Cellular Effects of T-2 Toxin on Primary Hepatic Cell Culture Models of Chickens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cellular Metabolic Activity
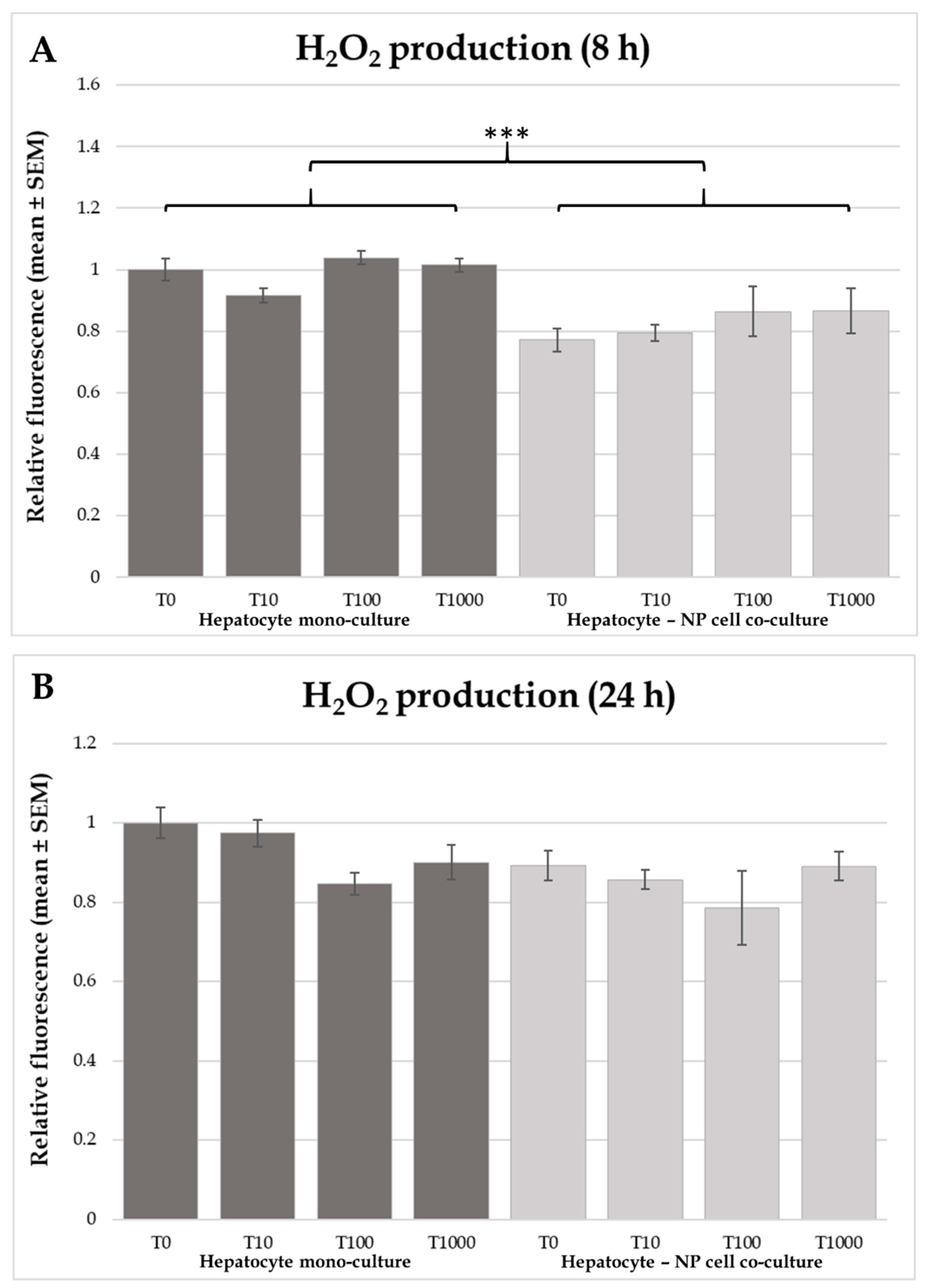
2.2. H2O2 Production
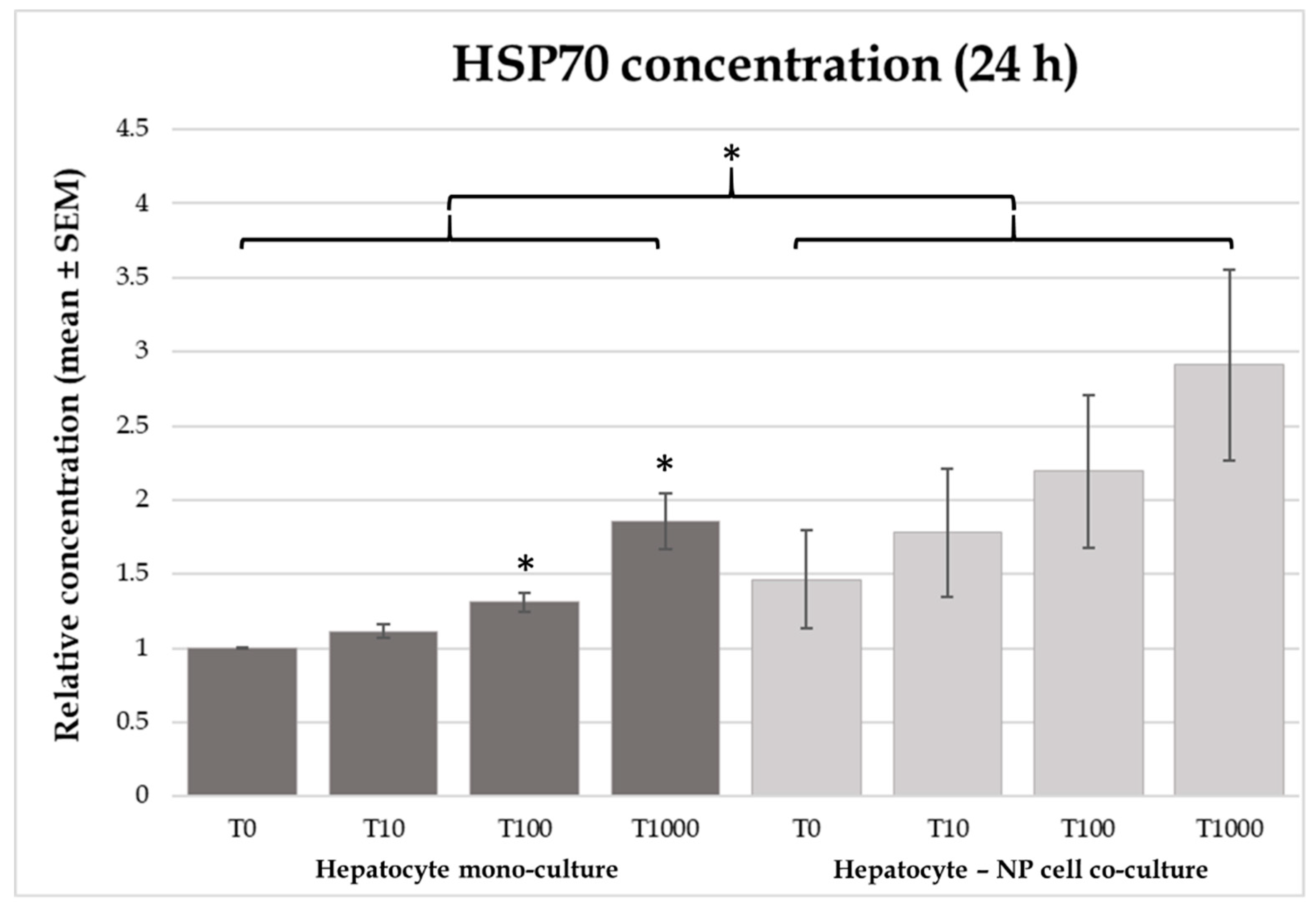
2.3. Heat Shock Protein 70 (HSP70) Concentrations
2.4. Interleukin-6 and -8 Concentrations
3. Discussion
4. Materials and Methods
4.1. Isolation and Culturing of Hepatocytes and Nonparenchymal Cells
4.2. Treatments and Measurements
4.3. Statistics and Calculations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Filtenborg, O.; Frisvad, J.; Thrane, U. Moulds in food spoilage. Int. J. Food Microbiol. 1996, 33, 85–102. [Google Scholar] [CrossRef]
- Moss, M.O. Mycotoxin review—Fusarium. Mycologist 2002, 16, 158–161. [Google Scholar] [CrossRef]
- Milićević, D.R.; Skrinjar, M.; Baltić, T. Real and Perceived Risks for Mycotoxin Contamination in Foods and Feeds: Challenges for Food Safety Control. Toxins 2010, 2, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Cope, R.B. Trichothecenes. Vet. Toxicol. 2018, 1, 1043–1053. [Google Scholar]
- Nährer, K.; Kovalsky, P. A Summary of the Major Threats. In BIOMIN Mycotoxin Survey; Biomin Holding GmbH: Getzersdorf, Austria, 2018. [Google Scholar]
- Wannemacher, R.W.; Wiener, S.L.; Sidell, F.R.; Takafuji, E.T.; Franz, D.R. Trichothecene mycotoxins. In Medical Aspects of Chemical and Biological Warfare; Zajtchuk, R., Ed.; Office of the Surgeon General Department of the Army: Washington, DC, USA, 1997; Volume 6, pp. 655–676. [Google Scholar]
