Saporin from Saponaria officinalis as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience

 ,
,
Abstract
1. Introduction
2. Short Characteristics of Ribotoxins
3. Peptide-Saporin Conjugates
4. IgG-Saporin Conjugates
5. Saporin Conjugates as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience
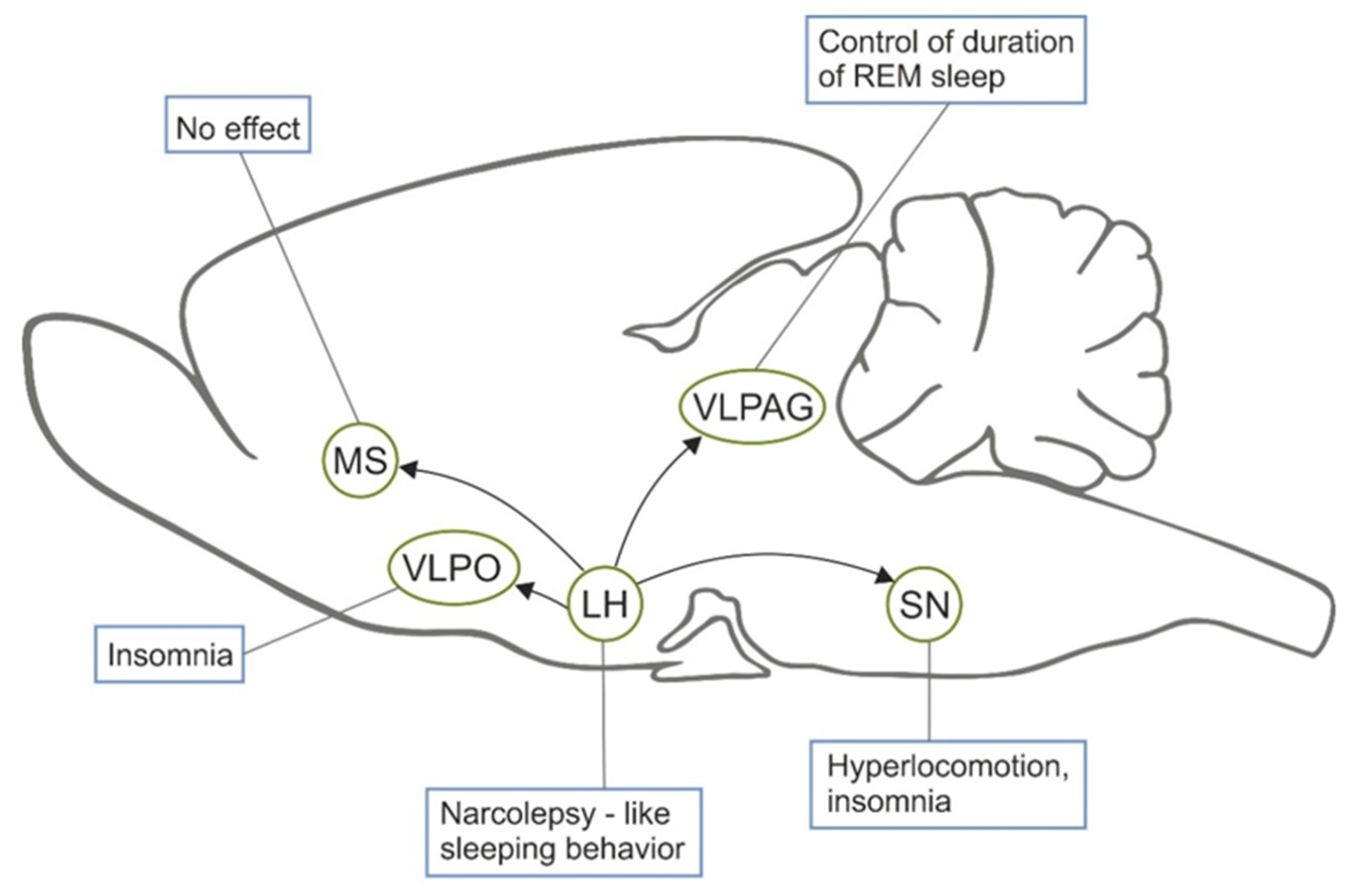
5.1. Sleep
5.2. General Anesthesia
5.3. Epilepsy
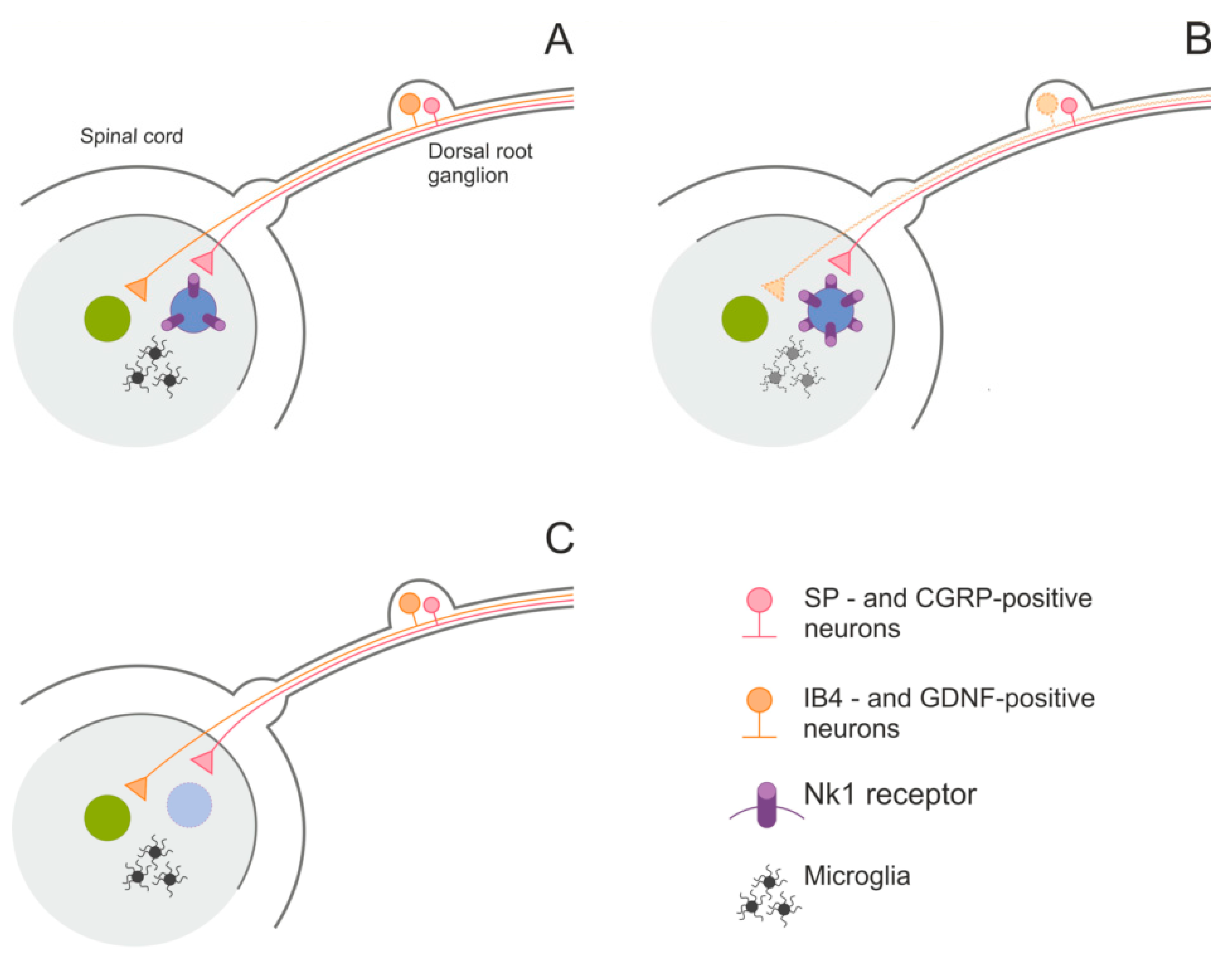
5.4. Pain
5.5. Anxiety and Autism Spectrum Disorders
5.6. Parkinson’s Disease
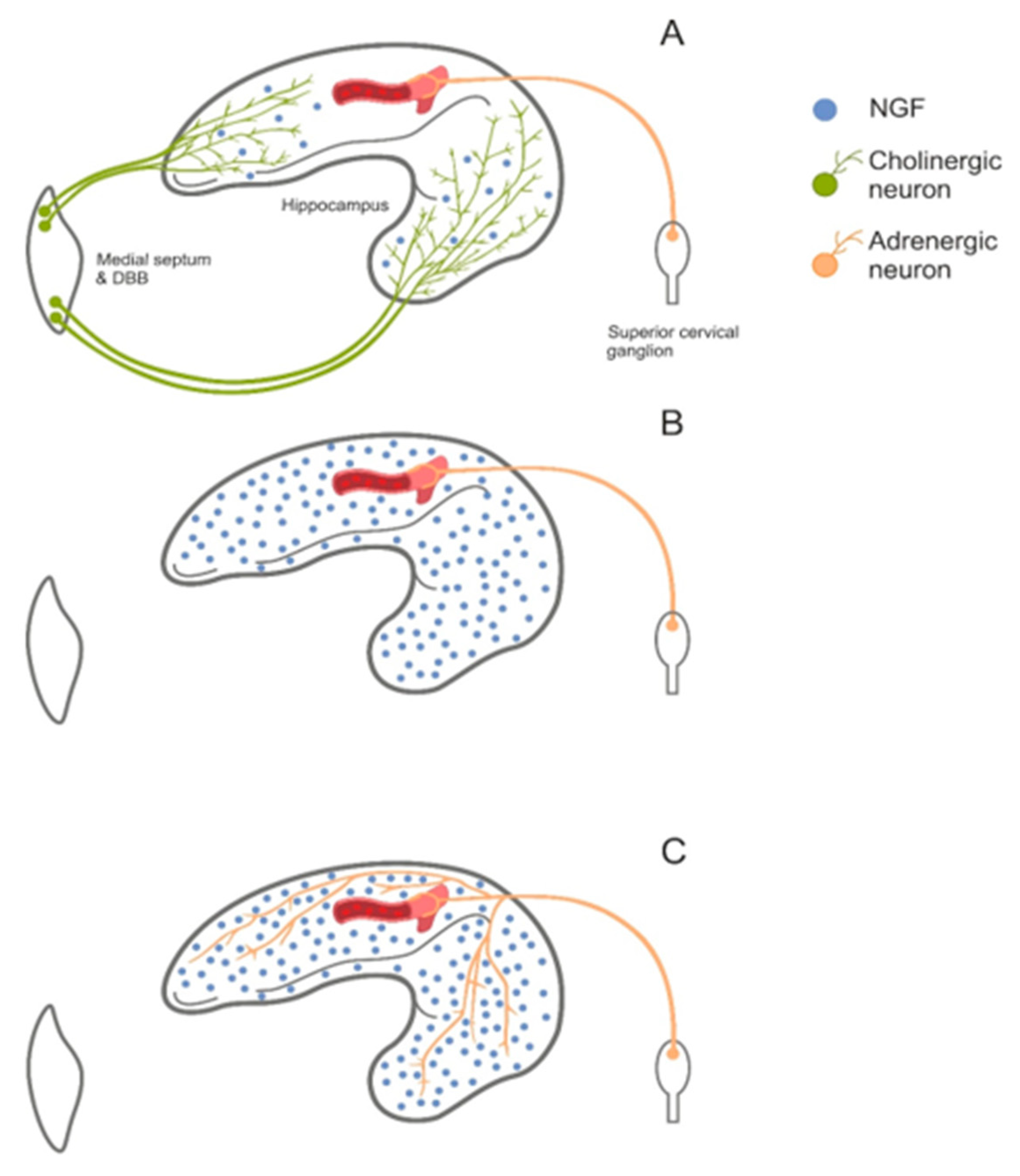
5.7. Alzheimer’s Disease
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giusti, S.A.; Vercelli, C.A.; Vogl, A.M.; Kolarz, A.W.; Pino, N.S.; Deussing, J.M.; Refojo, D. Behavioral phenotyping of Nestin-Cre mice: Implications for genetic mouse models of psychiatric disorders. J. Psychiatr. Res. 2014, 55, 87–95. [Google Scholar] [CrossRef]
- Declercq, J.; Brouwers, B.; Pruniau, V.P.E.G.; Stijnen, P.; De Faudeur, G.; Tuand, K.; Meulemans, S.; Serneels, L.; Schraenen, A.; Schuit, F.; et al. Metabolic and behavioural phenotypes in Nestin-Cre mice are caused by hypothalamic expression of human growth hormone. PLoS ONE 2015, 10, e0135502. [Google Scholar] [CrossRef] [PubMed]
- Mamad, O.; McNamara, H.M.; Reilly, R.B.; Tsanov, M. Medial septum regulates the hippocampal spatial representation. Front. Behav. Neurosci. 2015, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Mercatelli, D.; Battelli, M.G.; Bolognesi, A. Saporin-S6: A useful tool in cancer therapy. Toxins 2013, 5, 1698–1722. [Google Scholar] [CrossRef]
- Giansanti, F.; Flavell, D.J.; Angelucci, F.; Fabbrini, M.S.; Ippoliti, R. Strategies to improve the clinical utility of saporin-based targeted toxins. Toxins 2018, 10, 82. [Google Scholar] [CrossRef]
- Gilabert-Oriol, R.; Weng, A.; Mallinckrodt, B.; Melzig, M.; Fuchs, H.; Thakur, M. Immunotoxins Constructed with ribosome-inactivating proteins and their enhancers: A lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 2014, 20, 6584–6643. [Google Scholar] [CrossRef]
- Walsh, M.J.; Dodd, J.E.; Hautbergue, G.M. Ribosome-inactivating proteins potent poisons and molecular tools. Virulence 2013, 4, 774–784. [Google Scholar] [CrossRef]
- de Zaeytijd, J.; Van Damme, E.J.M. Extensive evolution of cereal ribosome-inactivating proteins translates into unique structural features, activation mechanisms, and physiological roles. Toxins 2017, 9, 123. [Google Scholar] [CrossRef]
- Lappi, D.A.; Esch, F.S.; Barbieri, L.; Stirpe, F.; Soria, M. Characterization of a Saponaria officinalis seed ribosome-inactivating protein: Immunoreactivity and sequence homologies. Biochem. Biophys. Res. Commun. 1985, 129, 934–942. [Google Scholar] [CrossRef]
- Pizzo, E.; Di Maro, A. A new age for biomedical applications of Ribosome Inactivating Proteins (RIPs): From bioconjugate to nanoconstructs. J. Biomed. Sci. 2016, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Seth, D.; Batra, J.K. The cytotoxic activity of ribosome-inactivating protein saporin-6 is attributed to its rRNA N-glycosidase and internucleosomal DNA fragmentation activities. J. Biol. Chem. 2003, 278, 4813–4820. [Google Scholar] [CrossRef] [PubMed]
- Sikriwal, D.; Ghosh, P.; Batra, J.K. Ribosome inactivating protein saporin induces apoptosis through mitochondrial cascade, independent of translation inhibition. Int. J. Biochem. Cell Biol. 2008, 40, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.G.; Oeltmann, T.N.; Lappi, D.A. Immunolesioning: Selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res. 1991, 562, 149–153. [Google Scholar] [CrossRef]
- Stirpe, F.; Barbieri, L.; Battelli, M.G.; Soria, M.; Lappi, D.A. Ribosome-inactivating proteins from plants: Present status and future prospects. Bio/Technology 1992, 10, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Alpár, A.; Kacza, J.; Caleo, M.; Verderio, C.; Giani, A.; Martens, H.; Chaudhry, F.A.; Allegra, M.; Grosche, J.; et al. Cracking down on inhibition: Selective removal of GABAergic interneurons from hippocampal networks. J. Neurosci. 2012, 32, 1989–2001. [Google Scholar] [CrossRef]
- Siena, S.; Lappi, D.A.; Bregni, M.; Formosa, A.; Villa, S.; Soria, M.; Bonadonna, G.; Gianni, A.M. Synthesis and characterization of an antihuman T-lymphocyte saporin immunotoxin (OKT1-SAP) with in vivo stability into nonhuman primates. Blood 1988, 72, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, J.; Dwyer, Z.; Beauchamp, S.; Rodriguez, R.; Fortin, T.; Hayley, S. Quantum dot conjugated saporin activates microglia and induces selective substantia nigra degeneration. Neurotoxicology 2020, 76, 153–161. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Rogers, S.D.; Honore, P.; Allen, B.J.; Ghilardi, J.R.; Li, J.; Daughters, R.S.; Lappi, D.A.; Wiley, R.G.; Simone, D.A. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 1997, 278, 275–279. [Google Scholar] [CrossRef]
