Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays



Abstract
1. Introduction
2. Results
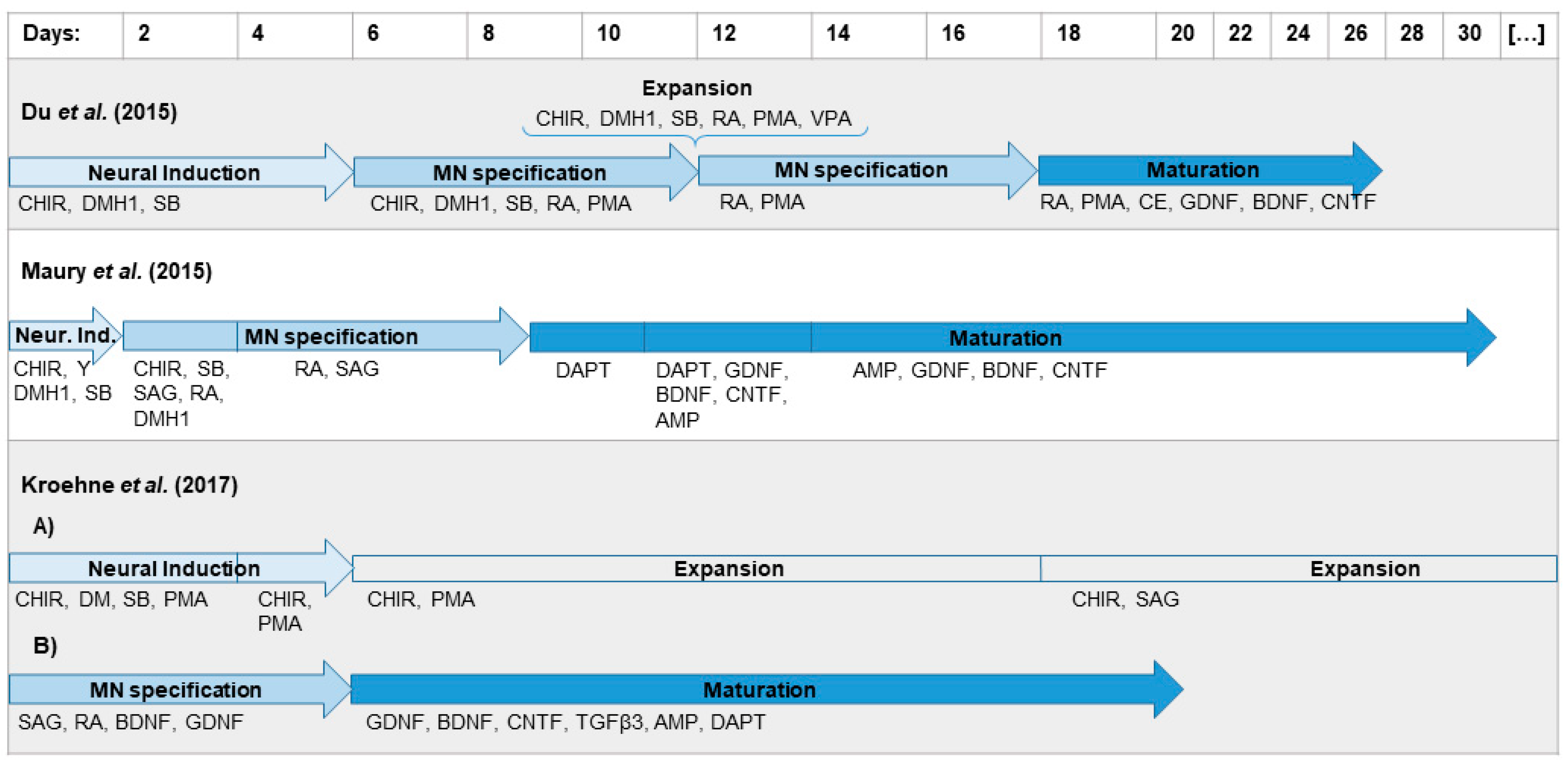
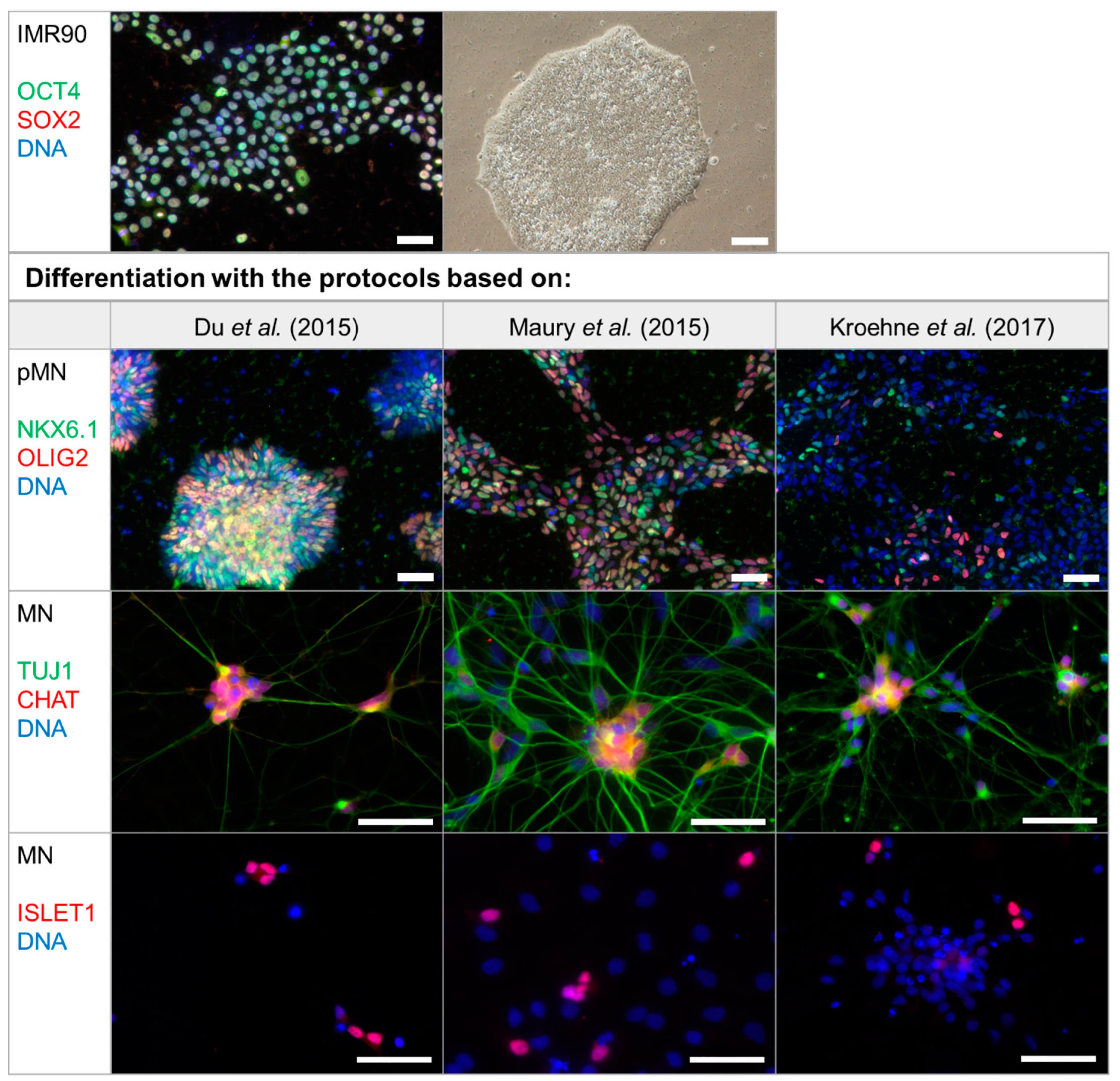
2.1. Differentiation Protocol by Du el al. for Differentiation of iPSCs to MNs Has the Highest MN Yield
2.2. MNs Differentiated In Vitro Exhibit the Full Panel of BoNT Serotype-Specific Substrates and Receptors
2.3. Potency Estimation of BoNT/A with MNs Differentiated In Vitro and SiMa Cells
3. Discussion
3.1. Differentiation Protocol Based on Du el al. for Differentiation of iPSCs to MNs Has the Highest MN Yield
3.2. Sensitivity for BoNTs
4. Materials and Methods
4.1. Cell Culture
4.2. Differentiation by Du et al.
4.3. Differentiation by Maury et al.
4.4. Differentiation by Kroehne et al.
4.5. Cultivation and Differentiation of SiMa Cells
4.6. Quantitative RT-qPCR
4.7. Immunocytochemistry
4.8. Analysis of SNAP25 Cleavage
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Supplement | Supplier | Stock Solution |
|---|---|---|
| Y-27632 (Y) | Bertin Pharma #T1725 | 10 mM in H2O |
| bFGF | Gibco #13256029 | 5 µg/mL in 10 mM Tris, pH 7.6 with 0.1% BSA |
| Dorsomorphin (DM) | abcam #ab120843 | 10 mM in DMSO |
| CHIR99021 (CHIR) | Axon MedChem #1386 | 6 mM in DMSO |
| Valproic acid (VPA) | Sigma #P6273 | 10 mM in DMEM/F12 |
| SB431542 (SB) | Stemcell #72232 | 40 mM in DMSO |
| L-Ascorbic acid (AA) | Sigma #A4544 | 150 mM in H2O |
| TGFß3 | Sigma #SRP3171 | 10 µg/mL in 5 mM citric acid with 0.1% BSA |
| DMH1 | Bertin Pharma, #16679 | 10 mM in DMF |
| Purmorphamine (PMA) | Stemcell #72202 | 10 mM in DMSO |
| Compound E (CE) | Bertin Pharma, #15579 | 10 mM in DMSO |
| GDNF | Peprotech, #450-10 | 10 µg/mL in 0.1% BSA |
| BDNF | Peprotech, #450-02 | 10 µg/mL in 0.1% BSA |
| CNTF | Peprotech, #450-13 | 10 µg/mL in 0.1% BSA |
| SAG | TargetMol #T1779 | 2.5 mM in DMSO |
| DAPT | Cayman Chemicals #Cay13197-5 | 10 mM in DMSO |
| dbcAMP (AMP) | Sigma #D0627 | 100 mM in H2O |
| Retinoic acid (RA) | Sigma #R2625 | 10 mM in DMSO, protect from light |
| Gene | Forward Primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| PPIA NM_001300981.2 | GCCAAGACTGAGTGGTTGGAT | GGCCTCCACAATATTCATGCC |
| RPS23 NM_001025.5 | ACAGGATGGGCAAGTGTCGT | CACTTCTGGTCTCGTCGGTG |
| SNAP25 NM_001322902.2 | AGCCTGGGGCAATAATCAGG | GGCATCATTTGTTACCCTGCG |
| STX1A NM_001165903.2 | CAACCCCGATGAGAAGACGA | GGCGTTGTACTCCGACATGA |
| STX1B NM_052874.5 | GAAGGACCACCACCAACGAA | ATCTCTCCCTGGCTCTCTACG |
