Consequences of Metabolic Interactions during Staphylococcus aureus Infection



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Host Responses to Acute Infection
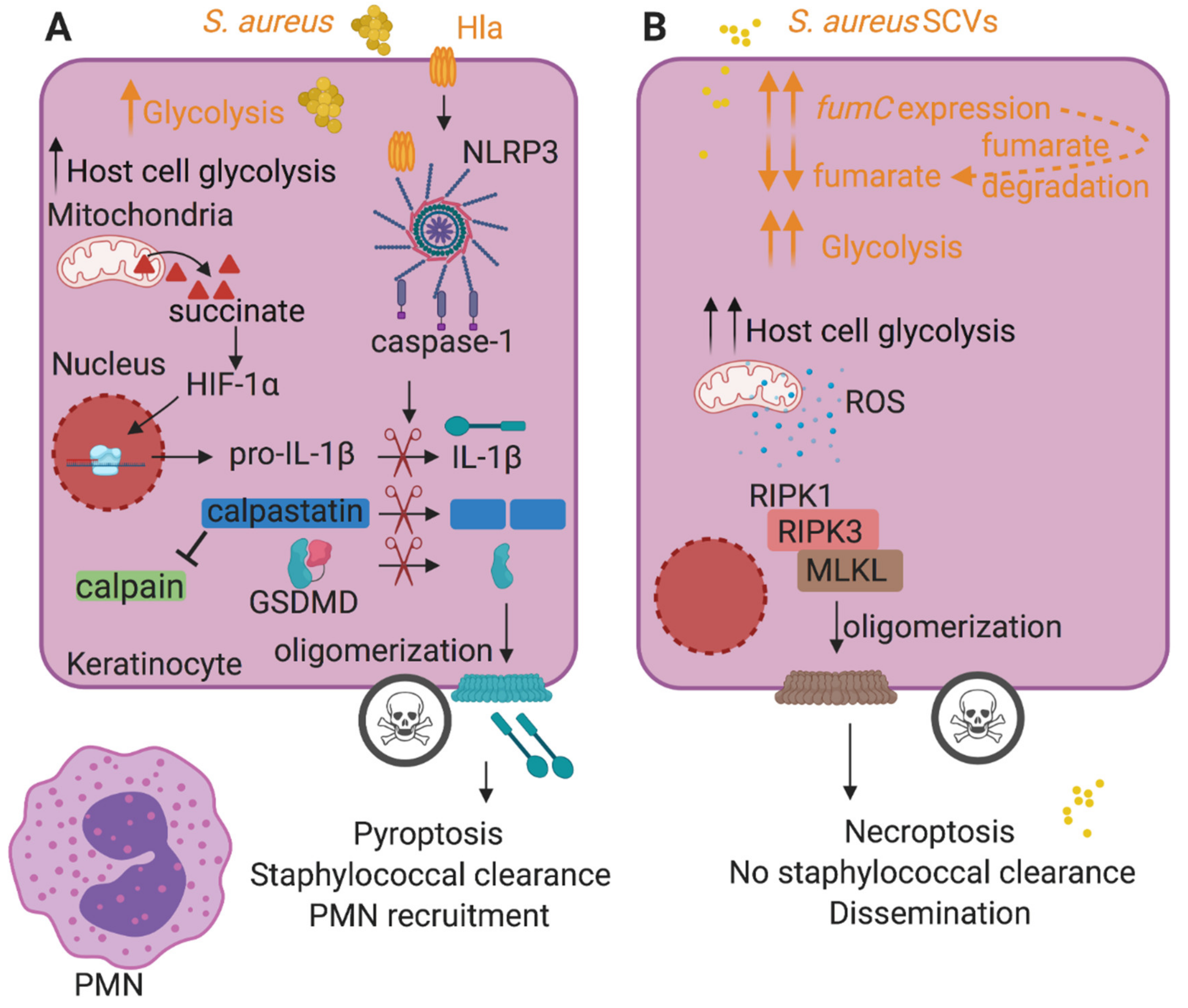
2.1. Inflammasome Activation and Pyroptosis
2.2. Consequences of S. aureus-Induced Necroptosis
3. Adaptive Metabolic Changes during Chronic S. aureus Infection
4. Host Metabolism Promotes Infection
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J.C. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Wong Fok Lung, T.; Prince, A. Pulmonary Pathogens Adapt to Immune Signaling Metabolites in the Airway. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Acker, K.P.; Wong Fok Lung, T.; West, E.; Craft, J.; Narechania, A.; Smith, H.; O’Brien, K.; Moustafa, A.M.; Lauren, C.; Planet, P.J.; et al. Strains of Staphylococcus aureus that Colonize and Infect Skin Harbor Mutations in Metabolic Genes. iScience 2019, 19, 281–290. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Wickersham, M.; Wachtel, S.; Wong Fok Lung, T.; Soong, G.; Jacquet, R.; Richardson, A.; Parker, D.; Prince, A. Metabolic Stress Drives Keratinocyte Defenses against Staphylococcus aureus Infection. Cell Rep. 2017, 18, 2742–2751. [Google Scholar] [CrossRef]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat. Microbiol. 2020, 5, 141–153. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Szafranska, A.K.; Oxley, A.P.A.; Chaves-Moreno, D.; Horst, S.A.; Roßlenbroich, S.; Peters, G.; Goldmann, O.; Rohde, M.; Sinha, B.; Pieper, D.H.; et al. High-Resolution Transcriptomic Analysis of the Adaptive Response of Staphylococcus aureus during Acute and Chronic Phases of Osteomyelitis. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Potter, A.D.; Butrico, C.E.; Ford, C.A.; Curry, J.M.; Trenary, I.A.; Tummarakota, S.S.; Hendrix, A.S.; Young, J.D.; Cassat, J. Host nutrient milieu drives an essential role for aspartate biosynthesis during invasive Staphylococcus aureus infection. Proc. Natl. Acad. Sci. USA 2020, 117, 12394–12401. [Google Scholar] [CrossRef] [PubMed]
- Gabryszewski, S.J.; Wong Fok Lung, T.; Annavajhala, M.K.; Tomlinson, K.L.; Riquelme, S.A.; Khan, I.N.; Noguera, L.P.; Wickersham, M.; Zhao, A.; Mulenos, A.M.; et al. Metabolic Adaptation in Methicillin-Resistant Staphylococcus aureus Pneumonia. Am. J. Respir. Cell Mol. Boil. 2019, 61, 185–197. [Google Scholar] [CrossRef]
