1. Introduction
Snakes use their venom for defense and to immobilize and digest their prey. These functions are accomplished by enzymatic and/or non-enzymatic components of the venom and are usually characterized by high target specificity and effectiveness, thermal stability, and resistance to proteolysis [
1]. The venoms of
Viperidae snakes are rich in proteins that strongly affect the hemostatic system by promoting or inhibiting platelet aggregation, blood coagulation, and fibrinolysis [
2]. Members of the two families of snake venom enzymes, serine proteases (SVSPs) and metalloproteases (SVMPs), exert procoagulant activity by activating one or more coagulation factors in the blood coagulation cascade [
3]. Depending on their principal target in the coagulation system, they are usually thrombin-like enzymes (TLEs), activators of prothrombin, factor V (FV), and FX. TLE and FV activators are exclusively SVSPs, while activators of FX and prothrombin can be either SVSPs or SVMPs [
4].
SVSPs are abundantly present in viperid venoms, mostly as monomeric glycoproteins of 26 to 67 kDa. Their mass depends on the extent of either
N- or
O-glycosylation. SVSPs are classified within the clan PA as subclan S, family S1 (chymotrypsin), or subfamily A of the proteolytic enzymes [
5]. Such proteases are characterized by a typical chymotrypsin fold and two six-stranded β-barrels. Their active sites lie in the cleft between the latter and include the canonical catalytic triad His-Asp-Ser [
6]. Despite having a 50% to 80% shared identity of their amino acid sequences, SVSPs differ substantially in their substrate specificity, and consequently, in their pharmacological activity [
7]. SVSPs that affect hemostasis express trypsin-like activity, hydrolyzing the BApNA (N
α-benzoyl-
L-arginine 4-nitroanilide) substrate. They are not inhibited by endogenous serine protease inhibitors (serpins) in the presence of heparin [
8]. Consequently, SVSPs can be used in medical treatments and the diagnosis of blood coagulation irregularities [
9]. The two best-known SVSP-based medical products are Viprinex
® and Defibrase
®. Viprinex
® (also known as Ancrod) is a defibrinogenating agent derived from Malayan pit viper venom and has been investigated for the treatment of acute ischemic stroke [
10]. Defibrase
® is a therapeutic product based on a procoagulant SVSP called batroxobin from
Bothrops atrox venom. It is used to decrease fibrinogen levels, leading to the inhibition of thrombogenesis and the prevention of thrombotic diseases. Batroxobin, sold as Reptilase
®, is also used to determine the blood clotting time (Reptilase
® time) [
11].
The nose-horned viper (
Vipera ammodytes ammodytes—
Vaa), the most venomous snake in Europe, exhibits diverse hematotoxic and coagulopathic effects in vivo [
12,
13] and in vitro [
14].
Vaa venom contains diverse types of enzymes, such as secreted phospholipases A
2 (sPLA
2s), SVMPs, L-amino acid oxidases (LAAOs), and SVSPs. Although the latter are highly abundant in the venom, they have not been the most widely studied.
Four
Vaa SVSPs have so far been purified and characterized: two kallikrein-like enzymes [
15]; a fibrin(ogen)ase with an unconventional catalytic triad, VaSP1 [
16]; and an anticoagulant, which is the enzymatically inactive SVSP homolog
VaaSPH-1 [
12]. Comprehensive transcriptomic and proteomic analysis of
Vaa venom has revealed the existence of at least ten other SVSP molecules [
17]. In this study, one of these SVSPs has been purified and characterized.
VaaSP-VX, as we named this molecule, is a potent procoagulant. We have shown that its blood-coagulation-promoting effect is likely due to its FV- and FX-activating activities. The activation of the precursors of both components that form the prothrombinase complex by a single venom protease would be unique and would endow
VaaSP-VX with promising medical potential.
3. Discussion
Vaa venom is a rich source of proteins that act on the hemostatic system [
14]. In the present study, we purified and characterized a procoagulant SP from this venom and named it
VaaSP-VX.
