo, C.; Perchat, S.; Lereclus, D.; Coquet, L.; Jouenne, T.; Ramarao, N. InhA1-Mediated Cleavage of the Metalloprotease NprA Allows Bacillus cereus to Escape From Macrophages. Front. Microbiol. 2018, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.L.; Guillemet, E.; Ngo-Camus, M.; Clybouw, C.; Puhar, A.; Moris, A.; Gohar, M.; Lereclus, D.; Ramarao, N. Hemolysin II is a Bacillus cereus virulence factor that induces apoptosis of macrophages. Cell Microbiol. 2011, 13, 92–108. [Google Scholar] [CrossRef]
- Guillemet, E.; Lereec, A.; Tran, S.L.; Royer, C.; Barbosa, I.; Sansonetti, P.; Lereclus, D.; Ramarao, N. The bacterial DNA repair protein Mfd confers resistance to the host nitrogen immune response. Sci. Rep. 2016, 6, 29349. [Google Scholar] [CrossRef]
- Guillemet, E.; Cadot, C.; Tran, S.L.; Guinebretiere, M.H.; Lereclus, D.; Ramarao, N. The InhA metalloproteases of Bacillus cereus contribute concomitantly to virulence. J. Bacteriol. 2010, 192, 286–294. [Google Scholar] [CrossRef]
- Darrigo, C.; Guillemet, E.; Dervyn, R.; Ramarao, N. The Bacterial Mfd Protein Prevents DNA Damage Induced by the Host Nitrogen Immune Response in a NER-Independent but RecBC-Dependent Pathway. PLoS ONE 2016, 11, e0163321. [Google Scholar] [CrossRef]
- Asano, S.; Nukumizu, Y.; Bando, H.; Iizuka, T.; Yamamoto, T. Cloning of novel enterotoxin genes from Bacillus cereus and Bacillus thuringiensis. Appl. Environ. Microbiol. 1997, 63, 1054–1057. [Google Scholar] [CrossRef]
- Tran, S.L.; Guillemet, E.; Gohar, M.; Lereclus, D.; Ramarao, N. CwpFM (EntFM) is a Bacillus cereus potential cell wall peptidase implicated in adhesion, biofilm formation and virulence. J. Bacteriol. 2010, 192, 2638–2642. [Google Scholar] [CrossRef]
- Shinagawa, K.; Sugiyama, J.; Terada, T.; Matsusaka, N.; Sugii, S. Improved methods for purification of an enterotoxin produced by Bacillus cereus. FEMS Microbiol. Lett. 1991, 80, 1–6. [Google Scholar] [CrossRef]
- Boonchai, N.; Asano, S.; Bando, H.; Wiwat, C. Study on cytotoxicity and nucleotide sequences of Enterotoxin FM of Bacillus cereus isolated from various food sources. J. Med. Assoc. Thail. 2008, 91, 1425–1432. [Google Scholar]
- Chon, J.W.; Yim, J.H.; Kim, H.S.; Kim, D.H.; Kim, H.; Oh, D.H.; Kim, S.K.; Seo, K.H. Quantitative Prevalence and Toxin Gene Profile of Bacillus cereus from Ready-to-Eat Vegetables in South Korea. Foodborne Pathog. Dis. 2015, 12, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Kim, J.M.; Cho, S.H.; Oh, H.S.; Choi, N.J.; Oh, D.H. Toxin genes profiles and toxin production ability of Bacillus cereus isolated from clinical and food samples. J. Food Sci. 2011, 76, T25–T29. [Google Scholar] [CrossRef] [PubMed]
- Ngamwongsatit, P.; Buasri, W.; Puianariyanon, P.; Pulsrikarn, C.; Ohba, M.; Assavanig, A.; Panbangreb, W. Broad distribution of enterotoxin genes (hblCDA, nheABC, cytK, and entFM) among Bacillus thuringiensis and Bacillus cereus as shown by novel primers. Int. J. Food Microbiol. 2008, 121, 352–356. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Abdubek, P.; Astakhova, T.; Axelrod, H.L.; Bakolitsa, C.; Cai, X.; Carlton, D.; Chen, C.; Chiu, H.J.; Chiu, M.; et al. Structure of the gamma-D-glutamyl-L-diamino acid endopeptidase YkfC from Bacillus cereus in complex with L-Ala-gamma-D-Glu: Insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66 Pt 10, 1354–1364. [Google Scholar] [CrossRef]