- Maciejewski-Lenoir, D.; Heinrichs, S.C.; Liu, X.J.; Ling, N.; Tucker, A.; Xie, Q.; Lappi, D.A.; Grigoriadis, D.E. Selective impairment of corticotropin-releasing factor1 (CRF1) receptor-mediated function using CRF coupled to saporin. Endocrinology 2000, 141, 498–504. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Salin-Pascual, R.; Shiromani, P.J. Effects of hypocretin-saporin injections into the medial septum on sleep and hippocampal theta. Brain Res. 2001, 913, 106–115. [Google Scholar] [CrossRef]
- Bugarith, K.; Dinh, T.T.; Li, A.J.; Speth, R.C.; Ritter, S. Basomedial hypothalamic injections of neuropeptide y conjugated to saporin selectively disrupt hypothalamic controls of food intake. Endocrinology 2005, 146, 1179–1191. [Google Scholar] [CrossRef]
- Baskin, D.G.; Kim, F.; Gelling, R.W.; Russell, B.J.; Schwartz, M.W.; Morton, G.J.; Simhan, H.N.; Moralejo, D.H.; Blevins, J.E. A new oxytocin-saporin cytotoxin for lesioning oxytocin-receptive neurons in the rat hindbrain. Endocrinology 2010, 151, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Keimpema, E.; Kang, Z.; Shantaram Barde, S.; Berghuis, P.; Dobszay, M.B.; Schnell, R.; Mulder, J.; Luiten, P.G.M.; David Xu, Z.; Runesson, J.; et al. GABAergic terminals are a source of galanin to modulate cholinergic neuron development in the neonatal forebrain. Cereb. Cortex 2014, 24, 3277–3288. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.G.; Zhao, Z.Q.; Meng, X.L.; Yin, J.; Liu, X.Y.; Chen, Z.F. Cellular basis of itch sensation. Science 2009, 325, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn-Smith, I.J.; Martin, C.L.; Arnolda, L.F.; Minson, J.B. Tracer-toxins: Cholera toxin B-saporin as a model. J. Neurosci. Methods 2000, 103, 83–90. [Google Scholar] [CrossRef]
- Davis, T.L.; Wiley, R.G. Anti-Thy-1 immunotoxin, OX7-saporin, destroys cerebellar purkinje cells after intraventricular injection in rats. Brain Res. 1989, 504, 216–222. [Google Scholar] [CrossRef]
- Berger-Sweeney, J.; Stearns, N.A.; Murg, S.L.; Floerke-Nashner, L.R.; Lappi, D.A.; Baxter, M.G. Selective immunolesions of cholinergic neurons in mice: Effects on neuroanatomy, neurochemistry, and behavior. J. Neurosci. 2001, 21, 8164–8173. [Google Scholar] [CrossRef]
- Fine, A.; Hoyle, C.; Maclean, C.J.; LeVatte, T.L.; Baker, H.F.; Ridley, R.M. Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience 1997, 81, 331–343. [Google Scholar] [CrossRef]
- Wiley, R.G.; Harrison, M.B.; Levey, A.I.; Lappi, D.A. Destruction of midbrain dopaminergic neurons by using immunotoxin to dopamine transporter. Cell. Mol. Neurobiol. 2003, 23, 839–850. [Google Scholar] [CrossRef]
- Picklo, M.J.; Wiley, R.G.; Lappi, D.A.; Robertson, D. Noradrenergic lesioning with an anti-dopamine β-hydroxylase immunotoxin. Brain Res. 1994, 666, 195–200. [Google Scholar] [CrossRef]
- Radley, J.J.; Gosselink, K.L.; Sawchenko, P.E. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J. Neurosci. 2009, 29, 7330–7340. [Google Scholar] [CrossRef] [PubMed]
- Kanai, T.; Watanabe, M.; Okazawa, A.; Sato, T.; Yamazaki, M.; Okamoto, S.; Ishii, H.; Totsuka, T.; Iiyama, R.; Okamoto, R.; et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology 2001, 121, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Sloviter, R.S. Focal inhibitory interneuron loss and principal cell hyperexcitability in the rat hippocampus after microinjection of a neurotoxic conjugate of saporin and a peptidase-resistant analog of Substance P. J. Comp. Neurol. 2001, 436, 127–152. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular architecture of the mouse nervous system. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Tasic, B.; Yao, Z.; Graybuck, L.T.; Smith, K.A.; Nguyen, T.N.; Bertagnolli, D.; Goldy, J.; Garren, E.; Economo, M.N.; Viswanathan, S.; et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 2018, 563, 72–78. [Google Scholar] [CrossRef]
- Tasic, B.; Menon, V.; Nguyen, T.N.T.; Kim, T.T.K.; Jarsky, T.; Yao, Z.; Levi, B.B.; Gray, L.T.; Sorensen, S.A.; Dolbeare, T.; et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016, 19, 335–346. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef]
- Sathyamurthy, A.; Johnson, K.R.; Matson, K.J.E.; Dobrott, C.I.; Li, L.; Ryba, A.R.; Bergman, T.B.; Kelly, M.C.; Kelley, M.W.; Levine, A.J. Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep. 2018, 22, 2216–2225. [Google Scholar] [CrossRef]
- Cembrowski, M.S.; Bachman, J.L.; Wang, L.; Sugino, K.; Shields, B.C.; Spruston, N. Spatial gene-expression gradients underlie prominent heterogeneity of CA1 pyramidal neurons. Neuron 2016, 89, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Pintus, R.; Riggi, M.; Cannarozzo, C.; Valeri, A.; de Leo, G.; Romano, M.; Gulino, R.; Leanza, G. Essential role of hippocampal noradrenaline in the regulation of spatial working memory and TDP-43 tissue pathology. J. Comp. Neurol. 2018, 526, 1131–1147. [Google Scholar] [CrossRef] [PubMed]