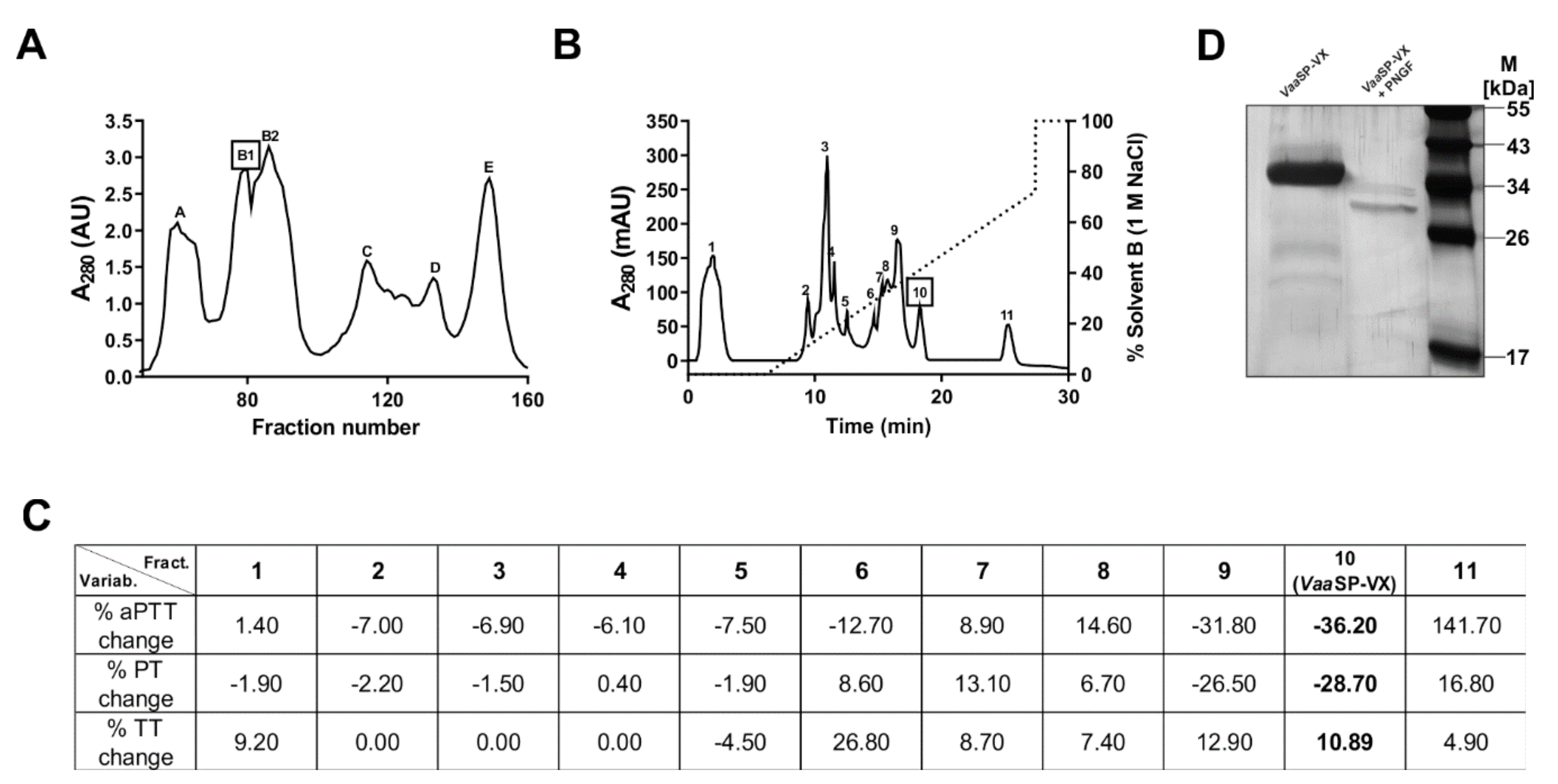
VaaSP-VX was isolated in a two-step chromatographic procedure, in which gel filtration was followed by cation-exchange chromatography (
Figure 1).