| VAMP1 NM_001297438.2 | CAGTTCCGTCCACTTCAGCC | CTGGAGCAGACATTTTTCTGACA |
| VAMP2 NM_001330125.1 | CCAAACCTCACCAGTAACAGGA | CTCATGATGTCCACCACCTCA |
| SV2A NM_001278719.1 | CCTCAGACAAGAGGACCACAG | GCCCTAGAGACCCCTTCACT |
| SV2B NM_001167580.3 | CCACCAACATGGGAAACTTGTG | GTGCTCGTAGAGGTCTGTGTT |
| SV2C NM_001297716.2 | TCGGGATTGGAGGAGCCATA | ATGCTGAAGCTCCACCCGTA |
| SYT1 NM_001135805.2 | GGATGTGGGTGGCTTATCCG | CCACCTGCACTTTCTGGATTTG |
| SYT2 NM_001136504.1 | CTTCAAGGTGCCATACCAGGA | CTCCACTCCTCAATGGGCTG |
| ST3GAL2 NM_006927.3 | TGAGAGTGCCAAGAACCTGC | CTGGGGCGTAGGTGAATCG |
| ST3GAL3 NM_001270459.1 | ATCTTCCCCCGGTTCTCCAA | CGAACTCCCGGATTCTAGCC |
| Antibody | Host and Type | Source | Dilution |
|---|---|---|---|
| OCT4 | Mouse monoclonal | Santa Cruz #sc-5279 | 1:50 |
| SOX2 | Rabbit monoclonal | Cell Signaling #3579 | 1:200 |
| OLIG2 | Rabbit polyclonal | Merck/Millipore #AB9610 | 1:250 |
| NKX6.1 | Mouse monoclonal | DSHB #F55A10 | 5 µg/mL |
| TUJ1 | Rabbit polyclonal | Sigma #T2200 | 1:100 |
| TUJ1 | Mouse monoclonal | Covance #MMS-435P | 1:750 |
| ISLET1 | Rabbit polyclonal | Abcam #ab20670 | 1:125 |
| CHAT | Goat polyclonal | Millipore #AB144P | 1:50 |
| SNAP25 | Rabbit polyclonal | Abcam #41455 | 1:100 |
| VAMP2 | Rabbit monoclonal | Cell signaling #D601A | 1:250 |
| SV2 (SV2A/B/C) | Mouse monoclonal | DSHB #SV2 | 10 µg/mL |
| GT1b | Mouse monoclonal | DSHB #GT1b-1 | 10 µg/µL |
| GD1a | Mouse monoclonal | DSHB #GD1a-1 | 10 µg/mL |
| β-Actin (HRP-coupled) | Mouse monoclonal | Santa Cruz #sc-47778 | 1:4000 |
| Antibody | Host and Type | Source | Dilution |
|---|---|---|---|
| Anti-Goat (Alexa Fluor 568) | Rabbit polyclonal | Invitrogen #A11079 | 1:1000 |
| Anti-Mouse (Alexa Fluor 488) | Goat polyclonal | Invitrogen #A11001 | 1:1500 |
| Anti-Rabbit (Alexa Fluor 568) | Goat polyclonal | Invitrogen #A11008 | 1:1000 |
| Anti-Rabbit (HRP) | Goat polyclonal | Sigma #A0545 | 1:20,000 |
References
- von Berg, L.; Stern, D.; Pauly, D.; Mahrhold, S.; Weisemann, J.; Jentsch, L.; Hansbauer, E.-M.; Müller, C.; Avondet, M.A.; Rummel, A.; et al. Functional detection of botulinum neurotoxin serotypes A to F by monoclonal neoepitope-specific antibodies and suspension array technology. Sci. Rep. 2019, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Bercsenyi, K.; Giribaldi, F.; Schiavo, G. The elusive compass of clostridial neurotoxins: Deciding when and where to go? Curr. Top. Microbiol. Immunol. 2013, 364, 91–113. [Google Scholar] [PubMed]