- Safandeev, V.V.; Kolacheva, A.A.; Ivanov, D.E.; Ugryumov, M.V. Detection of the latent functional insufficiency of dopaminergic neurons in the nigrostriatal system in a chronic model of Parkinson’s disease. Neurochem. J. 2017, 11, 290–295. [Google Scholar] [CrossRef]
- Ono, D.; Yamanaka, A. Hypothalamic regulation of the sleep/wake cycle. Neurosci. Res. 2017, 118, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Liblau, R.S.; Vassalli, A.; Seifinejad, A.; Tafti, M. Hypocretin (orexin) biology and the pathophysiology of narcolepsy with cataplexy. Lancet Neurol. 2015, 14, 318–328. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Blanco-Centurion, C.; Greco, M.A.; Shiromani, P.J. Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience 2003, 116, 223–235. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Kohls, M.D.; Greco, M.A.; Waleh, N.S.; Salin-Pascual, R.; Kilduff, T.S.; Lappi, D.A.; Shiromani, P.J. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J. Neurosci. 2001, 21, 7273–7283. [Google Scholar] [CrossRef]
- Kaur, S.; Thankachan, S.; Begum, S.; Liu, M.; Blanco-Centurion, C.; Shiromani, P.J. Hypocretin-2 saporin lesions of the ventrolateral periaquaductal gray (vlPAG) increase REM sleep in hypocretin knockout mice. PLoS ONE 2009, 4, e6346. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Blanco-Centurion, C.A.; Miller, J.D.; Shiromani, P.J. Insomnia following hypocretin2-saporin lesions of the substantia nigra. Neuroscience 2006, 137, 29–36. [Google Scholar] [CrossRef]
- Eikermann, M.; Vetrivelan, R.; Grosse-Sundrup, M.; Henry, M.E.; Hoffmann, U.; Yokota, S.; Saper, C.B.; Chamberlin, N.L. The ventrolateral preoptic nucleus is not required for isoflurane general anesthesia. Brain Res. 2011, 1426, 30–37. [Google Scholar] [CrossRef]
- Gvilia, I. Underlying brain mechanisms that regulate sleep-wakefulness cycles. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2010; Volume 93, pp. 1–21. [Google Scholar]
- Blanco-Centurion, C.; Gerashchenko, D.; Shiromani, P.J. Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J. Neurosci. 2007, 27, 14041–14048. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Rodriguez, E.; Liu, M.; Blanco-Centurion, C.; Shiromani, P.J. Effects of hypocretin (orexin) neuronal loss on sleep and extracellular adenosine levels in the rat basal forebrain. Eur. J. Neurosci. 2008, 28, 1191–1198. [Google Scholar] [CrossRef]
- Kalinchuk, A.V.; Porkka-Heiskanen, T.; Mccarley, R.W.; Basheer, R. Cholinergic neurons of the basal forebrain mediate biochemical and electrophysiological mechanisms underlying sleep homeostasis. Eur. J. Neurosci. 2015, 41, 182–195. [Google Scholar] [CrossRef]
- Kalinchuk, A.V.; McCarley, R.W.; Stenberg, D.; Porkka-Heiskanen, T.; Basheer, R. The role of cholinergic basal forebrain neurons in adenosine-mediated homeostatic control of sleep: Lessons from 192 IgG-saporin lesions. Neuroscience 2008, 157, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Rodríguez, E.; Millán-Aldaco, D.; Palomero-Rivero, M.; Morales-Lara, D.; Mechoulam, R.; Drucker-Colín, R. Cannabidiol partially blocks the excessive sleepiness in hypocretindeficient rats: Preliminary data. CNS Neurol. Disord. Drug Targets 2019, 18, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Shinde, A.; Mohammed, A.R.; Badange, R.K.; Reballi, V.; Bandyala, T.R.; Saraf, S.K.; Bojja, K.; Manchineella, S.; Achanta, P.K.; et al. Discovery and development of N-[4-(1-Cyclobutylpiperidin-4-yloxy)phenyl]-2-(morpholin-4-yl)acetamide Dihydrochloride (SUVN-G3031): A novel, potent, selective, and orally active histamine H 3 receptor inverse agonist with robust wake-promoting activity. J. Med. Chem. 2019, 62, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; Chen, J.; Han, B.; Meng, Q.C.; Veasey, S.C.; Beck, S.G.; Kelz, M.B. Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Curr. Biol. 2012, 22, 2008–2016. [Google Scholar] [CrossRef]
- Luo, T.; Leung, L.S. Involvement of tuberomamillary histaminergic neurons in isoflurane anesthesia. Anesthesiology 2011, 115, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pain, L.; Jeltsch, H.; Lehmann, O.; Lazarus, C.; Laalou, F.Z.; Cassel, J.C. Central cholinergic depletion induced by 192 IgG-Saporin alleviates the sedative effects of propofol in rats. Br. J. Anaesth. 2000, 85, 869–873. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laalou, F.Z.; De Vasconcelos, A.P.; Oberling, P.; Jeltsch, H.; Cassel, J.C.; Pain, L. Involvement of the basal cholinergic forebrain in the mediation of general (propofol) anesthesia. Anesthesiology 2008, 108, 888–896. [Google Scholar] [CrossRef]