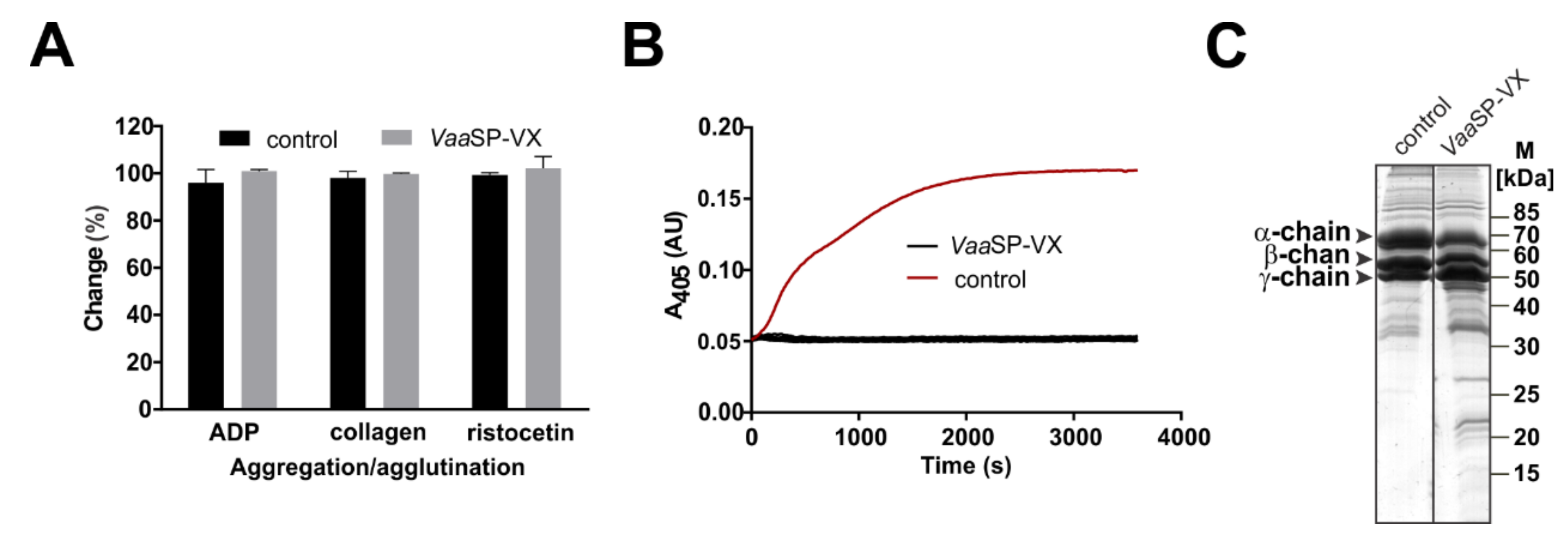
VaaSP-VX was found in the fraction, which substantially shortened aPTT and PT without interfering with the platelet function (
Figure 1C and
Figure 2A).
VaaSP-VX was found to be a single-chain
N-glycosylated protein with an apparent molecular mass of 34 kDa. The mass of the glycan moiety was about 5 kDa (
Figure 1D), representing approximately 15% of the total mass of the molecule. It was demonstrated that glycosylation affected the enzymatic activity and specificity of SVSPs by defining target recognition and by binding inhibitors [
22,
23,
24,
25]. Therefore, it may be expected that carbohydrates in
VaaSP-VX have similar functions.
The partial amino acid sequence of
VaaSP-VX was determined using Edman and MS/MS sequencing. The sequenced part of
VaaSP-VX, which covered about 40% of the predicted mature protein, exhibited a high sequence identity with another SVSP from the
Vaa venom, namely
VaaSP-6 (MG958495), with unknown functions. These two proteins differed in the sequenced parts by only three amino acid residues, and therefore, could be regarded as isoforms (
Figure 5). This conclusion is supported by a 2D-PAGE-based proteomic study of the
Vaa venom [
17], in which
VaaSP-6 isoforms were identified in 21 spots positioned in the pI range from 6 to 9. Moreover, in 3 of these 21 spots, a peptide
92EYTMWDKDIMLIR
104 was detected, which was identified as a part of nikobin, which is an SVSP from the closely related snake
V. berus nikolskii (E5AJX2) (
Figure 5), but may also have originated from
VaaSP-VX. The first part of this peptide,
92EYTMWDK
98, which was only identified in the case of
VaaSP-VX, was the same in the two molecules, as was the sequence preceding it, namely
85FFCLSSK
91. Nikobin and
VaaSP-VX were therefore identical in the region
85FFCLSSKEYTMWDK
98, while
VaaSP-6 was distinguished in this part from the other two SPs by the unique
92E/N substitution. In the non-redundant NCBI database, nikobin was the most similar protein to
VaaSP-VX and
VaaSP-6, displaying a 97% sequence identity (
Figure 5). However, some
VaaSPs may differ just in their glycosylation patterns, representing glycoforms of identical amino acid sequences.