- Rodriguez, J.; Haydinger, C.D.; Peet, D.J.; NGuyen, L.K.; von Kriegsheim, A. Asparagine hydroxylation is likely to be a reversible post-translational modification. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ehling-SChulz, M.; Lereclus, D.; Koehler, T. The Bacillus cereus group-Bacillus species with pathogenic potential. Microbiol. Spect. 2019, 7, 10. [Google Scholar]
- Ramarao, N.; Tran, S.L.; Marin, M.; Vidic, J. Advanced method for detection of Bacillus cereus and its pathogenic factors. Sensors 2020, 20, 2667. [Google Scholar] [CrossRef]
- Martinez, M.; Alzari, P.M.; André-Leroux, G. Signalling mechanism in Procaryotes. In Bacterial Membranes: Structural and Molecular Biology; Remaut, H., Fronzes, R., Eds.; Honrizon Scientific Press: Poole, UK, 2014. [Google Scholar]
- Do, T.; Schaefer, K.; Santiago, A.G.; Coe, K.A.; Fernandes, P.B.; Kahne, D.; Pinho, M.G.; Walker, S. Staphylococcus aureus cell growth and division are regulated by an amidase that trims peptides from uncrosslinked peptidoglycan. Nat. Microbiol. 2020, 5, 291–303. [Google Scholar] [CrossRef]
- Mitkowski, P.; Jagielska, E.; Nowak, E.; Bujnicki, J.M.; Stefaniak, F.; Niedzialek, D.; Bochtler, M.; Sabala, I. Structural bases of peptidoglycan recognition by lysostaphin SH3b domain. Sci. Rep. 2019, 9, 5965. [Google Scholar] [CrossRef]
- Xu, Q.; Mengin-Lecreulx, D.; Liu, X.W.; Patin, D.; Farr, C.L.; Grant, J.C.; Chiu, H.J.; Jaroszewski, L.; Knuth, M.W.; Godzik, A.; et al. Insights into Substrate Specificity of NlpC/P60 Cell Wall Hydrolases Containing Bacterial SH3 Domains. MBio 2015, 6, e02327-14. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Delgado, L.S.; Walters-Morgan, H.; Salamaga, B.; Robertson, A.J.; Hounslow, A.M.; Jagielska, E.; Sabala, I.; Williamson, M.P.; Lovering, A.L.; Mesnage, S. Two-site recognition of Staphylococcus aureus peptidoglycan by lysostaphin SH3b. Nat. Chem. Biol. 2020, 16, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.O.; Kong, M.; Kim, I.; Bai, J.; Cha, S.; Kim, B.; Ryu, K.S.; Ryu, S.; Suh, J.Y. Structural Basis for Cell-Wall Recognition by Bacteriophage PBC5 Endolysin. Structure 2019, 27, 1355–1365.e4. [Google Scholar] [CrossRef] [PubMed]
- Benesik, M.; Novacek, J.; Janda, L.; Dopitova, R.; Pernisova, M.; Melkova, K.; Tisakova, L.; Doskar, J.; Zidek, L.; Hejatko, J.; et al. Role of SH3b binding domain in a natural deletion mutant of Kayvirus endolysin LysF1 with a broad range of lytic activity. Virus Genes 2018, 54, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Margot, P.; Pagni, M.; Karamata, D. Bacillus subtilis 168 gene lytF encodes a gamma-D-glutamate-meso-diaminopimelate muropeptidase expressed by the alternative vegetative sigma factor, sigmaD. Microbiology 1999, 145 Pt 1, 57–65. [Google Scholar] [CrossRef]