- Leung, L.S.; Petropoulos, S.; Shen, B.; Luo, T.; Herrick, I.; Rajakumar, N.; Ma, J. Lesion of cholinergic neurons in nucleus basalis enhances response to general anesthetics. Exp. Neurol. 2011, 228, 259–269. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Epilepsy: A Public Health Imperative; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-151593-1. [Google Scholar]
- Chun, E.; Bumanglag, A.V.; Burke, S.N.; Sloviter, R.S. Targeted hippocampal GABA neuron ablation by stable substance P-saporin causes hippocampal sclerosis and chronic epilepsy in rats. Epilepsia 2019, 60, e52–e57. [Google Scholar] [CrossRef] [PubMed]
- Blümcke, I.; Thom, M.; Aronica, E.; Armstrong, D.D.; Bartolomei, F.; Bernasconi, A.; Bernasconi, N.; Bien, C.G.; Cendes, F.; Coras, R.; et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE commission on diagnostic methods. Epilepsia 2013, 54, 1315–1329. [Google Scholar] [CrossRef]
- Pascual, J.; Heinrichs, S.C. Olfactory neophobia and seizure susceptibility phenotypes in an animal model of epilepsy are normalized by impairment of brain corticotropin releasing factor. Epilepsia 2007, 48, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.H.; Lim, C.E.; Heinrichs, S.C. Antisocial and seizure susceptibility phenotypes in an animal model of epilepsy are normalized by impairment of brain corticotropin-releasing factor. Epilepsy Behav. 2007, 10, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef]
- Castro, A.R.; Pinto, M.; Lima, D.; Tavares, I. Secondary hyperalgesia in the monoarthritic rat is mediated by GABAB and NK1 receptors of spinal dorsal horn neurons: A behavior and c-fos study. Neuroscience 2006, 141, 2087–2095. [Google Scholar] [CrossRef]
- Vierck, C.J., Jr.; Kline, R.H.; Wiley, R.G. Intrathecal substance P-saporin attenuates operant escape from nociceptive thermal stimuli. Neuroscience 2003, 119, 223–232. [Google Scholar] [CrossRef]
- Brown, D.C.; Agnello, K. Intrathecal substance p-saporin in the dog efficacy in bone cancer pain. Anesthesiology 2013, 119, 1178–1185. [Google Scholar] [CrossRef]
- Vulchanova, L.; Olson, T.H.; Stone, L.S.; Riedl, M.S.; Elde, R.; Honda, C.N. Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience 2001, 108, 143–155. [Google Scholar] [CrossRef]
- Alvarez, P.; Gear, R.W.; Green, P.G.; Levine, J.D. IB4-saporin attenuates acute and eliminates chronic muscle pain in the rat. Exp. Neurol. 2012, 233, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.W.; Pawlowski, S.A.; Ribeiro-da-Silva, A. Limited changes in spinal lamina I dorsal horn neurons following the cytotoxic ablation of non-peptidergic C-fibers. Mol. Pain 2015, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.; Chen, X.; Bogen, O.; Green, P.G.; Levine, J.D. IB4(+) nociceptors mediate persistent muscle pain induced by GDNF. J. Neurophysiol. 2012, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pinto, L.G.; Souza, G.R.; Kusuda, R.; Lopes, A.H.; Sant’Anna, M.B.; Cunha, F.Q.; Ferreira, S.H.; Cunha, T.M. Non-peptidergic nociceptive neurons are essential for mechanical inflammatory hypersensitivity in mice. Mol. Neurobiol. 2019, 56, 5715–5728. [Google Scholar] [CrossRef]
- Araldi, D.; Khomula, E.V.; Ferrari, L.F.; Levine, J.D. Fentanyl induces rapid onset hyperalgesic priming: Type I at peripheral and type II at central nociceptor terminals. J. Neurosci. 2018, 38, 2226–2245. [Google Scholar] [CrossRef]
- Streit, W.J. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4). J. Histochem. Cytochem. 1990, 38, 1683–1686. [Google Scholar] [CrossRef]
- Manzhulo, I.V.; Ogurtsova, O.S.; Tyrtyshnaia, A.A.; Dyuizen, I.V. Neuro-microglial interactions in the spinal centers of pain modulation in the neuropathic pain syndrome. Neurochem. J. 2017, 11, 161–167. [Google Scholar] [CrossRef]
- Echeverry, S.; Shi, X.Q.; Yang, M.; Huang, H.; Wu, Y.; Lorenzo, L.-E.; Perez-Sanchez, J.; Bonin, R.P.; De Koninck, Y.; Zhang, J. Spinal microglia are required for long-term maintenance of neuropathic pain. Pain 2017, 158, 1792–1801. [Google Scholar] [CrossRef]
- Wiley, R.G. Substance P receptor-expressing dorsal horn neurons: Lessons from the targeted cytotoxin, substance P-saporin. Pain 2008, 136, 7–10. [Google Scholar] [CrossRef]
- Akiyama, T.; Nguyen, T.; Curtis, E.; Nishida, K.; Devireddy, J.; Delahanty, J.; Carstens, M.I.; Carstens, E. A central role for spinal dorsal horn neurons that express neurokinin-1 receptors in chronic itch. Pain 2015, 156, 1240–1246. [Google Scholar] [CrossRef]
- Han, N.; Zu, J.Y.; Chai, J. Spinal bombesin-recognized neurones mediate more nonhistaminergic than histaminergic sensation of itch in mice. Clin. Exp. Derm. 2012, 37, 290–295. [Google Scholar] [CrossRef]
- Hummel, M.; Cummons, T.; Lu, P.; Mark, L.; Harrison, J.E.; Kennedy, J.D.; Whiteside, G.T. Pain is a salient “stressor” that is mediated by corticotropin-releasing factor-1 receptors. Neuropharmacology 2010, 59, 160–166. [Google Scholar] [CrossRef]