The comparable effects of
VaaSP-VX on aPTT and PT (
Figure 1C) and clotting of FVII-deficient plasma by
VaaSP-VX (
Figure 3C) suggested that the action of this venom protease was directed toward the common pathway of the blood coagulation, in which FXa, as a component of the prothrombinase complex, activates prothrombin to thrombin, which then cleaves fibrinogen to form a fibrin clot [
26]. We have shown that VaaSP-VX did not have the same effect as thrombin because it could not directly induce the clotting of fibrinogen (
Figure 2B). Accordingly, the TT was also not shortened in its presence (
Figure 1C), but moreover, it was even slightly extended. Such an effect may be explained by the defibrinogenating effect of
VaaSP-VX due to its partial cleavage of the α chain of fibrinogen (
Figure 2C). Since we showed that
VaaSP-VX also did not possess FXa-like activity, its effect on blood plasma must be associated with the activity of the prothrombinase complex assembled from the activated forms of FV and FX. This suggested that
VaaSP-VX was involved in the activation of these blood-clotting factors. To test this hypothesis, we treated FV and FX with
VaaSP-VX and analyzed the cleavage products.
FV circulates in the blood as a 330 kDa protein comprised of six domains, A1, A2, A3, B, C1, and C2. In the process of its physiological activation, it is cleaved by thrombin to release the B domain. A heterodimeric FV is formed, in which the A1 and A2 domains constitute the HC and A3 and the C1 and C2 domains the LC, respectively [
27]. This form of FV is further trimmed by FXa or by thrombin (FIIa). FXa cleaves FV at Arg
348 in the A2 domain and at Arg
1753 in the A3 domain, which produces its fully activated form (
Figure 4C) [
18,
19,
20]. Bovine FV was incubated with
VaaSP-VX and the degradation products were N-terminally sequenced. In this way, we established that
VaaSP-VX cleaved FV at the same positions as FXa did. The report that treatment of human FV with FXa induced the FVa activity, which is associated with the cleavage at Arg
1765 (this is Arg
1753 in bovine FV) [
28], strongly suggests that
VaaSP-VX could convert not only bovine but also human FV into its activated form (
Figure 4A,C). FV has already been identified as a target of SVSPs. Two well-studied examples are the FV activators from the venoms of
Daboia snakes, 26 kDa RVV-V from
D. siamensis and 28.4 kDa LVV-V from
V. (M.) lebetina [
29,
30]. However, they activate human FV in a thrombin-like manner, cleaving it at Arg
1545 (which corresponds to Arg
1536 in bovine FV;
Figure 4C) [
31]. Both enzymes are single-chain glycoproteins. Regarding
VaaSP-VX, RVV-V appears in several isoforms [
29]. Two of them, which have been completely sequenced, differ from each other only by six amino acid residues.
Mature FX is a vitamin-K-dependent SP zymogen, which circulates in the blood as a disulphide-linked two-chain molecule of 59 kDa [
32]. It is activated by FIXa or FVIIa, which are enzymatic components of the intrinsic or extrinsic tenase complex. In both cases, FXa is produced by cleavage of the Arg
194–Ile
195 peptide bond in the HC of FX. As we demonstrated,
VaaSP-VX cleaved FX in the same way as both physiological activators (
Figure 4B,D), and thus likely converted it into its activated form. Such an assumption was then also confirmed by the results of experiments using a specific chromogenic substrate of FXa. A variety of FX activators have already been found in snake venoms [
4]. While FX activators from
Viperidae venoms are mainly SVMPs, FX-activating SVSPs have only been described so far in two
Elapidae venoms [
33,
34]. Like
VaaSP-VX, they are single-chain glycoproteins but with a much higher molecular mass (62 and 70 kDa). No structural data are available for any of them; therefore, the first structural data of FX-activating SVSP is provided in this paper. The procoagulant activity of
VaaSP-VX was thus the consequence of the simultaneous activation of FV and FX in a physiological manner by this venom enzyme.