- Simpson, L. Identification of the characteristics that underlie botulinum toxin potency: Implications for designing novel drugs. Biochimie 2000, 82, 943–953. [Google Scholar] [CrossRef]
- Capek, P.; Dickerson, T.J. Sensing the deadliest toxin: Technologies for botulinum neurotoxin detection. Toxins 2010, 2, 24–53. [Google Scholar] [CrossRef]
- Binz, T.; Rummel, A. Cell entry strategy of clostridial neurotoxins. J. Neurochem. 2009, 109, 1584–1595. [Google Scholar] [CrossRef]
- Tehran, D.A.; Pirazzini, M. Novel Botulinum Neurotoxins: Exploring Underneath the Iceberg Tip. Toxins 2018, 10, 190. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef]
- Montecucco, C. How do tetanus and botulinum toxins bind to neuronal membranes? Trends Biochem. Sci. 1986, 11, 314–317. [Google Scholar] [CrossRef]
- Connan, C.; Popoff, M.R. Uptake of Clostridial Neurotoxins into Cells and Dissemination. Curr. Top. Microbiol. Immunol. 2017, 406, 39–78. [Google Scholar]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.I.; Martinez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 14130. [Google Scholar] [CrossRef]
- Davies, J.R.; Liu, S.M.; Acharya, K.R. Variations in the Botulinum Neurotoxin Binding Domain and the Potential for Novel Therapeutics. Toxins 2018, 10, 421. [Google Scholar] [CrossRef]
- Rummel, A. Two Feet on the Membrane: Uptake of Clostridial Neurotoxins. Curr. Top. Microbiol. Immunol. 2017, 406, 1–37. [Google Scholar]
- Strotmeier, J.; Willjes, G.; Binz, T.; Rummel, A. Human synaptotagmin-II is not a high affinity receptor for botulinum neurotoxin B and G: Increased therapeutic dosage and immunogenicity. FEBS Lett. 2012, 586, 310–313. [Google Scholar] [CrossRef]
- Dressler, D.; Benecke, R. Pharmacology of therapeutic botulinum toxin preparations. Disabil. Rehabil. 2007, 29, 1761–1768. [Google Scholar] [CrossRef]
- Bengtson, I.A. Direct Inoculation Test for B. Botulinus Toxin: Determination of the Presence of B. Botulinus Toxin by Intraperitoneal Inoculation of Laboratory Animals with Suspected Foods. Public Health Rep. (1896–1970) 1921, 36, 1665–1671. [Google Scholar] [CrossRef]
- Sesardic, D.; Leung, T.; Gaines Das, R. Role for standards in assays of botulinum toxins: International collaborative study of three preparations of botulinum type A toxin. Biologicals 2003, 31, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Johnson, E.A. Critical Analysis of Neuronal Cell and the Mouse Bioassay for Detection of Botulinum Neurotoxins. Toxins 2019, 11, 713. [Google Scholar] [CrossRef] [PubMed]