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Lipkin, E.; Marvin, A.R.; Law, J.K.; Lipkin, P.H. Anxiety and mood disorder in children with autism spectrum disorder and ADHD. Pediatrics 2018, 141, e20171377. [Google Scholar] [CrossRef]
- Truitt, W.A.; Sajdyk, T.J.; Dietrich, A.D.; Oberlin, B.; McDougle, C.J.; Shekhar, A. From anxiety to autism: Spectrum of abnormal social behaviors modeled by progressive disruption of inhibitory neuronal function in the basolateral amygdala in Wistar rats. Psychopharmacol 2007, 191, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Rajkumar, R.; Dawe, G.S. Selective lesioning of nucleus incertus with corticotropin releasing factor-saporin conjugate. Brain Res. 2014, 1543, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.R.; Rajkumar, R.; Lee, L.C.; Dawe, G.S. Nucleus incertus contributes to an anxiogenic effect of buspirone in rats: Involvement of 5-HT1A receptors. Neuropharmacology 2016, 110, 1–14. [Google Scholar] [CrossRef]
- Lyons, A.M.; Thiele, T.E. Neuropeptide y conjugated to saporin alters anxiety-like behavior when injected into the central nucleus of the amygdala or basomedial hypothalamus in BALB/cJ mice. Peptides 2010, 31, 2193–2199. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: The dual hit theory revisited. Ann. N. Y. Acad. Sci. 2009, 1170, 615–622. [Google Scholar] [CrossRef]
- Ostock, C.Y.; Lindenbach, D.; Goldenberg, A.A.; Kampton, E.; Bishop, C. Effects of noradrenergic denervation by anti-DBH-saporin on behavioral responsivity to l-DOPA in the hemi-parkinsonian rat. Behav. Brain Res. 2014, 270, 75–85. [Google Scholar] [CrossRef][Green Version]
- Shin, E.; Rogers, J.T.; Devoto, P.; Björklund, A.; Carta, M. Noradrenaline neuron degeneration contributes to motor impairments and development of L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Exp. Neurol. 2014, 257, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Falquetto, B.; Moreira, T.S.; Takakura, A.C. Orexinergic neurons are involved in the chemosensory control of breathing during the dark phase in a Parkinson’s disease model. Exp. Neurol. 2018, 309, 107–118. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Gannon, M.; Wang, Q. Complex noradrenergic dysfunction in Alzheimer’s disease: Low norepinephrine input is not always to blame. Brain Res. 2019, 1702, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Borodovitsyna, O.; Flamini, M.; Chandler, D. Noradrenergic modulation of cognition in health and disease. Neural Plast. 2017, 2017, 6031478. [Google Scholar] [CrossRef] [PubMed]
- Leanza, G.; Gulino, R.; Zorec, R. Noradrenergic hypothesis linking neurodegeneration-based cognitive decline and astroglia. Front. Mol. Neurosci. 2018, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V.; Bobkova, N.V.; Kolosova, N.G.; Samokhin, A.N.; Stepanichev, M.Y.; Stefanova, N.A. Molecular and cellular mechanisms of sporadic Alzheimer’s disease: Studies on rodent models in vivo. Biochemistry 2017, 82, 1088–1102. [Google Scholar] [CrossRef]
- Hanin, I. The AF64A model of cholinergic hypofunction: An update. Life Sci. 1996, 58, 1955–1964. [Google Scholar] [CrossRef]
- Taniuchi, M.; Schweitzer, J.B.; Johnson, E.M. Nerve growth factor receptor molecules in rat brain. Proc. Natl. Acad. Sci. USA 1986, 83, 1950–1954. [Google Scholar] [CrossRef]
- Richardson, P.M.; Verge Issa, V.M.K.; Riopelle, R.J. Distribution of neuronal receptors for nerve growth factor in the rat. J. Neurosci. 1986, 6, 2312–2321. [Google Scholar] [CrossRef]
- Moreau, P.H.; Cosquer, B.; Jeltsch, H.; Cassel, J.C.; Mathis, C. Neuroanatomical and behavioral effects of a novel version of the cholinergic immunotoxin mu p75-saporin in mice. Hippocampus 2008, 18, 610–622. [Google Scholar] [CrossRef]
- Hunter, C.L.; Quintero, E.M.; Gilstrap, L.; Bhat, N.R.; Granholm, A.C. Minocycline protects basal forebrain cholinergic neurons from mu p75-saporin immunotoxic lesioning. Eur. J. Neurosci. 2004, 19, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Roβner, S.; Bigl, V. Chapter 25 Immunolesion by 192IgG-saporin of rat basal forebrain cholinergic system: A useful tool to produce cortical cholinergic dysfunction. Prog. Brain Res. 1996, 109, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.G.; Bucci, D.J.; Gorman, L.K.; Wiley, R.G.; Gallagher, M. Selective immunotoxic lesions of basal forebrain cholinergic cells: Effects on learning and memory in rats. Behav. Neurosci. 2013, 127, 619–627. [Google Scholar] [CrossRef]
- Baxter, M.G.; Gallagher, M. Intact spatial learning in both young and aged rats following selective removal of hippocampal cholinergic input. Behav. Neurosci. 1996, 110, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; McMahan, R.; Chiba, A.; Gallagher, M. A re-examination of the role of basal forebrain cholinergic neurons in spatial working memory. Neuropharmacology 1998, 37, 481–487. [Google Scholar] [CrossRef]