- Yang, S.; Li, Y.; Cao, X.; Hu, D.; Wang, Z.; Wang, Y.; Shen, J.; Zhang, S. Metabolic Pathways of T-2 Toxin in in Vivo and in Vitro Systems of Wistar Rats. J. Agric. Food Chem. 2013, 61, 9734–9743. [Google Scholar] [CrossRef] [PubMed]
- Kuca, K.; Dohnal, V.; Jezkova, A.; Jun, D. Metabolic pathways of T-2 toxin. Curr. Drug Metab. 2008, 9, 77–82. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Beier, R.C.; Shen, J.; De Smet, D.; De Saeger, S.; Zhang, S. T-2 Toxin, a Trichothecene Mycotoxin: Review of Toxicity, Metabolism, and Analytical Methods. J. Agric. Food Chem. 2011, 59, 3441–3453. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol: Toxicity, mechanisms and animal health risks. Anim. Feed Sci. Technol. 2007, 137, 283–298. [Google Scholar] [CrossRef]
- Chaudhari, M.; Jayaraj, R.; Bhaskar, A.; Rao, P.L. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 2009, 262, 153–161. [Google Scholar] [CrossRef]
- Chaudhary, M.; Rao, P.L. Brain oxidative stress after dermal and subcutaneous exposure of T-2 toxin in mice. Food Chem. Toxicol. 2010, 48, 3436–3442. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol-Induced Proinflammatory Gene Expression: Mechanisms and Pathological Sequelae. Toxins 2010, 2, 1300–1317. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.H.; Wang, X.; Yang, W.; Nussler, A.K.; Xiong, L.Y.; Kuca, K.; Dohnal, V.; Zhang, X.J.; Yuan, Z.H. Oxidative stress-mediated cytotoxicity and metabolism of T-2 toxin and deoxynivalenol in animals and humans: An update. Arch. Toxicol. 2014, 88, 1309–1326. [Google Scholar] [CrossRef] [PubMed]