- Bitz, S. The botulinum neurotoxin LD50 test - problems and solutions. Altex 2010, 27, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique; Methuen & Co.” Ltd.: London, UK, 1959. [Google Scholar]
- Taylor, K.; Gericke, C.; Alvarez, L.R. Botulinum toxin testing on animals is still a Europe-wide issue. ALTEX 2019, 36, 81–90. [Google Scholar] [CrossRef]
- Adler, S.; Bicker, G.; Bigalke, H.; Bishop, C.; Blumel, J.; Dressler, D.; Fitzgerald, J.; Gessler, F.; Heuschen, H.; Kegel, B.; et al. The current scientific and legal status of alternative methods to the LD50 test for botulinum neurotoxin potency testing. The report and recommendations of a ZEBET Expert Meeting. Altern. Lab. Anim. 2010, 38, 315–330. [Google Scholar] [CrossRef]
- Hobbs, R.J.; Thomas, C.A.; Halliwell, J.; Gwenin, C.D. Rapid Detection of Botulinum Neurotoxins-A Review. Toxins 2019, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Kiris, E.; Kota, K.P.; Burnett, J.C.; Soloveva, V.; Kane, C.D.; Bavari, S. Recent developments in cell-based assays and stem cell technologies for botulinum neurotoxin research and drug discovery. Expert Rev. Mol. Diagn. 2014, 14, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Johnson, E.A. Botulinum neurotoxins A, B, C, E, and F preferentially enter cultured human motor neurons compared to other cultured human neuronal populations. FEBS Lett. 2019, 593, 2675–2685. [Google Scholar] [CrossRef] [PubMed]
- Fonfria, E.; Maignel, J.; Lezmi, S.; Martin, V.; Splevins, A.; Shubber, S.; Kalinichev, M.; Foster, K.; Picaut, P.; Krupp, J. The Expanding Therapeutic Utility of Botulinum Neurotoxins. Toxins 2018, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Genz, L.; Püschel, G.P. Botulinum neurotoxin dose-dependently inhibits release of neurosecretory vesicle-targeted luciferase from neuronal cells. ALTEX 2015, 32, 297–306. [Google Scholar] [PubMed]
- Rasetti-Escargueil, C.; Machado, C.B.; Preneta-Blanc, R.; Fleck, R.A.; Sesardic, D. Enhanced sensitivity to Botulinum type A neurotoxin of human neuroblastoma SH-SY5Y cells after differentiation into mature neuronal cells. Botulinum J. 2011, 2, 30–48. [Google Scholar] [CrossRef]
- Tegenge, M.A.; Böhnel, H.; Gessler, F.; Bicker, G. Neurotransmitter Vesicle Release from Human Model Neurons (NT2) is Sensitive to Botulinum Toxin A. Cell. Mol. Neurobiol. 2012, 32, 1021–1029. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O. Challenges in searching for therapeutics against Botulinum Neurotoxins. Expert Opin. Drug Discov. 2017, 12, 497–510. [Google Scholar] [CrossRef]