- McMahan, R.; Sobel, T.; Baxter, M. Selective immunolesions of hippocampal cholinergic input fail to impair spatial working memory. Hippocampus 1997, 7, 130–136. [Google Scholar] [CrossRef]
- Gibbs, R.B.; Johnson, D.A. Cholinergic lesions produce task-selective effects on delayed matching to position and configural association learning related to response pattern and strategy. Neurobiol. Learn. Mem. 2007, 88, 19–32. [Google Scholar] [CrossRef][Green Version]
- Gibbs, R.B. Basal forebrain cholinergic neurons are necessary for estrogen to enhance acquisition of a delayed matching-to-position T-maze task. Horm. Behav. 2002, 42, 245–257. [Google Scholar] [CrossRef]
- Johnson, D.A.; Zambon, N.J.; Gibbs, R.B. Selective lesion of cholinergic neurons in the medial septum by 192 IgG-saporin impairs learning in a delayed matching to position T-maze paradigm. Brain Res. 2002, 943, 132–141. [Google Scholar] [CrossRef]
- Lehmann, O.; Jeltsch, H.; Lehnardt, O.; Pain, L.; Lazarus, C.; Cassel, J.C. Combined lesions of cholinergic and serotonergic neurons in the rat brain using 192 IgG-saporin and 5,7-dihydroxytryptamine: Neurochemical and behavioural characterization. Eur. J. Neurosci. 2000, 12, 67–79. [Google Scholar] [CrossRef]
- Lehmann, O.; Jeltsch, H.; Lazarus, C.; Tritschler, L.; Bertrand, F.; Cassel, J.C. Combined 192 IgG-saporin and 5,7-dihydroxytryptamine lesions in the male rat brain: A neurochemical and behavioral study. Pharm. Biochem. Behav. 2002, 72, 899–912. [Google Scholar] [CrossRef]
- Leanza, G.; Muir, J.; Nilsson, O.G.; Wiley, R.G.; Dunnett, S.B.; Björklund, A. Selective immunolesioning of the basal forebrain cholinergic system disrupts short-term memory in rats. Eur. J. Neurosci. 1996, 8, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Leanza, G.; Nilsson, O.G.; Wiley, R.G.; Björklund, A. Selective lesioning of the basal forebrain cholinergic system by intraventricular 192 IgG–saporin: Behavioural, biochemical and stereological studies in the rat. Eur. J. Neurosci. 1995, 7, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, D.P.; Thal, L.J.; Winkler, J. Mnemonic deficits in animals depend upon the degree of cholinergic deficit and task complexity. Exp. Neurol. 2002, 177, 292–305. [Google Scholar] [CrossRef]
- Calza, L.; Giuliani, A.; Fernandez, M.; Pirondi, S.; D’Intino, G.; Aloe, L.; Giardino, L. Neural stem cells and cholinergic neurons: Regulation by immunolesion and treatment with mitogens, retinoic acid, and nerve growth factor. Proc. Natl. Acad. Sci. USA 2003, 100, 7325–7330. [Google Scholar] [CrossRef]
- Cooper-Kuhn, C.M.; Winkler, J.; Kuhn, H.G. Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. J. Neurosci. Res. 2004, 77, 155–165. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef]
- Roher, A.E.; Kuo, Y.M.; Potter, P.E.; Emmerling, M.R.; Durham, R.A.; Walker, D.G.; Sue, L.I.; Honer, W.G.; Beach, T.G. Cortical cholinergic denervation elicits vascular Aβ deposition. Ann. N. Y. Acad. Sci. 2000, 903, 366–373. [Google Scholar] [CrossRef]
- Yao, X.Q.; Jiao, S.S.; Saadipour, K.; Zeng, F.; Wang, Q.H.; Zhu, C.; Shen, L.L.; Zeng, G.H.; Liang, C.R.; Wang, J.; et al. P75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol. Psychiatry 2015, 20, 1301–1310. [Google Scholar] [CrossRef]
- Kelly, S.C.; McKay, E.C.; Beck, J.S.; Collier, T.J.; Dorrance, A.M.; Counts, S.E. Locus coeruleus degeneration induces forebrain vascular pathology in a transgenic rat model of Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 70, 369–386. [Google Scholar] [CrossRef]
- Dobryakova, Y.V.; Volobueva, M.N.; Manolova, A.O.; Medvedeva, T.M.; Kvichansky, A.A.; Gulyaeva, N.V.; Markevich, V.A.; Stepanichev, M.Y.; Bolshakov, A.P. Cholinergic deficit induced by central administration of 192IgG-saporin is associated with activation of microglia and cell loss in the dorsal hippocampus of rats. Front. Neurosci. 2019, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Paban, V.; Chambon, C.; Farioli, F.; Alescio-Lautier, B. Gene regulation in the rat prefrontal cortex after learning with or without cholinergic insult. Neurobiol. Learn. Mem. 2011, 95, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, Y.V.; Kasianov, A.; Zaichenko, M.I.; Stepanichev, M.Y.; Chesnokova, E.A.; Kolosov, P.M.; Markevich, V.A.; Bolshakov, A.P. Intracerebroventricular administration of 192IgG-saporin alters expression of microglia-associated genes in the dorsal but not ventral hippocampus. Front. Mol. Neurosci. 2018, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Paban, V.; Farioli, F.; Romier, B.; Chambon, C.; Alescio-Lautier, B. Gene expression profile in rat hippocampus with and without memory deficit. Neurobiol. Learn. Mem. 2010, 94, 42–56. [Google Scholar] [CrossRef]
