Isolation of an Anti–tumour Disintegrin: Dabmaurin–1, a Peptide Lebein–1–like, from Daboia mauritanica Venom

 , ,
, , 
Abstract
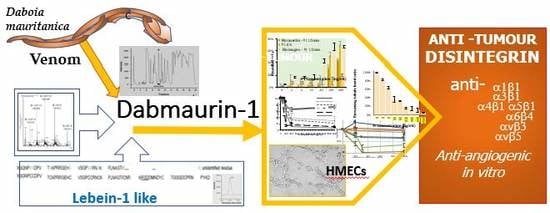
1. Introduction
2. Results
2.1. Active Peptide Isolation Managed by Anti–proliferative Activity on HMECs
2.2. Identification of a P1 Moiety
2.3. Effect on HMEC Cell Viability and Concentration Dependent Proliferation
2.4. Effect on Angiogenesis
2.4.1. HMEC Adhesion Assays
2.4.2. HMEC Migration Assays in Boyden Chamber
2.4.3. Video Microscopy of Locomotion of HMECs
2.4.4. Tubulogenesis Assays
2.5. Identification Assays of Cell Targets of the Venom Peptide P1
2.5.1. Tumour Cell Adhesion Assays in Presence of the Venom Peptide
2.5.2. Adhesion Assays to Anti–integrins in Presence of the Venom Peptide
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Separation and Analysis of the Venom Fractions
5.2.1. Venom Source
5.2.2. Separation
5.2.3. Analysis by Mass–Spectrometry
5.2.4. N–terminal Edman Sequencing
5.2.5. Quantification
5.3. Cell Proliferation Assays and Viability Measures with HMEC
5.3.1. Cell Proliferation
5.3.2. Measure of Viable Cells
5.3.3. Cytotoxicity Assays
5.4. Tubulogenesis Assays
5.5. Cell Migration Assays
5.5.1. Cell Migration in Modified Boyden Chamber
5.5.2. Videomicroscopy
5.6. Cell Adhesion Assays
5.6.1. Cell Adhesion Assay to ECM
5.6.2. Cell Adhesion Assay to Anti–integrin Antibodies
5.6.3. Cell Adhesion Assay to Peptide P1
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oukkache, N.; Eljaoudi, R.; Larreche, S.; Chakir, S.; Fouad, C.; Hmyene, A.; Mion, G. Snake bites in morocco: Progress and challenges. Adv. Toxicol. Toxic Effects 2019, 3, 9–14. [Google Scholar] [CrossRef]
- Chakir, S.; Daoudi, K.; Darkaoui, B.; Lafnoune, A.; Hmyene, A.; Oukkache, N. Research article screening of active biomolecules from the venom of the moroccan viper Daboia mauritanica. EC Pharmacol. Toxicol. 2019, 7, 144–149. [Google Scholar]
- Chaisakul, J.; Hodgson, W.C.; Kuruppu, S.; Prasongsook, N. Effects of Animal Venoms and Toxins on Hallmarks of Cancer. J. Cancer 2016, 7, 1571–1578. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Ingbert, D.E. Integrating with Integrins. Mol. Biol. Cell 1994, 5, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Koretz, K.; Schlag, P.; Boumsell, L.; Moller, P. Expression of VLA–α2, VLA–α6 and VLA–β1 chains in normal mucosa and adenomas of the colon and in colon carcinomas and their liver metastases. Am. J. Pathol. 1991, 138, 741–750. [Google Scholar] [PubMed]
- Kawano, K.; Kantak, S.S.; Murai, M.; Yao, C.C.; Kramer, R.H. Integrin α3β1 engagement disrupts intercellular adhesion. Exp. Cell Res. 2001, 262, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Rust, W.L.; Carper, S.W.; Plopper, G.E. The promise of integrins as effective targets for anticancer agents. J. Biomed. Biotechnol. 2002, 2, 124–130. [Google Scholar] [CrossRef]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Gomes, A.; Bhattacharjee, P.; Mishra, R.; Biswas, A.K.; Dasgupta, S.C.; Giri, B. Anticancer potential of animal venoms and toxins. Indian J. Exp. Biol. 2010, 48, 93–103. [Google Scholar]
- Rupp, P.A.; Little, C.D. Integrins in vascular development. Circ. Res. 2001, 89, 566–572. [Google Scholar] [CrossRef]