- Pellett, S. Progress in cell based assays for botulinum neurotoxin detection. Curr. Top. Microbiol. Immunol. 2013, 364, 257–285. [Google Scholar]
- Kiris, E.; Nuss, J.E.; Burnett, J.C.; Kota, K.P.; Koh, D.C.; Wanner, L.M.; Torres-Melendez, E.; Gussio, R.; Tessarollo, L.; Bavari, S. Embryonic stem cell-derived motoneurons provide a highly sensitive cell culture model for botulinum neurotoxin studies, with implications for high-throughput drug discovery. Stem Cell Res. 2011, 6, 195–205. [Google Scholar] [CrossRef]
- Rossetto, O. The binding of botulinum neurotoxins to different peripheral neurons. Toxicon 2018, 147, 27–31. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Li, X.J.; Hu, B.Y.; Jones, S.A.; Zhang, Y.S.; Lavaute, T.; Du, Z.W.; Zhang, S.C. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells 2008, 26, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Williams, L.A.; Klim, J.R.; Eggan, K. How to make spinal motor neurons. Development 2014, 141, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Bruijn, L.I.; Chandran, S.; Eggan, K.; Ho, R.; Klim, J.R.; Livesey, M.R.; Lowry, E.; Macklis, J.D.; Rushton, D.; et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 2016, 19, 542–553. [Google Scholar] [CrossRef]
- Du, Z.W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.L.; Zhong, X.; Fan, F.; Zhang, S.C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef]
- Maury, Y.; Come, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Kroehne, V.; Tsata, V.; Marrone, L.; Froeb, C.; Reinhardt, S.; Gompf, A.; Dahl, A.; Sterneckert, J.; Reimer, M.M. Primary Spinal OPC Culture System from Adult Zebrafish to Study Oligodendrocyte Differentiation In Vitro. Front. Cell. Neurosci. 2017, 11, 284. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Verderio, C.; Rossetto, O.; Grumelli, C.; Frassoni, C.; Montecucco, C.; Matteoli, M. Entering neurons: Botulinum toxins and synaptic vesicle recycling. EMBO Rep. 2006, 7, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Li, D.; Louis, K.R.; Li, X.; Yang, H.; Sun, Q.; Crandall, S.R.; Tsang, S.; Zhou, J.; Cox, C.L.; et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 2014, 5, 3449. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Dressler, D.; Saberi, F.A.; Barbosa, E.R. Botulinum toxin: Mechanisms of action. Arq. Neuropsiquiatr. 2005, 63, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Willison, H.J. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction. J. Physiol. 2009, 587, 3979–3999. [Google Scholar] [CrossRef]