- Lemke, R.; Härtig, W.; Rossner, S.; Bigl, V.; Schliebs, R. Interleukin-6 is not expressed in activated microglia and in reactive astrocytes in response to lesion of rat basal forebrain cholinergic system as demonstrated by combined in situ hybridization and immunocytochemistry. J. Neurosci. Res. 1998, 51, 223–236. [Google Scholar] [CrossRef]
- Hartlage-Rübsamen, M.; Schliebs, R. Sequential upregulation of cell adhesion molecules in degenerating rat basal forebrain cholinergic neurons and in phagocytotic microglial cells. Brain Res. 2001, 897, 20–26. [Google Scholar] [CrossRef]
- Seeger, G.; Hartig, W.; Rossner, S.; Schliebs, R.; Bruckner, G.; Bigl, V.; Brauer, K. Electron microscopic evidence for microglial phagocytic activity and cholinergic cell death after administration of the immunotoxin 192IgG-saporin in rat. J. Neurosci. Res. 1997, 48, 465–476. [Google Scholar] [CrossRef]
- Lemke, R.; Roßner, S.; Schliebs, R. Leukemia inhibitory factor expression is not induced in activated microglia and reactive astrocytes in response to rat basal forebrain cholinergic lesion. Neurosci. Lett. 1999, 267, 53–56. [Google Scholar] [CrossRef]
- Lemke, R.; Hartlage-Rübsamen, M.; Schliebs, R. Differential injury-dependent glial expression of interleukins-1 alpha, beta, and interleukin-6 in rat brain. Glia 1999, 27, 75–87. [Google Scholar] [CrossRef]
- Volobueva, M.N.; Dobryakova, Y.V.; Manolova, A.O.; Stepanichev, M.Y.; Kvichansky, A.A.; Gulyaeva, N.V.; Markevich, V.A.; Bolshakov, A.P. Intracerebroventricular administration of 192IgG-saporin alters the state of microglia in the neocortex. Neurochem. J. 2020, 14, 37–42. [Google Scholar] [CrossRef]
- Coradazzi, M.; Gulino, R.; Fieramosca, F.; Falzacappa, L.V.; Riggi, M.; Leanza, G. Selective noradrenaline depletion impairs working memory and hippocampal neurogenesis. Neurobiol. Aging 2016, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Barefoot, H.C.; Maclean, C.J.; Pugh, P.; Baker, H.F. Different effects on learning ability after injection of the cholinergic immunotoxin ME20.4IgG-saporin into the diagonal band of Broca, basal nucleus of Meynert, or both in monkeys. Behav. Neurosci. 1999, 113, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Roßner, S.; Wörtwein, G.; Gu, Z.; Yu, J.; Schliebs, R.; Bigl, V.; Perez-Polo, J.R. Cholinergic control of nerve growth factor in adult rats: Evidence from cortical cholinergic deafferentation and chronic drug treatment. J. Neurochem. 1997, 69, 947–953. [Google Scholar] [CrossRef]
- Gelfo, F.; Tirassa, P.; De Bartolo, P.; Caltagirone, C.; Petrosini, L.; Angelucci, F. Brain and serum levels of nerve growth factor in a rat model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 25, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Valle-Leija, P.; Cancino-Rodezno, A.; Sánchez-Tafolla, B.M.; Arias, E.; Elinos, D.; Feria, J.; Zetina, M.E.; Morales, M.A.; Cifuentes, F. Presence of functional neurotrophin TrkB receptors in the rat superior cervical ganglion. Front. Physiol. 2017, 8, 474. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.E.; McKinnon, D. Expression of the trk gene family of neurotrophin receptors in prevertebral sympathetic ganglia. Dev. Brain Res. 1994, 77, 177–182. [Google Scholar] [CrossRef]
- Nelson, A.R.; Kolasa, K.; McMahon, L.L. Noradrenergic sympathetic sprouting and cholinergic reinnervation maintains non-amyloidogenic processing of AβPP. J. Alzheimer’s Dis. 2014, 38, 867–879. [Google Scholar] [CrossRef]
- Scheiderer, C.L.; McCutchen, E.; Thacker, E.E.; Kolasa, K.; Ward, M.K.; Parsons, D.; Harrell, L.E.; Dobrunz, L.E.; McMahon, L.L. Sympathetic sprouting drives hippocampal cholinergic reinnervation that prevents loss of a muscarinic receptor-dependent long-term depression at CA3-CA1 synapses. J. Neurosci. 2006, 26, 3745–3756. [Google Scholar] [CrossRef]
- Shin, J.; Kong, C.; Lee, J.; Choi, B.Y.; Sim, J.; Koh, C.S.; Park, M.; Na, Y.C.; Suh, S.W.; Chang, W.S.; et al. Focused ultrasound-induced blood-brain barrier opening improves adult hippocampal neurogenesis and cognitive function in a cholinergic degeneration dementia rat model. Alzheimer’s Res. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Zhang, C.; Feng, W.; Vodovozova, E.; Tretiakova, D.; Boldyrevd, I.; Li, Y.; Kürths, J.; Yu, T.; Semyachkina-Glushkovskaya, O.; Zhu, D. Photodynamic opening of the blood-brain barrier to high weight molecules and liposomes through an optical clearing skull window. Biomed. Opt. Express 2018, 9, 4850–4862. [Google Scholar] [CrossRef]
