Production of Recombinant Gelonin Using an Automated Liquid Chromatography System



Abstract
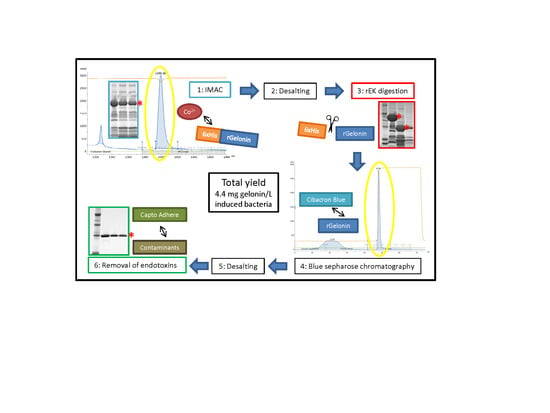
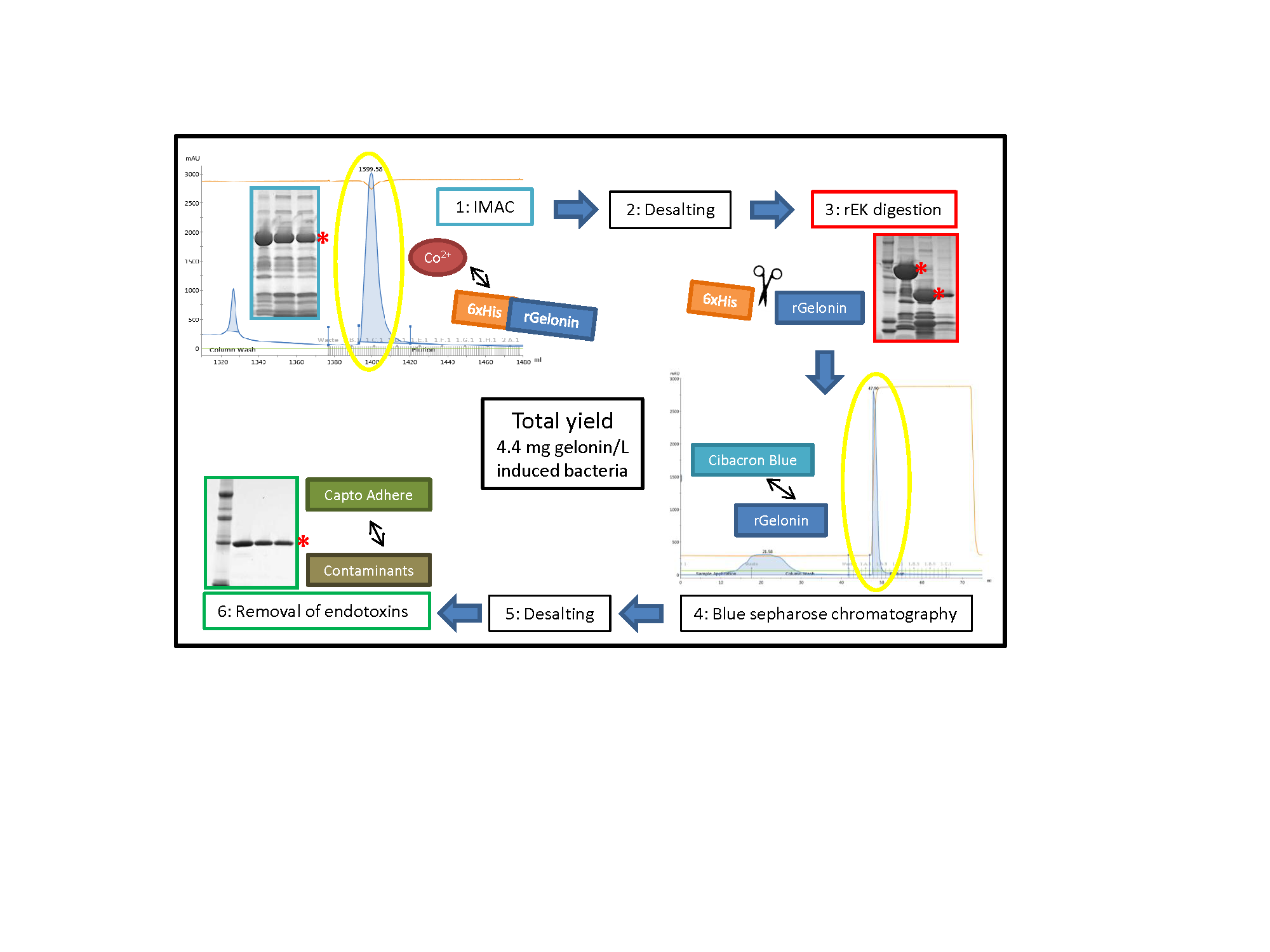{kind=link}
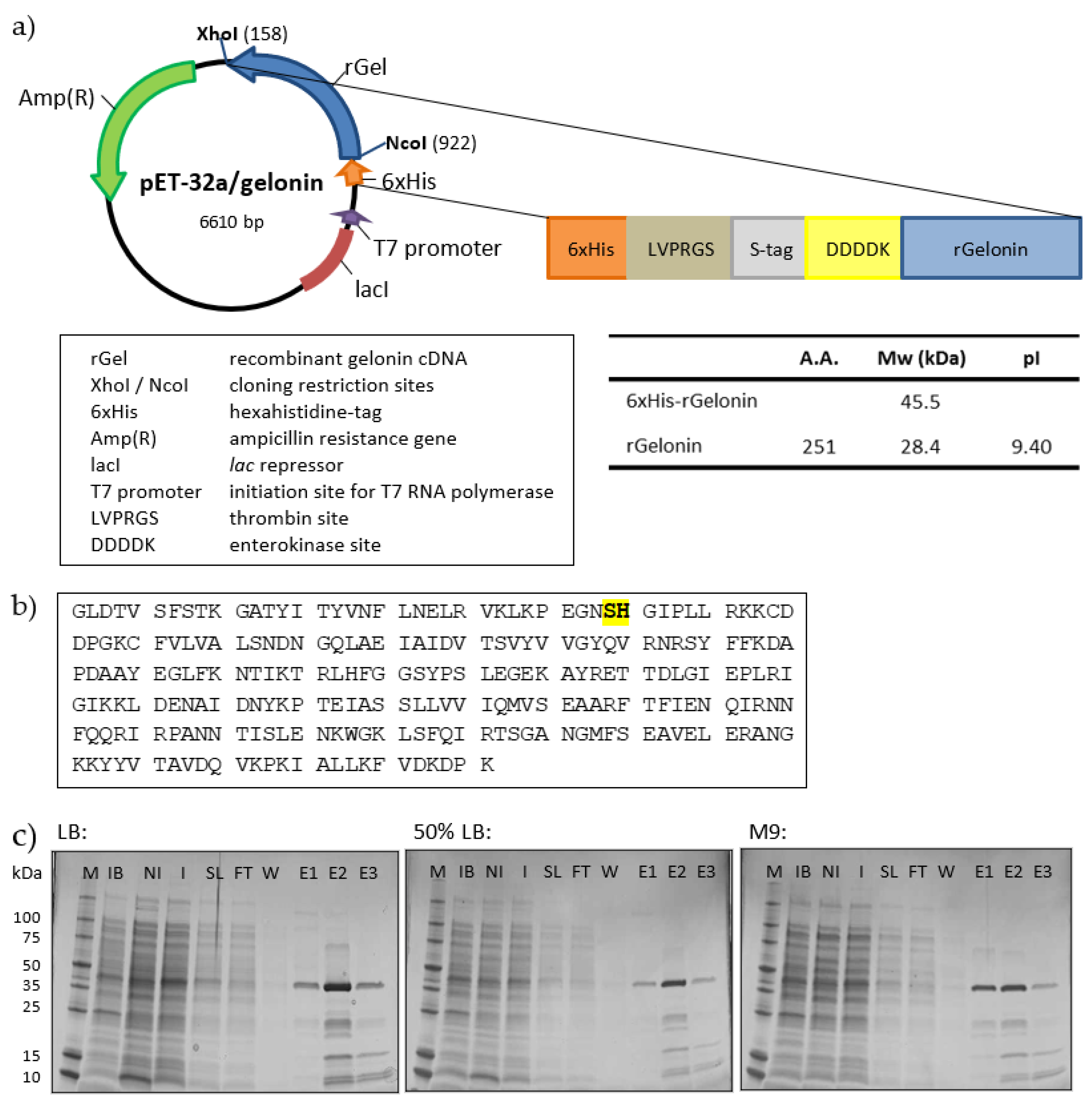{kind=link}
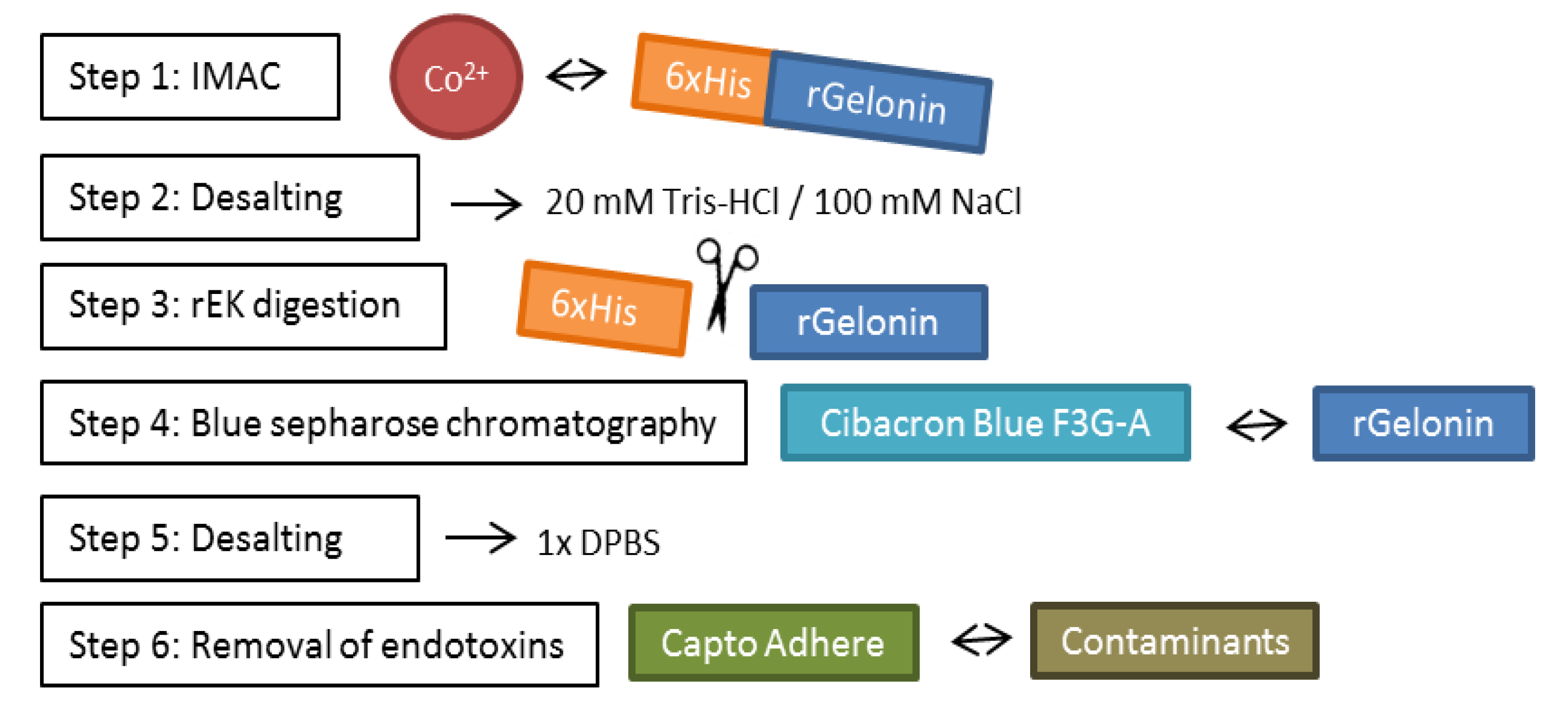{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Cloning, Expression and Small-Scale Production of rGel
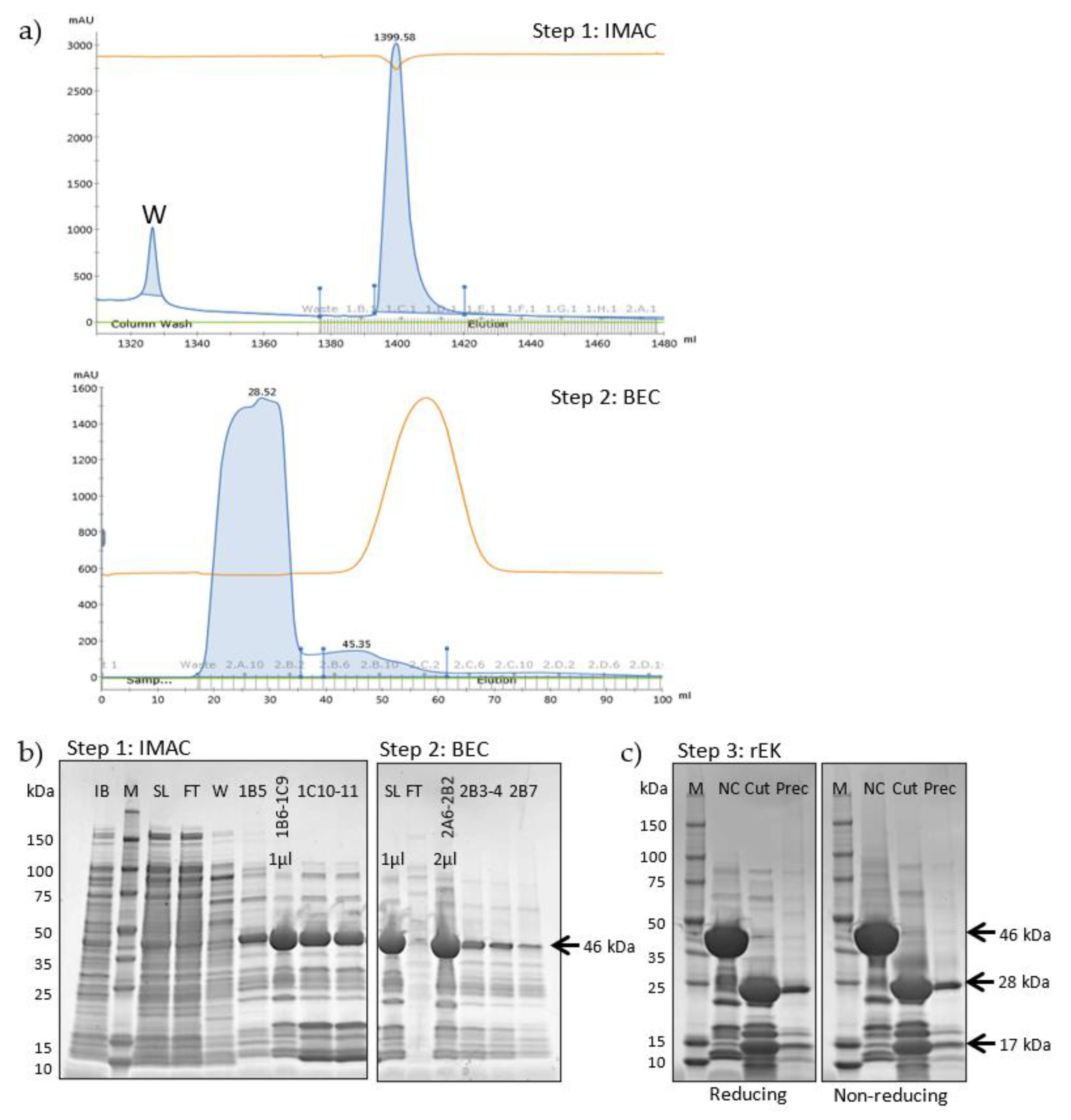
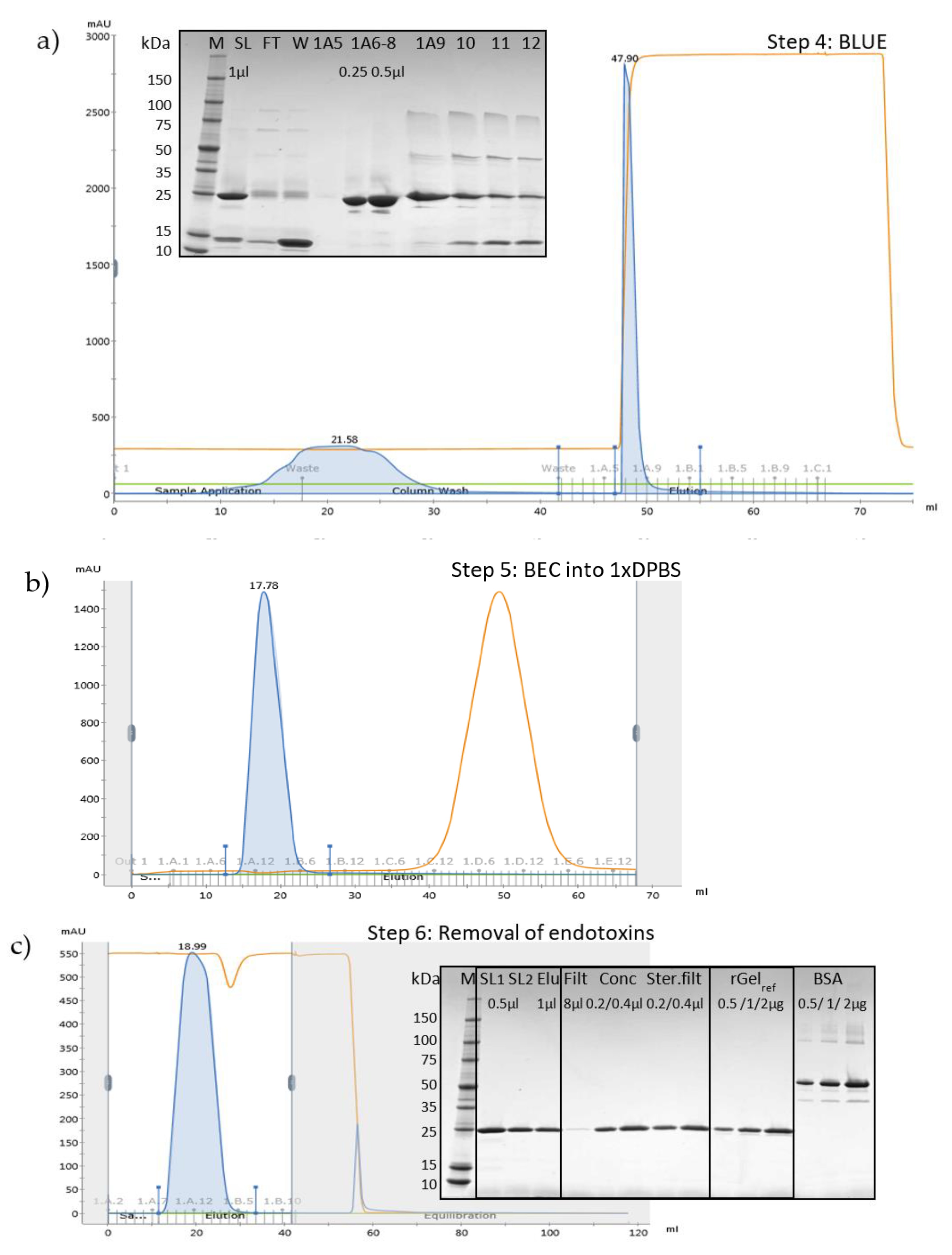
2.2. Large-Scale Expression and Purification
2.3. Removal of Endotoxins by Capto Adhere
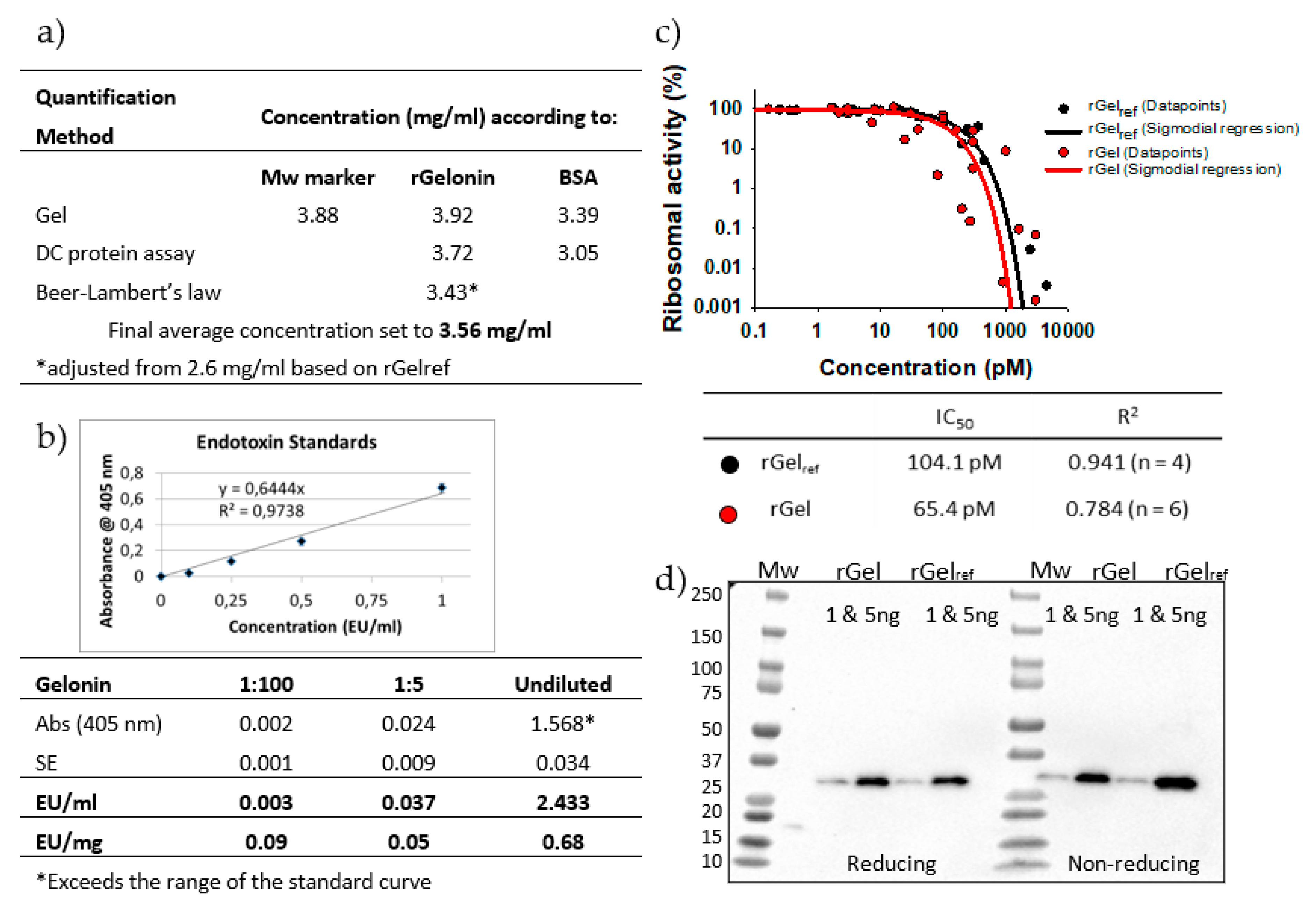
2.4. Qantification of Final Purified Product and Endotoxin Levels
2.5. Biological Activity and Identity of rGel
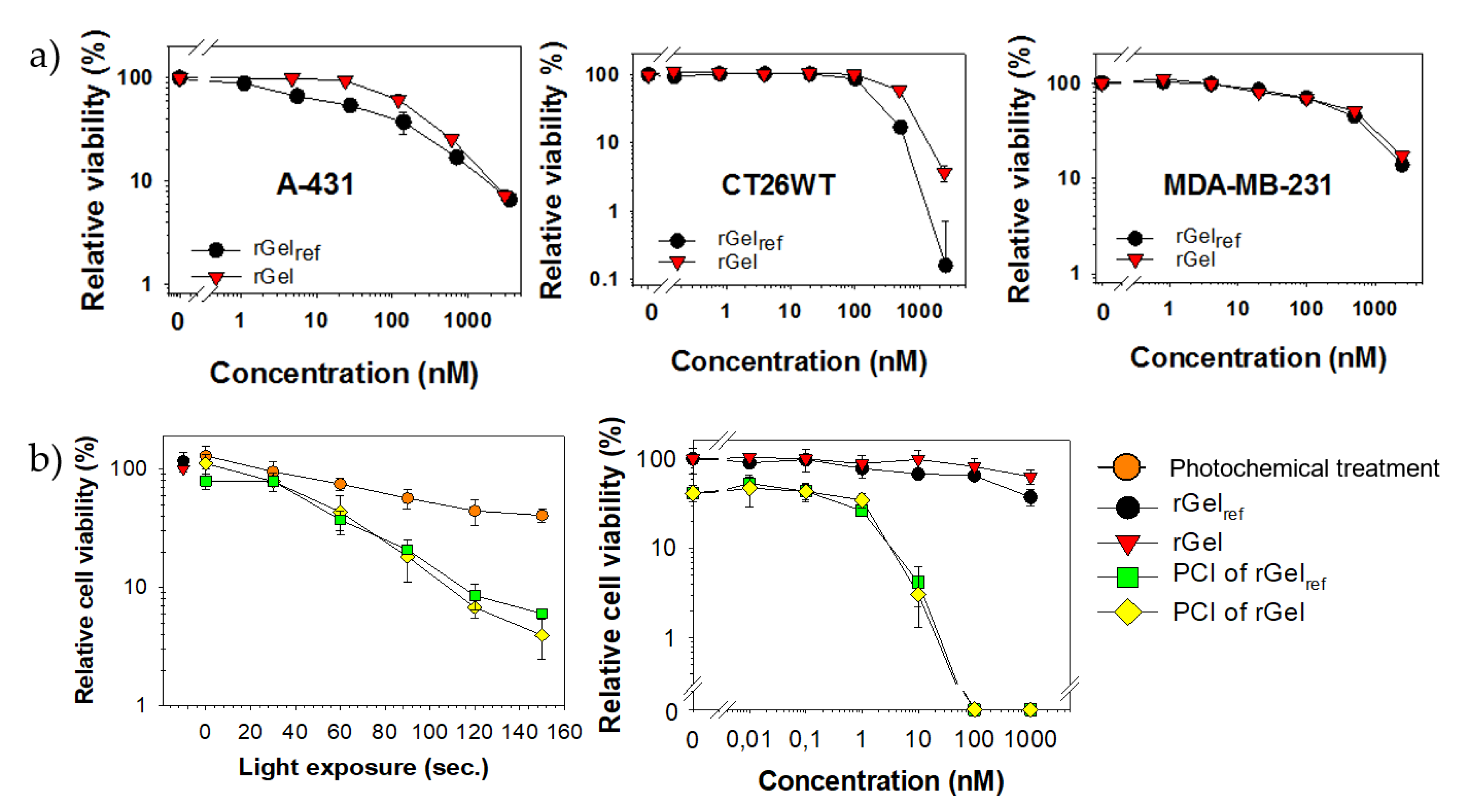
2.6. Cytotoxicity of rGel
3. Conclusions
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. pET-32a/rGel Plasmid Construction
4.3. Protein Expression
4.4. Protein Purification
4.5. Endotoxin Removal and Detection
4.6. Verification of Concentration and Identity
4.7. Protein Translation Inhbition Assay
4.8. Cytotoxicity Assays
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Polito, L.; Djemil, A.; Bortolotti, M. Plant Toxin-Based Immunotoxins for Cancer Therapy: A Short Overview. Biomedicines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Freedman, L.P.; Gibson, M.C. The impact of preclinical irreproducibility on drug development. Clin. Pharmacol. Ther. 