- Otrock, Z.K.; Mahfouz, R.A.R.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Null. Mol. Dis. 2007, 39, 2120–2220. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.M. In vitro assays of angiogenesis for assessment of angiogenic and anti–angiogenic agents. Microvasc. Res. 2007, 74, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Sepp, N.T.; Li, L.J.; Lee, K.H.; Brown, E.J.; Caughman, S.W.; Lawley, T.J.; Swerlick, R.A. Basic fibroblast growth factor increases expression of the αvβ3 integrin complex in human microvascular endothelial cells. J. Investig. Dermatol. 1994, 103, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Walton, H.L.; Corjay, M.H.; Mohamed, S.N.; Mousa, S.A.; Santomenna, L.D.; Reilly, T.M. Hypoxia induces differential expression of the integrin receptors αvβ3 and αvβ5 in cultured human endothelial cells. J. Cell Biochem. 2000, 78, 674–680. [Google Scholar] [CrossRef]
- Silletti, S.; Kessler, T.; Goldberg, J.; Boger, D.L.; Cheresh, D.A. Disruption of matrix metalloproteinase 2 binding to integrin alpha(v)beta(3) by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; D’Amico, G.; Hodivala–Dilke, K.M.; Reynolds, L.E. Integrins: The keys to unlocking angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1703–1713. [Google Scholar] [CrossRef]
- Senger, D.R.; Claffey, K.P.; Benes, J.E.; Perruzzi, C.A.; Sergiou, A.P.; Detmar, M. Angiogenesis promoted by vascular endothelial growth factor: Regulation through α1β1 and α2β1 integrins. Proc. Natl. Acad. Sci. USA 1997, 94, 13612–13617. [Google Scholar] [CrossRef]
- Hong, Y.K.; Lange–Asschenfeldt, B.; Velasco, P.; Hirakawa, S.; Kunstfeld, R.; Brown, L.F.; Bohlen, P.; Senger, D.R.; Detmar, M. VEGF–A promotes tissue repair–associated lymphatic vessel formation via VEGFR–2 and the α1β1 and α2β1 integrins. FASEB J. 2004, 18, 1111–1113. [Google Scholar] [CrossRef]
- Swenson, S.; Ramu, S.; Markland, F.S. Anti–angiogenesis and RGD–containing snake venom disintegrins. Curr. Pharm. Des. 2007, 13, 2860–2871. [Google Scholar] [CrossRef]
- For reviews on the subject: Chatterjee, B. Animal Venoms have Potential to Treat Cancer. Curr. Top. Med. Chem. 2018, 18, 2555–2566. [Google Scholar] [CrossRef]
- Ma, R.; Mahadevappa, R.; Kwok, H.F. Venom–based peptide therapy: Insights into anti–cancer mechanism. Oncotarget 2017, 8, 100908–100930. [Google Scholar] [CrossRef] [PubMed]
- Arruda Macedo, J.K.; Fox, J.W.; de Souza Castro, M. Disintegrins from Snake Venoms and their Applications in Cancer Research and Therapy. Curr. Protein Peptide Sci. 2015, 16, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Rádis–Baptista, G. Integrins, cancer and snake toxins (mini–review). J. Venom. Anim. Toxins Incl. Trop. Dis. 2005, 11, 3, 217–241. [Google Scholar] [CrossRef]
- Kerkkamp, H.; Bagowski, C.; Kool, J.; van Soolingen, B.; Vonk, F.J.; Vlecken, D. Whole snake venoms: Cytotoxic, anti–metastatic and anti–angiogenic properties. Toxicon 2018, 150, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Golubkov, V.; Hawes, D.; Markland, F.S. Anti–angiogenic activity of contortrostatin, a disintegrin from Agkistrodon contortrix contortrix snake venom. Angiogenesis 2003, 6, 213–224. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI–BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Gasmi, A.; Srairi, N.; Guermazi, S.; Dkhil, H.; Karoui, H.; El Ayeb, M. Amino acid structure and characterization of a heterodimeric disintegrin from Vipera lebetina venom. Biochim. Biophys. Acta 2001, 1547, 51–56. [Google Scholar] [CrossRef]
- Calvete, J.J. The continuing saga of snake venom disintegrins. Toxicon 2013, 62, 40–49. [Google Scholar] [CrossRef]
- BLAST. Available online: http://web.expasy.org/cgi–bin/protparam (accessed on 9 May 2017).