- Fukushima, T.; Afkham, A.; Kurosawa, S.; Tanabe, T.; Yamamoto, H.; Sekiguchi, J. A new D,L-endopeptidase gene product, YojL (renamed CwlS), plays a role in cell separation with LytE and LytF in Bacillus subtilis. J. Bacteriol. 2006, 188, 5541–5550. [Google Scholar] [CrossRef]
- Mesnage, S.; Dellarole, M.; Baxter, N.J.; Rouget, J.B.; Dimitrov, J.D.; Wang, N.; Fujimoto, Y.; Hounslow, A.M.; Lacroix-Desmazes, S.; Fukase, K.; et al. Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nat. Commun. 2014, 5, 4269. [Google Scholar] [CrossRef]
- Wong, J.E.; Alsarraf, H.M.; Kaspersen, J.D.; Pedersen, J.S.; Stougaard, J.; Thirup, S.; Blaise, M. Cooperative binding of LysM domains determines the carbohydrate affinity of a bacterial endopeptidase protein. FEBS J. 2014, 281, 1196–1208. [Google Scholar] [CrossRef]
- Carballido-Lopez, R.; Formstone, A.; Li, Y.; Ehrlich, S.D.; Noirot, P.; Errington, J. Actin homolog MreBH governs cell morphogenesis by localization of the cell wall hydrolase LytE. Dev. Cell 2006, 11, 399–409. [Google Scholar] [CrossRef]
- Damman, C.J.; Eggers, C.H.; Samuels, D.S.; Oliver, D.B. Characterization of Borrelia burgdorferi BlyA and BlyB proteins: A prophage-encoded holin-like system. J. Bacteriol. 2000, 182, 6791–6797. [Google Scholar] [CrossRef]
- Blackman, S.A.; Smith, T.J.; Foster, S.J. The role of autolysins during vegetative growth of Bacillus subtilis 168. Microbiology 1998, 144 Pt 1, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Chica, J.; Correa, M.M.; Aceves-Diez, A.E.; Rasschaert, G.; Heyndrickx, M.; Castaneda-Sandoval, L.M. Genomic and Toxigenic Heterogeneity of Bacillus cereus sensu lato Isolated from Ready-to-Eat Foods and Powdered Milk in Day Care Centers in Colombia. Foodborne Pathog. Dis. 2020, 17, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, J.; Fei, P.; Feng, H.; Wang, Y.; Ali, M.A.; Li, S.; Jing, H.; Yang, W. Prevalence, molecular characterization, and antibiotic susceptibility of Bacillus cereus isolated from dairy products in China. J. Dairy Sci. 2020, 103, 3994–4001. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Kim, H.J.; Jeong, M.; Koo, M. Enterotoxin Genes, Antibiotic Susceptibility, and Biofilm Formation of Low-Temperature-Tolerant Bacillus cereus Isolated from Green Leaf Lettuce in the Cold Chain. Foods 2020, 9, 249. [Google Scholar] [CrossRef]
- Gao, T.; Ding, Y.; Wu, Q.; Wang, J.; Zhang, J.; Yu, S.; Yu, P.; Liu, C.; Kong, L.; Feng, Z.; et al. Prevalence, Virulence Genes, Antimicrobial Susceptibility, and Genetic Diversity of Bacillus cereus Isolated From Pasteurized Milk in China. Front. Microbiol. 2018, 9, 533. [Google Scholar] [CrossRef]
- Chon, J.W.; Kim, J.H.; Lee, S.J.; Hyeon, J.Y.; Song, K.Y.; Park, C.; Seo, K.H. Prevalence, phenotypic traits and molecular characterization of emetic toxin-producing Bacillus cereus strains isolated from human stools in Korea. J Appl Microbiol 2012, 112, 1042–1049. [Google Scholar] [CrossRef]
- Yang, Y.; Gu, H.; Yu, X.; Zhan, L.; Chen, J.; Luo, Y.; Zhang, Y.; Lu, Y.; Jiang, J.; Mei, L. Genotypic heterogeneity of emetic toxin producing Bacillus cereus isolates from China. FEMS Microbiol. Lett. 2017, 364, fnw237. [Google Scholar] [CrossRef][Green Version]