- Poulain, B.; Popoff, M.R. Why Are Botulinum Neurotoxin-Producing Bacteria So Diverse and Botulinum Neurotoxins So Toxic? Toxins 2019, 11, 34. [Google Scholar] [CrossRef]
- Pennuto, M.; Bonanomi, D.; Benfenati, F.; Valtorta, F. Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol. Biol. Cell 2003, 14, 4909–4919. [Google Scholar] [CrossRef]
- Pang, Z.P.; Melicoff, E.; Padgett, D.; Liu, Y.; Teich, A.F.; Dickey, B.F.; Lin, W.; Adachi, R.; Sudhof, T.C. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J. Neurosci. 2006, 26, 13493–13504. [Google Scholar] [CrossRef]
- Jenkinson, S.P.; Grandgirard, D.; Heidemann, M.; Tscherter, A.; Avondet, M.A.; Leib, S.L. Embryonic Stem Cell-Derived Neurons Grown on Multi-Electrode Arrays as a Novel In vitro Bioassay for the Detection of Clostridium botulinum Neurotoxins. Front. Pharmacol. 2017, 8, 73. [Google Scholar] [CrossRef]
- Lau, C.G.; Takayasu, Y.; Rodenas-Ruano, A.; Paternain, A.V.; Lerma, J.; Bennett, M.V.; Zukin, R.S. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J. Neurosci. 2010, 30, 242–254. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A. Do we have brain to spare? Neurology 2005, 64, 2004–2005. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Hammond, L.A.; Kanjhan, R.; Bellingham, M.C.; Noakes, P.G. A method for the three-dimensional reconstruction of Neurobiotin-filled neurons and the location of their synaptic inputs. Front. Neural Circuits 2013, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Haas, G.; Langoth-Fehringer, N.; Püschel, G.P. Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins. Toxins 2018, 10, 360. [Google Scholar] [CrossRef] [PubMed]
- Bak, N.; Rajagopal, S.; Stickings, P.; Sesardic, D. SiMa Cells for a Serotype Specific and Sensitive Cell-Based Neutralization Test for Botulinum Toxin A and E. Toxins 2017, 9, 230. [Google Scholar]
- Fernandez-Salas, E.; Wang, J.; Molina, Y.; Nelson, J.B.; Jacky, B.P.; Aoki, K.R. Botulinum neurotoxin serotype A specific cell-based potency assay to replace the mouse bioassay. PLoS ONE 2012, 7, e49516. [Google Scholar] [CrossRef]
- Cai, B.B.; Francis, J.; Brin, M.F.; Broide, R.S. Botulinum neurotoxin type A-cleaved SNAP25 is confined to primary motor neurons and localized on the plasma membrane following intramuscular toxin injection. Neuroscience 2017, 352, 155–169. [Google Scholar] [CrossRef]