- Eble, J.E.; Bruckner, P.; Mayer, U. Vipera lebetina venom contains two disintegrins inhibiting laminin–binding β1 integrins. J. Biol. Chem. 2003, 278, 26488–26496. [Google Scholar] [CrossRef]
- Hadmouda, M.; Montenegro, M.; Sánchez–del–Campo, L.; Zakraoui, O.; Aloui, Z.; Riahi–Chebbi, I.; Karoui, H.; Rodríguez–López, J.; Essafi-Benkhadir, K. Lebein, a Snake Venom Disintegrin, Induces Apoptosis in Human Melanoma Cells. Toxins 2016, 8, 206–220. [Google Scholar] [CrossRef]
- Zakraoui, O.; Marcinkiewicz, C.; Aloui, Z.; Othman, H.; Grepin, R.; Haoues, M.; Essafi, M.; Srairi–Abid, N.; Gasmi, A.; Karoui, H.; et al. Lebein, a snake venom disintegrin, suppresses human colon cancer cells proliferation and tumor–induced angiogenesis through cell cycle arrest, apoptosis induction and inhibition of VEGF expression. Mol. Carcinog. 2017, 56, 18–35. [Google Scholar] [CrossRef]
- Herrera, Y.; Heras, N.; Cardoso, D. Adaptación a microplacas y validación de la técnica de Lowry. VacciMonitor 1999, 3, 7–11. [Google Scholar]
- Bradford, M.A. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sapan, C.V.; Lundblad, R.L.; Price, N.C. Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 1999, 2, 99–108. [Google Scholar] [CrossRef]
- Estimation using the internet tool: ic50.tk. Available online: http://www.ic50.tk (accessed on 7 March 2018).
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Haubner, R.; Weber, W.A.; Beer, A.J.; Vabuliene, E.; Reim, D.; Sarbia, M.; Becker, K.F.; Goebel, M.; Hein, R.; Wester, H.J.; et al. Noninvasive visualization of the activated alphavbeta3 integrin in cancer patients by positron emission tomography and [18F] Galacto–RGD. PLoS Med. 2005, 2, e70. [Google Scholar] [CrossRef]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial Cell Migration During Angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand–binding specificities of laminin–binding integrins: A comprehensive survey of laminin–integrin interactions using recombinant α3β1, α6β1, α7β1 and α6β4 integrins. Matrix. Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef]
- Eble, J.A.; Wucherpfennig, K.W.; Gauthier, L.; Dersch, P.; Krukonis, E.; Isberg, R.R.; Hemler, M.E. Recombinant soluble human α3b1 integrin: Purification, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry 1998, 37, 10945–10955. [Google Scholar] [CrossRef]
- Colognato, H.; Yurchenco, P.D. Form and function: The laminin family of heterotrimers. Dev. Dyn. 2000, 218, 213–234. [Google Scholar] [CrossRef]
- Smith, J.W. The structural basis of integrin–ligand (RGD) interaction. In Integrins: Molecular and Biological Responses to the Extracellular Matrix; Cheresh, D.A., Mecham, R.P., Eds.; Academic Press: San Diego, CA, USA, 1994; pp. 1–32. [Google Scholar]
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos–Sergiou, A.; Peruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the αϖβ integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305. [Google Scholar]
- Cheresh, D.A.; Spiro, R.C. Biosynthetic and functional properties of an Arg–Gly–Asp–directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J. Biol. Chem. 1987, 262, 17703–17711. [Google Scholar]
- Luis, J. (Aix—Marseille University, Marseille, France). Personal communication. 2017. [Google Scholar]
- Zhang, Z.; Tarone, G.; Turner, D.C. Expression of integrin α1β1 is regulated by nerve growth factor and dexamethasone in PC12 cells. J. Biol. Chem. 1993, 268, 5557–5565. [Google Scholar]
- Hemler, M.E.; Huang, C.; Takada, Y.; Schwarz, L.; Strominger, J.L.; Clabby, M.L. Characterization of the cell surface heterodimer VLA–4 and related peptides. J. Biol. Chem. 1987, 262, 11478–11485. [Google Scholar]