- Appadurai, R.; Uversky, V.N.; Srivastava, A. The Structural and Functional Diversity of Intrinsically Disordered Regions in Transmembrane Proteins. J. Membr. Biol. 2019, 252, 273–292. [Google Scholar] [CrossRef]
- Li, Q.; Chevalier, C.; Henry, C.; Richard, C.-A.; Moudjou, M.; Vidic, J. Shadoo binds lipid membranes and undergoes aggregation and fibrillization. Biochem. Biophys. Res. Commun. 2013, 438, 519–525. [Google Scholar] [CrossRef]
- Chevalier, C.; Al Bazzal, A.; Vidic, J.; Février, V.; Bourdieu, C.; Bouguyon, E.; Le Goffic, R.; Vautherot, J.-F.; Bernard, J.; Moudjou, M. PB1-F2 influenza A virus protein adopts a β-sheet conformation and forms amyloid fibers in membrane environments. J. Biol. Chem. 2010, 285, 13233–13243. [Google Scholar] [CrossRef]
- Vidic, J.; Richard, C.-A.; Péchoux, C.; Da Costa, B.; Bertho, N.; Mazerat, S.; Delmas, B.; Chevalier, C. Amyloid assemblies of influenza A virus PB1-F2 protein damage membrane and induce cytotoxicity. J. Biol. Chem. 2016, 291, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.A.; Teng, F.; Nallapareddy, S.R.; Murray, B.E. Pleiotrophic effects of 2 Enterococcus faecalis sagA-like genes, salA and salB, which encode proteins that are antigenic during human infection, on biofilm formation and binding to collagen type i and fibronectin. J. Infect. Dis. 2006, 193, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Chen, G.; Wang, W.; Xia, L.; Wang, Z.; Lu, Y. Identification of a cell-wall peptidase (NlpC/P60) from Nocardia seriolae which induces apoptosis in fathead minnow cells. J. Fish Dis. 2020, 43, 571–581. [Google Scholar] [CrossRef]
- Padhi, A.; Naik, S.K.; Sengupta, S.; Ganguli, G.; Sonawane, A. Expression of Mycobacterium tuberculosis NLPC/p60 family protein Rv0024 induce biofilm formation and resistance against cell wall acting anti-tuberculosis drugs in Mycobacterium smegmatis. Microbes Infect. 2016, 18, 224–236. [Google Scholar] [CrossRef]
- Gao, L.Y.; Pak, M.; Kish, R.; Kajihara, K.; Brown, E.J. A mycobacterial operon essential for virulence in vivo and invasion and intracellular persistence in macrophages. Infect. Immun. 2006, 74, 1757–1767. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Lun, S.; Guo, H.; Ammerman, N.C.; Geiman, D.E.; Bishai, W.R. Rv2190c, an NlpC/P60 family protein, is required for full virulence of Mycobacterium tuberculosis. PLoS ONE 2012, 7, e43429. [Google Scholar] [CrossRef]
- Sela-Abramovich, S.; Chitlaru, T.; Gat, O.; Grosfeld, H.; Cohen, O.; Shafferman, A. Novel and unique diagnostic biomarkers for Bacillus anthracis infection. Appl. Environ. Microbiol. 2009, 75, 6157–6167. [Google Scholar] [CrossRef]
- Wydau-Dematteis, S.; El Meouche, I.; Courtin, P.; Hamiot, A.; Lai-Kuen, R.; Saubamea, B.; Fenaille, F.; Butel, M.J.; Pons, J.L.; Dupuy, B.; et al. Cwp19 Is a Novel Lytic Transglycosylase Involved in Stationary-Phase Autolysis Resulting in Toxin Release in Clostridium difficile. MBio 2018, 9, e00648-18. [Google Scholar] [CrossRef]
- Boudreau, M.A.; Fisher, J.F.; Mobashery, S. Messenger functions of the bacterial cell wall-derived muropeptides. Biochemistry 2012, 51, 2974–2990. [Google Scholar] [CrossRef] [PubMed]