2015, 97, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The Economics of Reproducibility in Preclinical Research. PLoS Biol. 2015, 13, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Olsnes, S.; Pihl, A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J. Biol. Chem. 1980, 255, 6947–6953. [Google Scholar] [PubMed]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Rosenblum, M.G.; Kohr, W.A.; Beattie, K.L.; Beattie, W.G.; Marks, W.; Toman, P.D.; Cheung, L. Amino acid sequence analysis, gene construction, cloning, and expression of gelonin, a toxin derived from Gelonium multiflorum. J. Interferon Cytokine Res. 1995, 15, 547–555. [Google Scholar] [CrossRef]
- Nolan, P.A.; Garrison, D.A.; Better, M. Cloning and expression of a gene encoding gelonin, a ribosome-inactivating protein from Gelonium multiflorum. Gene 1993, 134, 223–227. [Google Scholar] [CrossRef]
- Hosur, M.V.; Nair, B.; Satyamurthy, P.; Misquith, S.; Surolia, A.; Kannan, K.K. X-ray structure of gelonin at 1.8 A resolution. J. Mol. Biol. 1995, 250, 368–380. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Cheung, L.H.; Liu, Y.; Marks, J.W., III. Design, expression, purification, and characterization, in vitro and in vivo, of an antimelanoma single-chain Fv antibody fused to the toxin gelonin. Cancer Res. 2003, 63, 3995–4002. [Google Scholar]