- Tan, X.; Coureuil, M.; Ramond, E.; Euphrasie, D.; Dupuis, M.; Tros, F.; Meyer, J.; Nemazanyy, I.; Chhuon, C.; Guerrera, I.C.; et al. Chronic Staphylococcus aureus Lung Infection Correlates With Proteogenomic and Metabolic Adaptations Leading to an Increased Intracellular Persistence. Clin. Infect. Dis. 2019, 69, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Bernardy, E.E.; Petit, R.A., 3rd; Raghuram, V.; Alexander, A.M.; Read, T.D.; Goldberg, J.B. Genotypic and Phenotypic Diversity of Staphylococcus aureus Isolates from Cystic Fibrosis Patient Lung Infections and Their Interactions with Pseudomonas aeruginosa. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Majerczyk, C.D.; Sadykov, M.R.; Luong, T.T.; Lee, C.; Somerville, G.A.; Sonenshein, A.L. Staphylococcus aureus CodY Negatively Regulates Virulence Gene Expression. J. Bacteriol. 2008, 190, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.S.; Kafer, J.M.; Wasserman, G.A.; Velickovic, L.; Mathema, B.; Holzman, R.S.; Knipe, T.A.; Becker, K.; Von Eiff, C.; Peters, G.; et al. Nasal Carriage as a Source of agr-Defective Staphylococcus aureus Bacteremia. J. Infect. Dis. 2012, 206, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.E.; Wagner, N.J.; Li, L.; Beam, J.E.; Wilkinson, A.D.; Radlinski, L.C.; Zhang, Q.; Miao, E.A.; Conlon, B.P. Reactive oxygen species induce antibiotic tolerance during systemic Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 282–290. [Google Scholar] [CrossRef]
- Yamada, K.J.; Heim, C.E.; Xi, X.; Attri, K.S.; Wang, D.; Zhang, W.; Singh, P.K.; Bronich, T.K.; Kielian, T. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PLoS Pathog. 2020, 16, e1008354. [Google Scholar] [CrossRef]
- Geoghegan, J.A.; Irvine, A.; Foster, T.J. Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol. 2018, 26, 484–497. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev.Dermatol. 2010, 5, 183–195. [Google Scholar] [CrossRef]
- Kagan, J.C.; Horng, T. NLRP3 inflammasome activation: CD36 serves double duty. Nat. Immunol. 2013, 14, 772–774. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Götz, F.; et al. Staphylococcus aureus Evades Lysozyme-Based Peptidoglycan Digestion that Links Phagocytosis, Inflammasome Activation, and IL-1β Secretion. Cell Host Microbe 2010, 7, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Soong, G.; Chun, J.; Parker, D.; Prince, A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J. Infect. Dis. 2012, 205, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Wardenburg, J.B.; McElvania-TeKippe, E.; Ting, J.P.; Duncan, J. Staphylococcus aureus α-Hemolysin Activates the NLRP3-Inflammasome in Human and Mouse Monocytic Cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Gieldon, L.; Mysore, V.; Nippe, N.; Taxman, D.J.; Duncan, J.A.; Broglie, P.M.; Marketon, K.; Austermann, J.; Vogl, T.; et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Boil. 2012, 92, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Melehani, J.H.; James, D.B.A.; Dumont, A.L.; Torres, V.J.; Duncan, J. Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog. 2015, 11, e1004970. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.S.; Núñez, G. A Critical Role for Hemolysins and Bacterial Lipoproteins in Staphylococcus aureus-Induced Activation of the Nlrp3 Inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci—An overview. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 2007, 179. [Google Scholar] [CrossRef]
- Leite, E.L.; Gautron, A.; Deplanche, M.; Nicolas, A.; Ossemond, J.; Nguyen, M.-T.; Carmo, F.L.R.D.; Gilot, D.; Azevedo, V.; Götz, F.; et al. Involvement of caspase-1 in inflammasomes activation and bacterial clearance in S. aureus-infected osteoblasts-like MG-63 cells. Cell. Microbiol. 2020, 22, e13204. [Google Scholar] [CrossRef]
- Haugwitz, U.; Bobkiewicz, W.; Han, S.-R.; Beckmann, E.; Veerachato, G.; Shaid, S.; Biehl, S.; Dersch, K.; Bhakdi, S.; Husmann, M. Pore-forming Staphylococcus aureus alpha-toxin triggers epidermal growth factor receptor-dependent proliferation. Cell. Microbiol. 2006, 8, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.M.; Prince, A. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus aureus Lung Damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.M.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.C.; Becker, K.; Löffler, B. Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin. Microbiol. Rev. 2016, 29, 401–427. [Google Scholar] [CrossRef]
- Wolter, D.J.; Onchiri, F.M.; Emerson, J.; Precit, M.R.; Lee, M.; McNamara, S.; Nay, L.; Blackledge, M.; Uluer, A.; Orenstein, D.M.; et al. Prevalence and clinical associations of Staphylococcus aureus small-colony variant respiratory infection in children with cystic fibrosis (SCVSA): A multicentre, observational study. Lancet Respir. Med. 2019, 7, 1027–1038. [Google Scholar] [CrossRef]
- Proctor, R.A.; Van Langevelde, P.; Kristjansson, M.; Maslow, J.N.; Arbeit, R.D. Persistent and Relapsing Infections Associated with Small-Colony Variants of Staphylococcus aureus. Clin. Infect. Dis. 1995, 20, 95–102. [Google Scholar] [CrossRef]
- Proctor, R.A.; Von Eiff, C.; Kahl, B.C.; Becker, K.; McNamara, P.; Herrmann, M.; Peters, G. Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 2006, 4, 295–305. [Google Scholar] [CrossRef]
- Kriegeskorte, A.; Grubmüller, S.; Huber, C.; Kahl, B.C.; Von Eiff, C.; Proctor, R.A.; Peters, G.; Eisenreich, W.; Becker, K. Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front. Microbiol. 2014, 4. [Google Scholar] [CrossRef]