- Schaub, R.E.; Perez-Medina, K.M.; Hackett, K.T.; Garcia, D.L.; Dillard, J.P. Neisseria gonorrhoeae PBP3 and PBP4 Facilitate NOD1 Agonist Peptidoglycan Fragment Release and Survival in Stationary Phase. Infect. Immun. 2019, 87, e00833-18. [Google Scholar] [CrossRef] [PubMed]
- Glasset, B.; Herbin, S.; Granier, S.; Cavalié, L.; Lafeuille, E.; Guérin, C.; Ruimy, R.; Casagrande, F.; Levast, M.; Chautemps, N.; et al. Bacillus cereus, a serious cause of nosocomial infections: Epidemiologic and genetic survey. PLoS ONE 2018, 13, e0194346. [Google Scholar] [CrossRef] [PubMed]
- Guinebretière, M.H.; Broussolle, V.; Nguyen-The, C. Enterotoxigenic profiles of food-poisoning and food-borne Bacillus cereus strains. J. Clin. Microbiol. 2002, 40, 3053–3056. [Google Scholar] [CrossRef] [PubMed]
- Kamar, R.; Gohar, M.; Jehanno, I.; Rejasse, A.; Kallassy, M.; Lereclus, D.; Sanchis, V.; Ramarao, N. Pathogenic potential of Bacillus cereus strains as revealed by phenotypic analysis. J. Clin. Microbiol. 2013, 51, 320–323. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; SØnderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Webb, B.; Salí, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar]
- Mézáros, B.; Erdös, G.; Dostányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acid Res. 2018, 46, 329–337. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Origin/Description | PanC Group | Ref Genome |
|---|---|---|---|---|
| B. cereus | ATCC 14579 | Isolated from a farmhouse in the United States, 1916 | IV | NC_004722.1 |
| B. cereus | ATCC 10987 | Isolated from dairy cheese in Canada, 1930 | III | NC_003909.8 |
| B. thuringiensis | Bt407 cry- | Soil isolate that has been cured of the plasmid that encodes the intesticidal crystalline toxin | IV | NC_018877.1 |
| B. thuringiensis | serovar konkukian str. 97–27 | Soil organism isolated from a severe tissue necrosis of a soldier severely wounded by a land mine explosion in former Yugoslavia, 1995 | III | NC_005957.1 |
| B. mycoides | ATCC 6462 | Also known as DSM 2048; isolated from soil | VI | NZ_CP009692.1 |
| B. pseudomycoides | DSM 12442 | Also known as NRRL B617; isolated from soil in Ghana, 1998 | I | NZ_CM000745.1 |
| B. weihenstephanensis | KBAB4 | Psychrotolerant soil isolate isolated in forest soil in France, 2000 | VI | NC_010184.1 |
| B. anthracis | Ames | Isolated from a dead cow in Texas, 1981 | III | NC_007530.2 |
| B. cytotoxicus | NVH 391-98 | Isolated from a food poisoning outbreak (vegetable puree) in a nursing home for elderly people in France, 1998 | VII | NC_009674.1 |
| B. toyonensis | BCT-7112 | Isolated for use as probiotics in animal nutrition in Japan, 1966 | V | NC_022781.1 |
| Species | Strain | Identities | Gaps | Strand | Gene Identification | Protein Name | Protein Reference | Max Score | Total Scorer | Query Cover | E Value | Per. Ident. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. cereus | ATCC 14579 | 1277/1293 (99%) | 12/1293 (0%) | Plus/Plus | BC_RS09755 | C40 family peptidase | WP_000755498.1 | 838 | 838 | 1.00 | 0.0 | 99.07% |