The activity of blood coagulation factors is tightly regulated by endogenous protein inhibitors (serpins), such as antithrombin [
35]. The serpin interaction site in thrombin includes its 44-loop and 148-loop (
Figure 5). As was experimentally demonstrated using RVV-V and LVV-V [
31], the absence of these two loops renders SVSPs resistant to serpins.
VaaSP-VX also lacks these two loops; therefore, it is expected not to be inhibited by serpins. As an activator of multiple blood coagulation factors that is resistant to inhibition by serpins,
VaaSP-VX is of potential interest regarding the development of an innovative clot-promoting drug for external use in patients suffering from hemophilia or other bleeding conditions [
36].
Additionally, the potential double-activating activity of
VaaSP-VX offers an elegant solution to replacing the unreliable diluted Russell‘s viper venom (dRVV) in clinical testing of the lupus anticoagulant (LA) [
37]. Two components of dRVV are important for this test: RVV-V, which is an SVSP that activates FV and RVV-X, which is an SVMP that activates FX. However, the problem with the dRVV reagent is its consistency since it is obtained from a natural source that is subject to considerable biological variations. Accordingly, the results of the LA test are frequently inconsistent. To improve the sensitivity and specificity of the test, the use of purified snake venom components for the activation of FV and FX has already been suggested [
38]. An even better solution would be to use
VaaSP-VX, which would simultaneously activate both FV and FX.
5. Materials and Methods
5.1. Purification of VaaSP-VX
Crude Vaa venom was provided by the Institute of Immunology, Zagreb, Croatia. Lyophilized venom was kept at −20 °C. Before the analysis, it was dissolved in 50 mM Tris, 2 mM CaCl2, 300 mM NaCl, pH 7.0 (buffer A). Fifteen milliliters of the crude venom solution (i.e., 1 g of the venom) was applied to a Sephacryl S-200 Superfine column (100 cm × 4 cm) (GE Healthcare BioSciences AB, Uppsala, Sweden). Gel chromatography was performed at a constant flow rate of 33.6 mL/h. The concentration of proteins in the mobile phase was monitored by measuring their absorbance at 280 nm (A280). The B1 fraction was dialyzed against 20 mM MES, 2 mM CaCl2, pH 6.5 (buffer B), and further fractionated on a Mono S 5/50 GL column of the FPLC system (ÄKTA, GE Healthcare Life-Science, Uppsala, Sweden), equilibrated with the same buffer. The column-retained material was eluted using a linear gradient of NaCl (0 to 1 M) in the buffer B. VaaSP-VX was eluted from the column, concentrated, and dialyzed against buffer B, at pH 7.5.
5.2. Polyacrylamide Gel Electrophoreses
One-dimensional polyacrylamide gel electrophoresis (PAGE) of the samples (as specified in the descriptions of figures showing the results) was performed under non-denaturing or denaturing conditions, i.e., in the absence and presence of the detergent sodium dodecyl sulphate (SDS-PAGE). For SDS-PAGE analysis, 12.5% (
m/
v) PA gels were used under non-reducing and reducing conditions. The proteins in gels were visualized via silver staining using the modified method of Morrissey, where protein fixation is accomplished by a mixture of water, ethanol (30% (
v/
v)), and acetic acid (10% (
v/
v)) [
39]. The molecular mass standards were from Fermentas (Thermo Fisher Scientific, Vilnius, Lithuania).
Two-dimensional gel electrophoresis (2DE) of 9 μg of
VaaSP-VX was performed as specified in Latinović et al. [
39].
5.3. N-Terminal Amino Acid Sequencing and Mass Spectrometry
N-terminal amino acid sequencing of the isolated VaaSP-VX was performed using automated Edman degradation on a Procise 492A Sequencing System (Applied Biosystems, Foster City, CA, USA).