- Cao, Y.; Marks, J.D.; Marks, J.W.; Cheung, L.H.; Kim, S.; Rosenblum, M.G. Construction and characterization of novel, recombinant immunotoxins targeting the Her2/neu oncogene product: In vitro and in vivo studies. Cancer Res. 2009, 69, 8987–8995. [Google Scholar] [CrossRef]
- Shin, M.C.; Zhang, J.; David, A.E.; Trommer, W.E.; Kwon, Y.M.; Min, K.A.; Kim, J.H.; Yang, V.C. Chemically and biologically synthesized CPP-modified gelonin for enhanced anti-tumor activity. J. Control Release 2013, 172, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Eiklid, K.; Olsnes, S.; Pihl, A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp. Cell Res. 1980, 126, 321–326. [Google Scholar] [CrossRef]
- Pirie, C.M.; Hackel, B.J.; Rosenblum, M.G.; Wittrup, K.D. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 2011, 286, 4165–4172. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhao, J.; Zhang, J.; Huang, Y.; He, H.; Wang, M.; Min, K.A.; Yang, V.C. Recombinant TAT-gelonin fusion toxin: Synthesis and characterization of heparin/protamine-regulated cell transduction. J. Biomed. Mater. Res. A 2015, 103, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Min, K.A.; Cheong, H.; Moon, C.; Huang, Y.; He, H.; Yang, V.C. Preparation and Characterization of Gelonin-Melittin Fusion Biotoxin for Synergistically Enhanced Anti-Tumor Activity. Pharm. Res. 2016, 33, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Pirie, C.M.; Liu, D.V.; Wittrup, K.D. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol. Cancer Ther. 2013, 12, 1774–1782. [Google Scholar]