| B. cereus | ATCC 10987 | 1238/1281 (97%) | 0/1281 (0%) | Plus/Plus | BCE_RS10070 | C40 family peptidase | WP_000755518.1 | 828 | 828 | 1.00 | 0.0 | 97.89% |
| B. thuringiensis | Bt407 cry- | 1268/1293 (98%) | 12/1293 (0%) | Plus/Plus | BTB_RS09950 | C40 family peptidase | WP_000755523.1 | 836 | 836 | 1.00 | 0.0 | 98.84% |
| B. thuringiensis | serovar konkukian str. 97-27 | 1215/1281 (95%) | 18/1281 (1%) | Plus/Plus | BT9727_RS09585 | C40 family peptidase | WP_000755548.1 | 800 | 800 | 1.00 | 0.0 | 95.54% |
| B. mycoides | ATCC 6462 | 1178/1312 (90%) | 32/1312 (2%) | Plus/Minus | BG05_RS21755 | C40 family peptidase | WP_003188709.1 | 667 | 667 | 1.00 | 0.0 | 88.99% |
| B. pseudomycoides | DSM 12442 | 997/1324 (75%) | 47/1324 (3%) | Plus/Plus | BPMYX0001_RS08705 | C40 family peptidase | WP_006094561.1 | 582 | 582 | 0.99 | 0.0 | 72.27% |
| B. weihenstephanensis | KBAB4 | 1168/1296 (90%) | 18/1296 (1%) | Plus/Plus | BCERKBAB4_RS09410 | C40 family peptidase | WP_002012346.1 | 672 | 672 | 1.00 | 0.0 | 91.44% |
| B. anthracis | Ames | 1211/1281 (95%) | 18/1281 (1%) | Plus/Plus | GBAA_RS09755 | C40 family peptidase | WP_000755532.1 | 796 | 796 | 1.00 | 0.0 | 95.07% |
| B. cytotoxicus | NVH 391-98 | 1050/1290 (81%) | 39/1290 (3%) | Plus/Plus | BCER98_RS07835 | C40 family peptidase | WP_012093983.1 | 644 | 644 | 0.99 | 0.0 | 81.65% |
| B. toyonensis | BCT-7112 | 1200/1309 (92%) | 35/1309 (2%) | Plus/Plus | BTOYO_RS22750 | C40 family peptidase | WP_016513917.1 | 711 | 711 | 1.00 | 0.0 | 91.45% |
| Strain | Collection | Origin/Description | Ref Genome | Identities | Gaps | Strand | Gene Identification | Protein Name | Protein Reference |
|---|---|---|---|---|---|---|---|---|---|
| NVH 0075/95 | FBO | Stew with vegetables, food poisoning outbreak of diarrheal syndrome in Norway, 1995 | LABM00000000.1 | 1236/1281(96%) | 0/1281(0%) | Plus/Plus | TU63_19225 | peptidase P60 | KMP84856.1 |
| G9842 | FBO | Human stool, outbreak that involved three individuals in the USA, 1996 | NC_011772.1 | 1273/1299(98%) | 18/1299(1%) | Plus/Plus | BCG9842_RS09225 | C40 family peptidase | WP_000755522.1 |
| AH187 | FBO | Human vomit of a person having previously eaten cooked rice in London, emetic outbreak in UK, 1972 | NC_011658.1 | 1236/1281(96%) | 0/1281(0%) | Plus/Plus | BCAH187_RS10030 | C40 family peptidase | WP_000755553.1 |
| NC7401 | FBO | Feces/vomit, food poisoning in Japan, 1994 | NC_016771.1 | 1234/1281(96%) | 0/1281(0%) | Plus/Plus | BCN_RS09705 | C40 family peptidase | WP_014297774.1 |
| NVH_141/1-01_V_C53 | FBO | Vegetarian pasta, diarrheal food poisoning outbreak in Norway, 2001 | FMJK00000000.1 | 1191/1299(92%) | 27/1299(2%) | Plus/Plus | BC141101_01248 | Enterotoxin | SCN16078.1 |
| NVH 0674-98 | FBO | Mashed swedes/scrambled eggs, diarrheal food poisoning in Norway, 1998 | FMJM00000000.1 | 1231/1281(96%) | 6/1281(0%) | Plus/Plus | BC067498_01849 | Enterotoxin | SCN44411.1 |
| HN001 | FBO | Human vomit, food poisoning in China, 2000 | NZ_CP011155.1 | 1272/1287(99%) | 6/1287(0%) | Plus/Plus | WR52_RS09190 | C40 family peptidase | WP_063536128.1 |