- Delamarre, E.; Taboubi, S.; Mathieu, S.; Bérenguer, C.; Rigot, V.; Lissitzky, J.-C.; Branger, D.F.; Ouafik, L.H.; Luis, J. Enhances Glioblastoma Progression. Am. J. Pathol. 2009, 175, 844–864. [Google Scholar] [CrossRef]
- Li, R.; Luo, M.; Ren, M.; Chen, N.; Xia, J.; Deng, X.; Zeng, M.; Yan, K.; Luo, T.; Wu, J. Vitronectin regulation of vascular endothelial growth factor–mediated angiogenesis. J. Vasc. Res. 2014, 51, 110–117. [Google Scholar] [CrossRef]
- Towler, M.C.; Kaufman, S.J.; Brodsky, F.M. Membrane traffic in skeletal muscle. Traffic 2004, 5, 129–139. [Google Scholar] [CrossRef]
- Clark, R.A.F. Wound repair. Overview and general considerations. In The Molecular and Cellular Biology of Wound Repair; Clark, R.A.F., Ed.; Plenum Press: New York, NY, USA, 1996; pp. 3–50. [Google Scholar] [CrossRef]
- Cruet–Hennequart, S.; Maubant, S.; Luis, J.; Gauduchon, P.; Staedel, C.; Dedhar, S. α v integrins regulate cell proliferation through integrin–linked kinase (ILK) in ovarian cancer cells. Oncogene 2003, 22, 1688–1702. [Google Scholar] [CrossRef]
- Calvete, J.J.; Marcinkiewicz, C.; Monle´on, D.; Esteve, V.; Celda, B.; Ju´arez, P.; Sanz, L. Snake venom disintegrins: Evolution of structure and function. Toxicon 2005, 45, 1063–1074. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef]
- Springer, T.A.; Wang, J.H. The three–dimensional structure of integrins and their ligands, and conformational regulation of cell adhesion. Adv. Protein Chem. 2004, 68, 29–63. [Google Scholar] [CrossRef]
- Mould, P.A.; Akiyama, S.K.; Humphries, M.J. Regulation of integrine α5β1 – fibronectine interactions by divalent cations. J. Biol. Chem. 1995, 270, 26270–26277. [Google Scholar] [CrossRef]
- Walsh, E.M.; Marcinkiewicz, C. Non–RGD–containing snake venom disintegrins, functional and structural relations. Toxicon 2011, 58, 355–362. [Google Scholar] [CrossRef]
- Rosenow, F.; Ossig, R.; Thormeyer, D.; Gasmann, P.; Schlüter, K.; Brunner, G.; Haier, J.; Eble, J.A. Integrin as antimetastatic targets of RGD–independent snake venom components in liver metastasis. Neoplasia 2008, 10, 168–176. [Google Scholar] [CrossRef]
- Gassmann, P.; Kang, M.-L.; Mees, S.T.; Haier, J. In vivo tumor cell adhesion in the pulmonary microvasculature is exclusively mediated by tumor cell – endothelial cell interaction. BMC Cancer 2010, 10, 177–188. [Google Scholar] [CrossRef]
- Yang, G.Y.; Xu, K.S.; Pan, Z.Q.; Zhang, Z.Y.; Mi, Y.T.; Wang, J.S.; Chen, R.; Niu, J. Integrin alpha v beta 6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver. Cancer Sci. 2008, 99, 879–887. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Niu, W.; Liu, E.; Wang, J.; Peng, C.; Lin, P.; Wang, B.; Khan, A.Q.; Gao, H.; et al. The beta6–integrin–ERK/MAP kinase pathway contributes to chemo resistance in colon cancer. Cancer Lett. 2013, 328, 325–334. [Google Scholar] [CrossRef]
- Zhengchuan, N.; Jiayong, W.; Jun, N. Protein expression of eIF4E and integrin αvβ6 in colon cancer can predict clinical significance, reveal their correlation and imply possible mechanism of interaction. Cell Biosci. 2014, 4, 23–31. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Xu, K.S.; Wang, J.S.; Yang, G.Y.; Wang, W.; Wang, J.Y.; Niu, W.B.; Liu, E.Y.; Mi, Y.T.; Niu, J. Integrin alpha v beta 6 acts as a prognostic indicator in gastric carcinoma. Clin. Ongol. 2008, 20, 61–66. [Google Scholar] [CrossRef]
- Akalu, A.; Cretu, A.; Brooks, P.C. Targeting integrins for the control of tumour angiogenesis. Expert Opin. Investig. Drugs 2005, 14, 1475–1486. [Google Scholar] [CrossRef]