For the mass spectrometry (MS) analysis, 2 μg of vacuum-dried
VaaSP-VX was dissolved in 10 μL of 50 mM NH
4HCO
3 supplemented with 6 M urea. The protein was reduced via the addition of 0.25 µL of 100 mM dithiothreitol (DTT) in 50 mM NH
4HCO
3 and incubation at 37 °C for 1 h. The mixture was then diluted with 10 µL of 50 mM NH
4HCO
3 and alkylated with 0.8 µL of 250 mM iodoacetamide in 50 mM NH
4HCO
3 at room temperature (RT) for 45 min in the dark. Alkylation was stopped by adding 2.5 µL of DTT and then 50 mM NH
4HCO
3 to 90 µL. Fragmentation of the reduced and alkylated
VaaSP-VX was accomplished by adding 0.1 μg of endoproteinase Lys-C and standing at 37 °C overnight. Fifty nanograms of trypsin in 10 µL of 50 mM NH
4HCO
3 was then added, and the mixture was left overnight for further fragmentation at 37 °C. Proteolysis was stopped via the addition of formic acid to the sample to a final concentration of 0.1% (
v/
v). The sample was desalted and concentrated in Vivapure C18 micro spin columns (Sartorius Stedim Biotech, Göttingen, Germany) according to the manufacturer’s instructions, with formic acid replacing trifluoroacetic acid. The
VaaSP-VX hydrolysate was analyzed on an ion trap mass spectrometer 1200 series HPLC-Chip-LC/MSD Trap XCT Ultra (Agilent Technologies, Waldbronn, Germany), as described in Leonardi et al. [
40]. MS and tandem MS (MS/MS) spectra were searched against the
Vaa venom gland cDNA transcripts library and the non-redundant NCBI (National Center for Biotechnology Information) database using the Spectrum Mill software (A.03.03.084, Agilent Technologies, Santa Clara, CA, USA).
5.4. Search for the mRNA Transcript of VaaSP-VX
The cDNA sequence of
VaaSP-VX was searched for using the
Vaa venom gland cDNA library of mRNA transcripts [
41], and PCR primers designed using two different approaches. First, we designed the sense primer deduced from an amino acid stretch from the N-terminal region of
VaaSP-VX and determined using Edman degradation (CNINEHPF; cac
gaattcATGTAACATAAATGAACATCCTTTC, where the additional
EcoRI restriction site is underlined). Second, we designed a sense primer with a broader specificity (CNINEHP; cac
gaattcATGTAACATAAATGAACATCCTTT). The antisense primer was the same in both cases and was deduced from the end of the coding region of other known
VaaSP mRNAs, including the stop codon and a part of the 5′ UTR region (tat
aagcTTTTCAAAAGTTT
TCACGGGG, with an additional
HindIII site underlined and a stop codon doubly underlined). In an attempt to obtain more specific binding of primers to template DNA, we employed different PCR protocols by using Taq or Q5 polymerase, changing the number of cycles and Tm, and lowering concentrations of primers and dNTPs.
5.5. N-Deglycosylation
Four micrograms of dry VaaSP-VX were dissolved in 10 μL of 1% (m/v) SDS and incubated at 100 °C for 5 min. The solution was mixed with 34 μL of 1.5% (m/v) CHAPS in 50 mM Na2HPO4, pH 7.5. A total of 3 U (3 µL) of the peptide N-glycosidase F (PNGF, Roche, Mannheim, Germany) was added to half of this sample. To the other half, just water was added and served as the control. After overnight incubation at 37 °C, both samples were analyzed using SDS-PAGE on a 12.5% gel under reducing conditions. The gel was stained with colloidal silver, as described above.
5.6. Prothrombin Time, Activated Partial Thromboplastin Time, Thrombin Time, and Re-Calcification Time Measurements
To 45 µL of normal human plasma (Diagnostica stago, Asnières-sur-Seine, France) in a cuvette, we added an aliquot of each fraction eluted from a Mono S column in 5 µL (volume adjustment was accomplished using buffer B) to obtain the final concentration of protein in the assay of 1 µM. In the control experiment, just 5 µL of buffer B was added to human plasma. Samples were incubated for 1 min at 37 °C. Then, the prothrombin time (PT), activated partial thromboplastin time (aPTT), and thrombin time (TT) were measured using the BFT II Coagulation Analyzer (Siemens, Munich, Germany). The measurement of PT was initiated by adding 100 µL of Thromborel S reagent (lyophilized human placental thromboplastin (≤60 g/L) and CaCl2 (approx. 1.5 g/L); Siemens, Munich, Germany). The measurement of aPTT was initiated by adding 50 µL of Pathromtin SL reagent (SiO2 particles (1.2 g/L), plant phospholipids (0.25 g/L), NaCl, HEPES, pH 7.6; Siemens, Munich, Germany). After 2 min of incubation, 50 µL of 25 mM CaCl2 was added and the clotting time was recorded as the aPTT. The addition of 100 µL of Thrombin reagent (source of α-thrombin; Siemens, Germany) to the sample induced the clotting of plasma in a time registered as the TT.