| NVH 0861-00 | FBO | Ice scream, diarrheal food poisoning in Norway, 2000 | FMBJ00000000.1 | 1221/1305(94%) | 27/1305(2%) | Plus/Plus | BC0861_01953 | Enterotoxin | SCC09072.1 |
| RIVM_BC120 | FBO | Human feces, diarrheal food poisoning in Netherlands | FMIJ00000000.1 | 1237/1281(97%) | 0/1281(0%) | Plus/Plus | BCRIVMBC120_02096 | Enterotoxin FM | SCL92408.1 |
| RIVM_BC126 | FBO | Human feces, diarrheal food poisoning in Netherlands | FMJJ00000000.1 | 1238/1287(96%) | 6/1287(0%) | Plus/Plus | BCRIVMBC126_01928 | Enterotoxin | SCN07222.1 |
| F2404B-79 | FBO | Diarrheal food poisoning outbreak in the UK | FMJG00000000.1 | 1231/1287(96%) | 6/1287(0%) | Plus/Plus | BCF24048_01956 | Enterotoxin | SCM94734.1 |
| 6/27/S | FBO | Human feces, diarrheal | NZ_LABV00000000.1 | 1278/1281(99%) | 0/1281(0%) | Plus/Plus | TU48_RS34500 | C40 family peptidase | WP_000755525.1 |
| F3175/03(D7) | FBO | Human feces, diarrheal | NZ_JYPI00000000.1 | 1214/1289(94%) | 13/1289(1%) | Plus/Plus | TU54_28630 | peptidase P60 | KMP31545.1 |
| F528/94 | FBO | Poisoning outbreak from beef and chow rice in the UK, 1994 | NZ_JYPH00000000.1 | 1207/1290(94%) | 27/1290(2%) | Plus/Plus | TU52_12565 | peptidase P60 | KMP35524.1 |
| F4429/71 | FBO | Vanilla pudding, diarrheal | NZ_JYPJ00000000.1 | 1214/1281(95%) | 18/1281(1%) | Plus/Minus | TU55_10935 | peptidase P60 | KMP45116.1 |
| RIVM BC 90 | FBO | Human feces, diarrheal, 1999 | LABN00000000.1 | 1236/1281(96%) | 0/1281(0%) | Plus/Plus | TU64_27355 | peptidase P60 | KMP79201.1 |
| 7/27/S | FBO | Human feces, diarrheal | NZ_LABW00000000.1 | 1232/1287(96%) | 6/1287(0%) | Plus/Plus | TU49_15830 | peptidase P60 | KMP18977.1 |
| FORC_005 | FBO | Korean side dish, food-borne illness in South Korea | NZ_CP009686.1 | 1215/1301(93%) | 25/1301(1%) | Plus/Plus | FORC5_RS09925 | C40 family peptidase | WP_044307235.1 |
| F4430/73 | FBO | Peas soup, diarrheal syndrome in Belgium, 1973 | JYPK00000000.1 | 1278/1281(99%) | 0/1281(0%) | Plus/Minus | TU56_09675 | peptidase P60 | KMP71144.1 |
| F837/76 | FBO | Food-borne outbreak in the UK, 1976 | NC_016779.1 | 1214/1281(95%) | 18/1281(1%) | Plus/Plus | BCF_RS09455 | C40 family peptidase | WP_000755546.1 |
| 09–13 | Clinical | Premature newborn, blood culture, 2009 | this study | 1277/1287 (99%) | 6/1287 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–14 | Clinical | Premature newborn, blood culture, 2009 | this study | 1235/1293 (96%) | 12/1293 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–33 | Clinical | New born, axilla, 2009 | ERS1507218 | 1215/1287 (94%) | 24/1287 (1%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 12–31 | Clinical | Premature newborn, blood culture, 2011 | this study | 1236/1281 (96%) | 0/1281 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 13–06 | Clinical | Intensive care unit, blood culture from catheter, 2011 | this study | 1236/1281 (96%) | 0/1281 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–11 | Clinical | Premature newborn, blood culture, 2009 | ERS1493302 | 1215/1293 (94%) | 30/1293 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–16 | Clinical | New born, Umbilicus, 