- Alghisi, G.C.; Rüegg, C. Vascular integrins in tumor angiogenesis: Mediators and therapeutic targets. Endothelium 2006, 13, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Yatohgo, T.; Izumi, M.; Kashiwagi, H.; Hayashi, M. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct. Funct. 1988, 13, 281–292. [Google Scholar] [CrossRef]
- Lehmann, M.; Rabenandrasana, C.; Tamura, R.; Lissitzky, J.C.; Quaranta, V.; Pichon, J.; Marvaldi, J. A monoclonal antibody inhibits adhesion to fibronectin and vitronectin of a colon carcinoma cell line and recognizes the integrins alpha v beta3, alpha v beta 5, and alpha v beta 6. Cancer Res. 1994, 54, 2102–2107. [Google Scholar] [PubMed]
- Fantini, J.; Abadie, B.; Tirard, A.; Remy, L.; Ripert, J.P.; El Battari, A.; Marvaldi, J. Spontaneous and induced dome formation by two clonal cell populations derived from a human adenocarcinoma cell line, HT29. J. Cell Sci. 1986, 83, 235–249. [Google Scholar] [PubMed]
- Ades, E.W.; Candal, F.J.; Swerlick, R.A.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC–1: Establishment of an immortalized human microvascular endothelial cell line. J. Investig. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Mekhalfi, M.; Puppo, C.; Avilan, L.; Lebrun, R.; Mansuelle, P.; Maberly, S.C.; Gontero, B. Glyceraldehyde–3–phosphate dehydrogenase is regulated by ferredoxin–NADP reductase in the diatom Asterionella Formosa. New Phytol. 2014, 203, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Sarray, S.; Delamarre, E.; Marvaldi, J.; El Ayeb, M.; Marrakchi, N.; Luis, J. Lebectin and lebecetin; two C–type lectins from snake venom; inhibit alpha5beta1 and alphaV–containing integrins. Matrix Biol. 2007, 26, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Berthet, V.; Rigot, V.; Champion, S.; Secchi, J.; Fouchier, F.; Marvaldi, J.; Luis, J. Role of endoproteolytic processing in the adhesive and signaling functions of αvβ5 integrin. J. Biol. Chem. 2000, 275, 33308–33313. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECM | IC50 of P1 | Inhibition Maxima 2 |
|---|---|---|
| Col IV | 189.2 ± 1.4 ng/mL (~13.5 nM) | 29 ± 6% |
| Ln | 205.4 ± 8.9 ng/mL (~15.2 nM) | 22 ± 1% |
| Fn | 307.1 ± 2.0 ng/mL (~21.9 nM) | 8 ± 1% |
| Fg | 127.6 ± 8.1 ng/mL (~9.6 nM) | 11 ± 2% |
| Vn | 200.8 ± 2.1 ng/mL (~14.4 nM) | 7 ± 1% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalier, F.; Mugnier, L.; Tarbe, M.; Aboudou, S.; Villard, C.; Kovacic, H.; Gigmes, D.; Mansuelle, P.; de Pomyers, H.; Luis, J.; et al. Isolation of an Anti–tumour Disintegrin: Dabmaurin–1, a Peptide Lebein–1–like, from Daboia mauritanica Venom. Toxins 2020, 12, 102. https://doi.org/10.3390/toxins12020102
Chalier F, Mugnier L, Tarbe M, Aboudou S, Villard C, Kovacic H, Gigmes D, Mansuelle P, de Pomyers H, Luis J, et al. Isolation of an Anti–tumour Disintegrin: Dabmaurin–1, a Peptide Lebein–1–like, from Daboia mauritanica Venom. Toxins. 2020; 12(2):102. https://doi.org/10.3390/toxins12020102
Chicago/Turabian StyleChalier, Florence, Laura Mugnier, Marion Tarbe, Soioulata Aboudou, Claude Villard, Hervé Kovacic, Didier Gigmes, Pascal Mansuelle, Harold de Pomyers, José Luis, and et al. 2020. "Isolation of an Anti–tumour Disintegrin: Dabmaurin–1, a Peptide Lebein–1–like, from Daboia mauritanica Venom" Toxins 12, no. 2: 102. https://doi.org/10.3390/toxins12020102
APA StyleChalier, F., Mugnier, L., Tarbe, M., Aboudou, S., Villard, C., Kovacic, H., Gigmes, D., Mansuelle, P., de Pomyers, H., Luis, J., & Mabrouk, K. (2020). Isolation of an Anti–tumour Disintegrin: Dabmaurin–1, a Peptide Lebein–1–like, from Daboia mauritanica Venom. Toxins, 12(2), 102. https://doi.org/10.3390/toxins12020102