- Bai, Y.; Gou, M.; Yi, T.; Yang, L.; Liu, L.; Lin, X.; Su, D.; Wei, Y.; Zhao, X. Efficient Inhibition of Ovarian Cancer by Gelonin Toxin Gene Delivered by Biodegradable Cationic Heparin-polyethyleneimine Nanogels. Int. J. Med. Sci. 2015, 12, 397–406. [Google Scholar] [CrossRef]
- Min, K.A.; He, H.; Yang, V.C.; Shin, M.C. Construction and characterization of gelonin and saporin plasmids for toxic gene-based cancer therapy. Arch. Pharm. Res. 2016, 39, 677–686. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjolsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar]
- Selbo, P.K.; Weyergang, A.; Hogset, A.; Norum, O.J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J. Control Release 2010, 148, 2–12. [Google Scholar] [CrossRef]
- Sultan, A.A.; Jerjes, W.; Berg, K.; Hogset, A.; Mosse, C.A.; Hamoudi, R.; Hamdoon, Z.; Simeon, C.; Carnell, D.; Forster, M.; et al. Disulfonated tetraphenyl chlorin (TPCS2a)-induced photochemical internalisation of bleomycin in patients with solid malignancies: A phase 1, dose-escalation, first-in-man trial. Lancet Oncol. 2016, 17, 1217–1229. [Google Scholar] [CrossRef]
- Selbo, P.K.; Sandvig, K.; Kirveliene, V.; Berg, K. Release of gelonin from endosomes and lysosomes to cytosol by photochemical internalization. Biochim. Biophys. Acta 2000, 1475, 307–313. [Google Scholar] [CrossRef]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. Photochemical