- Semyachkina-Glushkovskaya, O.; Kurths, J.; Borisova, E.; Sokolovski, S.; Mantareva, V.; Angelov, I.; Shirokov, A.; Navolokin, N.; Shushunova, N.; Khorovodov, A.; et al. Photodynamic opening of blood-brain barrier. Biomed. Opt. Express 2017, 8, 5040–5048. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Zhao, H.; Zhu, T.; Yang, Z.; Xu, H.; Fu, Y.; Lin, F.; Pan, X.; Li, L.; et al. Enhanced in vivo blood-brain barrier penetration by circular tau-transferrin receptor bifunctional aptamer for tauopathy therapy. J. Am. Chem. Soc. 2020, 142, 3862–3872. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, W.; Zhang, J.; Meng, F.; Zhong, Z. Protein toxin chaperoned by LRP-1-targeted virus-mimicking vesicles induces high-efficiency glioblastoma therapy in vivo. Adv. Mater. 2018, 30, e1800316. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, J.; Meng, F.; Zhong, Z. Apolipoprotein e peptide-directed chimeric polymersomes mediate an ultrahigh-efficiency targeted protein therapy for glioblastoma. ACS Nano 2018, 12, 11070–11079. [Google Scholar] [CrossRef]
- Field, R.H.; Gossen, A.; Cunningham, C. Prior pathology in the basal forebrain cholinergic system predisposes to inflammation-induced working memory deficits: Reconciling inflammatory and cholinergic hypotheses of delirium. J. Neurosci. 2012, 32, 6288–6294. [Google Scholar] [CrossRef]
- Ohara, P.T.; Kelley, K.; Jasmin, L. B fragment of cholera toxin conjugated to saporin. In Molecular Neurosurgery with Targeted Toxins; Humana Press: Totowa, NJ, USA, 2005; pp. 293–306. ISBN 9781588291998. [Google Scholar]
- Roy, S.; Axup, J.Y.; Forsyth, J.S.; Goswami, R.K.; Hutchins, B.M.; Bajuri, K.M.; Kazane, S.A.; Smider, V.V.; Felding, B.H.; Sinha, S.C. SMI-Ribosome inactivating protein conjugates selectively inhibit tumor cell growth. Chem. Commun. 2017, 53, 4234–4237. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Saporin Conjugate(s) | Molecular Target of Toxin | Cell Subpopulation Killed by Toxin | References |
|---|---|---|---|
| Substance P-saporin | Neurokinin 1 receptor | Interneurons of the hippocampus and neocortex; spinal cord lamina I neurons | [19] |
| CRF-saporin | CRHR1 and CRHR2 receptors | Majority of CNS neurons | [20] |
| Orexin B-saporin | OX1 and OX2 receptors | Majority of CNS neurons | [21] |
| NPY-saporin | NPY receptors | Majority of CNS neurons | [22] |
| Oxytocin-saporin | Oxytocin receptors | Somatostatin-positive interneurons, some pyramidal neurons on the hippocampus and neocortex, mossy cells in the dentate gyrus | [23] |
| Galanin-saporin | Galanin receptors | Somatostatin-positive interneurons, neurons in globus pallidus and thalamus | [24] |
| Bombesin-saporin | Bombesin receptors (NMBR, GRPR, and BRS3) | GPRP-positive interneurons in the hippocampus and neocortex; neurons in the superficial dorsal horn of the spinal cord. | [25] |
| Cholera toxin B-saporin | GM1-ganglioside | Majority of CNS neurons | [26] |
| Saporin Conjugate(s) | Molecular Target of Toxin | Cell Subpopulation Killed by Toxin | Model of Pathology | References |
|---|---|---|---|---|
| OX7-saporin | Thy-1 glycoprotein | Majority of CNS neurons, Cerebellar Purkinje neurons | - | [27] |
| 192IgG-saporin (rat) mu-saporin (mice) ME20.4-saporin (primates) | NGFR | NGFR-positive neurons; forebrain cholinergic neurons; upper cervical ganglion | Alzheimer’s disease | [14,28,29] |
| Anti-DAT-antibody-saporin | Dopamine transporter (DAT) | Dopaminergic neurons | Parkinson’s disease | [30] |
| anti-DβH-saporin | dopamine-β-hydroxylase (DβH) | Locus coeruleus/subcoeruleus | Alzheimer’s disease | [31] |
| anti-GAT1-saporin | GABA transporter 1 (GAT1, slc6a1) | GABAergic neurons | - | [32] |
| saporin-conjugated anti-VGAT-C antibodies (SAVA) | Vesicular GABA transporter (VGAT) | GABAergic neurons | - | [16] |
| Anti-mac1-saporin | Integrin αM (Mac-1, CD11b) | Macrophages; microglia in CNS | - | [33] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolshakov, A.P.; Stepanichev, M.Y.; Dobryakova, Y.V.; Spivak, Y.S.; Markevich, V.A. Saporin from Saponaria officinalis as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience. Toxins 2020, 12, 546. https://doi.org/10.3390/toxins12090546
Bolshakov AP, Stepanichev MY, Dobryakova YV, Spivak YS, Markevich VA. Saporin from Saponaria officinalis as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience. Toxins. 2020; 12(9):546. https://doi.org/10.3390/toxins12090546
Chicago/Turabian StyleBolshakov, Alexey P., Mikhail Yu. Stepanichev, Yulia V. Dobryakova, Yulia S. Spivak, and Vladimir A. Markevich. 2020. "Saporin from Saponaria officinalis as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience" Toxins 12, no. 9: 546. https://doi.org/10.3390/toxins12090546
APA StyleBolshakov, A. P., Stepanichev, M. Y., Dobryakova, Y. V., Spivak, Y. S., & Markevich, V. A. (2020). Saporin from Saponaria officinalis as a Tool for Experimental Research, Modeling, and Therapy in Neuroscience. Toxins, 12(9), 546. https://doi.org/10.3390/toxins12090546