The dependence of aPTT on the concentration of VaaSP-VX was also measured in an alternative way. Fresh human plasma, aPTT soluble activator (SA) reagent (Helena Laboratories, Beaumont, TX, USA), 25 mM CaCl2 (Helena Laboratories, USA), and solutions of VaaSP-VX in phosphate-buffered saline (PBS: 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) were pre-warmed at 37 °C. Different amounts of VaaSP-VX were added to the plasma, which was incubated for 10 min at 37 °C. The aPTT SA reagent (0.1 mM Ellagic acid with a suspension of phospholipids extracted from dehydrated rabbit brain) was then added and the mixture was incubated for another 5 min. Finally, the clotting of plasma was initiated by the addition of CaCl2. All solutions were always added in 30 µL volumes. The formation of the clot was followed by measuring the absorbance at 405 nm (A405) on an Infinite M200 microplate reader (Tecan, Männedorf, Switzerland).
The same procedure, except substituting aPTT SA reagent with PBS (i.e., the procedure without supplement of phospholipids), was used to assess the full plasma re-calcification time.
The blood was donated by healthy human volunteers according to permission no. 53/08/11 of the National Medical Ethics Committee of the Republic of Slovenia.
5.7. Platelet Aggregation/Agglutination Assays
Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) were prepared as described in Leonardi et al. [
42]. To 250 μL of PRP, 25 μL of
VaaSP-VX solution was added, resulting in a final concentration of 1 µM
VaaSP-VX; it was then pre-incubated for 5 min at 37 °C. In the control experiment, 25 μL of water was added to 250 μL of PRP. Twenty-five microliters of collagen solution (DiaMed, Cressier,, Switzerland), giving a final concentration of 10 μg/mL; ristocetin (Chrono-Log Corporation, Havertown, PA, USA), resulting in a final concentration of 1 mg/mL; or ADP (Chrono-Log Corporation, USA) at a final concentration of 10 μM, were lastly added and the change in the optical density was monitored for 5 min using a Model 700 Whole Blood/Optical Lumi-Aggregometer (Chrono-Log Corporation, Havertown, PA, USA). The cuvette containing PRP corresponded to 0% light transmission and the cuvette with PPP to 100% light transmission. Blood was donated by healthy human volunteers according to permission no. 53/08/11 of the National Medical Ethics Committee of the Republic of Slovenia.
5.8. FVIIa, FIXa, and FXa-like Activity Testing
To test whether VaaSP-VX acts in the same way as FVIIa, FIXa, or FXa, we used the following specific chromogenic substrates: S-2288TM (Diapharma, West Chester Township, OH, USA) for FVIIa, Spectrozyme® (American Diagnostics Inc., New York, NY, USA) for FIXa, and S-2765TM and S-2222TM (Diapharma, West Chester Township, OH, USA) for FXa. To 100 µL VaaSP-VX (diluted with PBS to final concentrations of 1 µM and 3 µM), 50 µL of Spectrozyme®, S-2288TM, S-2765TM, or S-2222TM were added to final concentrations of 1 mM. Substrate hydrolysis was then examined on an Infinite M200 microplate reader (Tecan, Switzerland) by measuring A405.
Additionally, to test the FXa-like activity of VaaSP-VX on prothrombin, we incubated 10 pmol of prothrombin with 20 pmol of VaaSP-VX in 20 μL of a buffer consisting of 50 mM HEPES, 2 mM CaCl2, and 150 mM NaCl (pH 7.4). After 10 min of incubation at room temperature, the reaction mixture was 10-fold diluted with the buffer and transferred to a microplate well. After supplementing the reaction mixture with 10 μM α-thrombin-specific fluorogenic substrate Boc-Val-Pro-Arg-AFC (Peptide Institute Inc., Osaka, Japan), the hydrolysis of the substrate was assessed on an Infinite M1000 microplate reader (Tecan, Switzerland) by activating fluorescence at 380 nm and detecting it at 500 nm.