internalisation increases the cytotoxic effect of the immunotoxin MOC31-gelonin. Int. J. Cancer 2000, 87, 853–859. [Google Scholar] [CrossRef]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int. J. Cancer 2001, 92, 761–766. [Google Scholar] [CrossRef]
- Prasmickaite, L.; Hogset, A.; Selbo, P.K.; Engesaeter, B.O.; Hellum, M.; Berg, K. Photochemical disruption of endocytic vesicles before delivery of drugs: A new strategy for cancer therapy. Br. J. Cancer 2002, 86, 652–657. [Google Scholar] [CrossRef]
- Berstad, M.B.; Cheung, L.H.; Berg, K.; Peng, Q.; Fremstedal, A.S.; Patzke, S.; Rosenblum, M.G.; Weyergang, A. Design of an EGFR-targeting toxin for photochemical delivery: In vitro and in vivo selectivity and efficacy. Oncogene 2015, 10, 5582–5592. [Google Scholar] [CrossRef]
- Olsen, C.E.; Cheung, L.H.; Weyergang, A.; Berg, K.; Vallera, D.A.; Rosenblum, M.G.; Selbo, P.K. Design, Characterization, and Evaluation of scFvCD133/rGelonin: A CD133-Targeting Recombinant Immunotoxin for Use in Combination with Photochemical Internalization. J. Clin. Med. 2019, 9, 68. [Google Scholar] [CrossRef]
- Ding, G.B.; Wu, G.; Li, B.; Yang, P.; Li, Z. High-yield expression in Escherichia coli, biophysical characterization, and biological evaluation of plant toxin gelonin. 3 Biotech 2019, 9, 19. [Google Scholar] [CrossRef]