2009 | ERS1494027 | 1215/1293 (94%) | 30/1293 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–12 | Clinical | Premature newborn, cerebrospinal fluid, 2009 | ERS1494026 | 1215/1293 (94%) | 30/1293 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–17 | Clinical | Surface of neonatology ward (window sill), 2009 | ERS1494028 | 1236/1281 (96%) | 0/1281 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| 09–34 | Clinical | Premature newborn, stomach tube feeding, 2009 | ERS1494031 | 1236/1281 (96%) | 0/1281 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| INRA PF | Non-pathogenic | Milk protein | this study | 1237/1281 (97%) | 0/1281 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| INRA 5 | Non-pathogenic | Pasteurized zucchini puree | FLZU01000000 | 1187/1309 (91%) | 29/1309 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| INRA C64 | Non-pathogenic | Pasteurized vegetables | this study | 1157/1294 (89%) | 29/1294 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| ADRIA I3 | Non-pathogenic | Cooked foods | this study | 1156/1300 (89%) | 35/1300 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| INRA A3 | Non-pathogenic | Starch | LABH01000000 | 1277/1287 (99%) | 6/1287 (0%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| INRA SL’ | Non-pathogenic | Soil | this study | 1184/1290 (92%) | 21/1290 (1%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| I21 | Non-pathogenic | Unknown | this study | 1170/1293 (90%) | 30/1293 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| ADRIA I21 | Non-pathogenic | Cooked foods | FMJF01000000 | 1170/1293 (90%) | 30/1293 (2%) | Plus/Plus | putative peptidoglycan endopeptidase LytE | ||
| WSBC 10204 | Non-pathogenic | Pasteurized milk | PRJNA258373 | 1187/1306 (91%) | 26/1306 (1%) | Plus/Plus | putative peptidoglycan endopeptidase LytE |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, S.-L.; Cormontagne, D.; Vidic, J.; André-Leroux, G.; Ramarao, N. Structural Modeling of Cell Wall Peptidase CwpFM (EntFM) Reveals Distinct Intrinsically Disordered Extensions Specific to Pathogenic Bacillus cereus Strains. Toxins 2020, 12, 593. https://doi.org/10.3390/toxins12090593
Tran S-L, Cormontagne D, Vidic J, André-Leroux G, Ramarao N. Structural Modeling of Cell Wall Peptidase CwpFM (EntFM) Reveals Distinct Intrinsically Disordered Extensions Specific to Pathogenic Bacillus cereus Strains. Toxins. 2020; 12(9):593. https://doi.org/10.3390/toxins12090593
Chicago/Turabian StyleTran, Seav-Ly, Delphine Cormontagne, Jasmina Vidic, Gwenaëlle André-Leroux, and Nalini Ramarao. 2020. "Structural Modeling of Cell Wall Peptidase CwpFM (EntFM) Reveals Distinct Intrinsically Disordered Extensions Specific to Pathogenic Bacillus cereus Strains" Toxins 12, no. 9: 593. https://doi.org/10.3390/toxins12090593
APA StyleTran, S.-L., Cormontagne, D., Vidic, J., André-Leroux, G., & Ramarao, N. (2020). Structural Modeling of Cell Wall Peptidase CwpFM (EntFM) Reveals Distinct Intrinsically Disordered Extensions Specific to Pathogenic Bacillus cereus Strains. Toxins, 12(9), 593. https://doi.org/10.3390/toxins12090593