- Jurasinski, C.V.; Lieth, E.; Dang Do, A.N.; Schengrund, C.L. Correlation of cleavage of SNAP-25 with muscle function in a rat model of Botulinum neurotoxin type A induced paralysis. Toxicon 2001, 39, 1309–1315. [Google Scholar] [CrossRef]
- Keller, J.E.; Neale, E.A. The role of the synaptic protein snap-25 in the potency of botulinum neurotoxin type A. J. Biol. Chem. 2001, 276, 13476–13482. [Google Scholar] [CrossRef]
- Marini, P.; MacLeod, R.A.; Treuner, C.; Bruchelt, G.; Bohm, W.; Wolburg, H.; Schweizer, P.; Girgert, R. SiMa, a new neuroblastoma cell line combining poor prognostic cytogenetic markers with high adrenergic differentiation. Cancer Genet. Cytogenet. 1999, 112, 161–164. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]






| BoNT | Ganglioside Receptor [12] | Protein Receptor [12] | Substrate [6] | Cleavage Site [6] |
|---|---|---|---|---|
| A | GT1b > GD1a = GD1b > GM1 | SV2C>SV2A>SV2B | SNAP25 | Q197–198R |
| B | GT1b > GD1a > GD1b | SYT1 >SYT2 * | VAMP1 VAMP2 | Q78–79F Q76–77F |
| C | GD1b > GT1b > GD1a > GM1a | Not determined | SNAP25 STX1A STX1B | R198–199A K253–254A K252–253A |
| DC | GM1a > GD1a > GD1b = GT1b> | SYT2 >SYT1 | VAMP1 VAMP2 | K61–62L K59–60L |
| D | GD2 > GT1b = GD1b | SV2B>SV2C>SV2A | VAMP1 VAMP2 | K61–62L K59–60L |
| E | GD1a/GQ1b/GT1b >> GM1 | SV2A>SV2B | SNAP25 | R180–181I |
| F | GT1b = GD1a >> GM3 >> GD1b/GM1 | SV2A>SV2C>SV2B | VAMP1 VAMP2 | Q60–61K Q58–59K |
| G | GT1b = GD1a > GD1b > GM3 > GM1 | SYT1, SYT2 | VAMP1 VAMP2 | A83–84A A81–82A |
| H/FA | Not determined | SV2 | VAMP1 VAMP2 | L56–57E L54–55E |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schenke, M.; Schjeide, B.-M.; Püschel, G.P.; Seeger, B. Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays. Toxins 2020, 12, 276. https://doi.org/10.3390/toxins12050276
Schenke M, Schjeide B-M, Püschel GP, Seeger B. Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays. Toxins. 2020; 12(5):276. https://doi.org/10.3390/toxins12050276
Chicago/Turabian StyleSchenke, Maren, Brit-Maren Schjeide, Gerhard P. Püschel, and Bettina Seeger. 2020. "Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays" Toxins 12, no. 5: 276. https://doi.org/10.3390/toxins12050276
APA StyleSchenke, M., Schjeide, B.-M., Püschel, G. P., & Seeger, B. (2020). Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays. Toxins, 12(5), 276. https://doi.org/10.3390/toxins12050276