- Europe Co. European Pharmacopeia 5.0. In Bacterial Endotoxins; Council of Europe: Strasbourg, Germany, 2005. [Google Scholar]
- Rosenblum, M.G.; Murray, J.L.; Cheung, L.; Rifkin, R.; Salmon, S.; Bartholomew, R. A specific and potent immunotoxin composed of antibody ZME-018 and the plant toxin gelonin. Mol. Biother. 1991, 3, 6–13. [Google Scholar]
- Masuda, K.; Hirano, K.; Takagishi, Y. Use of blue-sepharose for purification of immunotoxin containing type 1 ribosome-inactivating protein, gelonin. Biomed. Chromatogr. 1994, 8, 9–13. [Google Scholar] [CrossRef]
- Dawson, M.E. Endotoxin Testing, Pharmaceutical Dosage Forms—Parenteral Medications Third Edition: Volume 2: Facility Design, Sterilization and Processing; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2010; pp. 146–187. [Google Scholar]
- Dietze, A.; Peng, Q.; Selbo, P.K.; Kaalhus, O.; Muller, C.; Bown, S.; Berg, K. Enhanced photodynamic destruction of a transplantable fibrosarcoma using photochemical internalisation of gelonin. Br. J. Cancer 2005, 92, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Fremstedal, A.S.; Skarpen, E.; Peng, Q.; Mohamedali, K.A.; Eng, M.S.; Cheung, L.H.; Rosenblum, M.G.; Waltenberger, J.; Berg, K. Light-enhanced VEGF121/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Control Release 2018, 288, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Veenendaal, L.M.; Jin, H.; Ran, S.; Cheung, L.; Navone, N.; Marks, J.W.; Waltenberger, J.; Thorpe, P.; Rosenblum, M.G. In vitro and in vivo studies of a VEGF121/rGelonin chimeric fusion toxin targeting the neovasculature of solid tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7866–7871. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.H.; Min, K.A.; Shin, M.C. Molecular tumor targeting of gelonin by fusion with F3 peptide. Acta Pharmacol. Sin. 2017, 38, 897–906. [Google Scholar] [CrossRef][Green Version]
- Bull-Hansen, B.; Cao, Y.; Berg, K.; Skarpen, E.; Rosenblum, M.G.; Weyergang, A. Photochemical activation of the recombinant HER2-targeted fusion toxin MH3-B1/rGel; Impact of HER2 expression on treatment outcome. J. Control Release 2014, 182, 58–66. [Google Scholar] [CrossRef]
- Ran, S.; Mohamedali, K.A.; Luster, T.A.; Thorpe, P.E.; Rosenblum, M.G. The vascular-ablative agent VEGF(121)/rGel inhibits pulmonary metastases of MDA-MB-231 breast tumors. Neoplasia 2005, 7, 486–496. [Google Scholar] [CrossRef]
- Wong, J.J.W.; Berstad, M.B.; Fremstedal, A.S.V.; Berg, K.; Patzke, S.; Sørensen, V.; Peng, Q.; Selbo, P.K.; Weyergang, A. Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy. Cancers 2020, 12, 417. [Google Scholar] [CrossRef]
- Weyergang, A.; Cheung, L.H.; Rosenblum, M.G.; Mohamedali, K.A.; Peng, Q.; Waltenberger, J.; Berg, K. Photochemical internalization augments tumor vascular cytotoxicity and specificity of VEGF(121)/rGel fusion toxin. J. Control Release 2014, 180, 1–9. [Google Scholar] [CrossRef]