- Beavers, W.N.; Skaar, E.P. Neutrophil-generated oxidative stress and protein damage in Staphylococcus aureus. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Genet. 2005, 3, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of Protein A in the Evasion of Host Adaptive Immune Responses by Staphylococcus aureus. mBio 2013, 4, e00575-13. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 640–668. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Takahashi, H.; Takaya, A.; Inoue, Y.; Katayama, Y.; Kusuya, Y.; Shoji, T.; Takada, S.; Nakagawa, S.; Oguma, R.; et al. Staphylococcus Agr virulence is critical for epidermal colonization and associates with atopic dermatitis development. Sci. Transl. Med. 2020, 12, eaay4068. [Google Scholar] [CrossRef] [PubMed]
- Medina, L.M.P.; Becker, A.-K.; Michalik, S.; Yedavally, H.; Raineri, E.J.; Hildebrandt, P.; Salazar, M.G.; Surmann, K.; Pförtner, H.; Mekonnen, S.A.; et al. Metabolic Cross-Talk Between Human Bronchial Epithelial Cells and Internalized Staphylococcus aureus as a Driver for Infection. Mol. Cell. Proteom. 2019, 18, 892–908. [Google Scholar] [CrossRef]
- Suligoy, C.M.; Lattar, S.M.; Llana, M.N.; Gonzalez, C.D.; Alvarez, L.P.; Robinson, D.A.; Gómez, M.I.; Buzzola, F.R.; Sordelli, D.O. Mutation of Agr Is Associated with the Adaptation of Staphylococcus aureus to the Host during Chronic Osteomyelitis. Front. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Altman, D.R.; Sullivan, M.J.; Chacko, K.I.; Balasubramanian, D.; Pak, T.R.; Sause, W.E.; Kumar, K.; Sebra, R.; Deikus, G.; Attie, O.; et al. Genome Plasticity of agr-Defective Staphylococcus aureus during Clinical Infection. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; A Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.-C.; Wang, S.-Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Heim, C.E.; Bosch, M.E.; Yamada, K.J.; Aldrich, A.L.; Chaudhari, S.S.; Klinkebiel, D.; Gries, C.M.; Alqarzaee, A.A.; Li, Y.; Thomas, V.C.; et al. Lactate production by Staphylococcus aureus biofilm inhibits HDAC11 to reprogramme the host immune response during persistent infection. Nat. Microbiol. 2020, 1–14. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong Fok Lung, T.; Prince, A. Consequences of Metabolic Interactions during Staphylococcus aureus Infection. Toxins 2020, 12, 581. https://doi.org/10.3390/toxins12090581
Wong Fok Lung T, Prince A. Consequences of Metabolic Interactions during Staphylococcus aureus Infection. Toxins. 2020; 12(9):581. https://doi.org/10.3390/toxins12090581
Chicago/Turabian StyleWong Fok Lung, Tania, and Alice Prince. 2020. "Consequences of Metabolic Interactions during Staphylococcus aureus Infection" Toxins 12, no. 9: 581. https://doi.org/10.3390/toxins12090581
APA StyleWong Fok Lung, T., & Prince, A. (2020). Consequences of Metabolic Interactions during Staphylococcus aureus Infection. Toxins, 12(9), 581. https://doi.org/10.3390/toxins12090581