5.9. Testing of FIX and FX Activation
The activation of FIX and FX by VaaSP-VX was determined using the following specific chromogenic substrates: Spectrozyme® (American Diagnostics Inc., USA) for FIXa and S-2765TM (Diapharma, USA) for FXa. In a well of a 96-well plate, 50 µL of 1 µM VaaSP-VX was added to 50 µL of 100 nM FIX, and then incubated for 15 min at 37 °C. Furthermore, to 50 µL of 100 nM FX, 3 µM or even 10 µM VaaSP-VX was added before the incubation. In negative controls, VaaSP-VX was replaced with PBS. Activation of a particular blood coagulation factor by VaaSP-VX was detected using an Infinite M200 microplate reader (Tecan, Switzerland). The activation of FIX or FX was detected via an increase in A405 following the addition of 50 µL of Spectrozyme® (final concentration 1 mM) or S-2765TM (final concentration 0.65 mM) to the reaction mixture, respectively.
5.10. Characterization of Proteolytic Specificity
The proteolytic activity of VaaSP-VX was characterized by testing its ability to hydrolyze and/or activate blood coagulation factors and cofactors.
The fibrinogenolytic activity of
VaaSP-VX was examined as described in Sajevic et al. [
43] using a human fibrinogen to
VaaSP-VX mass ratio of 100:1. It was then tested to determine whether the cleavage of fibrinogen by
VaaSP-VX resulted in the formation of a stable fibrin clot. To this end, 40 µL of
VaaSP-VX in PBS (0.1 to 10 µM final concentration), or 40 µL of thrombin (final concentration 1 µM; Helena Laboratories, USA) in a positive control, was added to a well of the 96-well microtiter plate. Subsequently, 40 µL of fibrinogen (final concentration 2 µM; Sigma-Aldrich, St. Louis, MO, USA) was added to a well and the fibrin clot formation was monitored at 405 nm (A
405) using a microplate reader (Infinite M200, Tecan, Switzerland).
To examine the hydrolysis of FV and FX (Haematologic Technologies Inc., Essex Junction, VT, USA) by
VaaSP-VX, we used more easily accessible bovine FV, which appears to be functionally identical to human FV [
19] and human FX. Seven micrograms of each substrate was incubated with or without (control) 1 μg of
VaaSP-VX in 25 µL of 20 mM Tris/HCl, 5 mM CaCl
2, 140 mM NaCl, pH 7.5 at 37 °C. A 10 µL aliquot of the reaction mixture was withdrawn after 1, 3, 6, and 24 h, and analyzed using 12.5% (
m/
v) SDS-PAGE under reducing conditions. The gels were stained with PageBlue
® (Thermo Fisher Scientific, Vilnius, Lithuania). Protein bands were electroblotted from the gel to the PVDF membrane in Towbin transfer buffer (4 mM Tris/HCl, pH 7.5, 12 mM NaCl, and 20% (
v/
v) methanol) at 200 mA per gel for 90 min at room temperature. The PVDF membrane was immersed in 100% methanol for a few seconds, then moved to a 0.1% (
m/
v) Coomassie Blue R-250 (Thermo Fisher Scientific, Vilnius, Lithuania) + 1% (
v/
v) acetic acid + 40% (
v/
v) methanol solution for 1 min. The membrane was destained in 50% (
v/
v) methanol, then washed with water. Protein bands were excised and sequenced using automated Edman degradation on a Procise 492A Sequencing System (Applied Biosystems, Waltham, MH, USA).
5.11. Clotting of FVII-Deficient Plasma
Seventy-five microliters of commercial human plasma lacking FVII (HemosIL
®, Lexington, MA, USA), 95 µL of 25 mM CaCl
2 (Siemens, Germany), and 20 µL
VaaSP-VX in PBS to various final concentrations (refer to
Figure 3C for concentrations) were individually pre-warmed to 37 °C.
VaaSP-VX (or only PBS in the control experiment) was then added to the plasma and incubated for 2 min at 37 °C, followed by the addition of 25 mM CaCl
2 to induce clotting. Plasma clotting time was measured using the BFT II Coagulation Analyzer (Siemens, Germany).

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