- Grimsley, G.R.; Pace, C.N. Spectrophotometric determination of protein concentration. Curr. Protoc. Protein Sci. 2004, 33, 3.1.1–3.1.9. [Google Scholar] [CrossRef]
- Weyergang, A.; Selbo, P.K.; Berg, K. Photochemically stimulated drug delivery increases the cytotoxicity and specificity of EGF-saporin. J. Control Release 2006, 111, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Weyergang, A.; Bonsted, A.; Bown, S.G.; Berg, K. Photochemical internalization of therapeutic macromolecular agents: A novel strategy to kill multidrug-resistant cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berstad, M.E.B.; Cheung, L.H.; Weyergang, A. Production of Recombinant Gelonin Using an Automated Liquid Chromatography System. Toxins 2020, 12, 519. https://doi.org/10.3390/toxins12080519
Berstad MEB, Cheung LH, Weyergang A. Production of Recombinant Gelonin Using an Automated Liquid Chromatography System. Toxins. 2020; 12(8):519. https://doi.org/10.3390/toxins12080519
Chicago/Turabian StyleBerstad, Maria E. B., Lawrence H. Cheung, and Anette Weyergang. 2020. "Production of Recombinant Gelonin Using an Automated Liquid Chromatography System" Toxins 12, no. 8: 519. https://doi.org/10.3390/toxins12080519
APA StyleBerstad, M. E. B., Cheung, L. H., & Weyergang, A. (2020). Production of Recombinant Gelonin Using an Automated Liquid Chromatography System. Toxins, 12(8), 519. https://doi.org/10.3390/toxins12080519