- Bócsai, A.; Pelyhe, C.; Zándoki, E.; Ancsin, Z.; Erdélyi, M.; Mézes, M.; Balogh, K.; Szabó-Fodor, J. Short-term effects of T-2 toxin exposure on some lipid peroxide and glutathione redox parameters of broiler chickens. J. Anim. Physiol. Anim. Nutr. 2015, 100, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Bensassi, F.; El Golli-Bennour, E.; Abid-Essefi, S.; Bouaziz, C.; Hajlaoui, M.R.; Bacha, H. Pathway of deoxynivalenol-induced apoptosis in human colon carcinoma cells. Toxicology 2009, 264, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Rezar, V.; Frankic, T.; Narat, M.; Levart, A.; Salobir, J. Dose-Dependent Effects of T-2 Toxin on Performance, Lipid Peroxidation, and Genotoxicity in Broiler Chickens. Poult. Sci. 2007, 86, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- El Golli-Bennour, E.; Bacha, H. Hsp70 expression as biomarkers of oxidative stress: Mycotoxins’ exploration. Toxicology 2011, 287, 1–7. [Google Scholar] [CrossRef]
- Sehata, S.; Kiyosawa, N.; Atsumi, F.; Ito, K.; Yamoto, T.; Teranishi, M.; Uetsuka, K.; Nakayama, H.; Doi, K. Microarray analysis of T-2 toxin-induced liver, placenta and fetal liver lesions in pregnant rats. Exp. Toxicol. Pathol. 2005, 57, 15–28. [Google Scholar] [CrossRef]
- El Golli, E.; Hassen, W.; Bouslimi, A.; Bouaziz, C.; Ladjimi, M.M.; Bacha, H. Induction of Hsp 70 in Vero cells in response to mycotoxins. Toxicol. Lett. 2006, 166, 122–130. [Google Scholar] [CrossRef]
- Seeboth, J.; Solinhac, R.; Oswald, I.P.; Guzylack-Piriou, L. The fungal T-2 toxin alters the activation of primary macrophages induced by TLR-agonists resulting in a decrease of the inflammatory response in the pig. Vet. Res. 2012, 43, 35. [Google Scholar] [CrossRef]
- Kankkunen, P.; Rintahaka, J.; Aalto, A.; Leino, M.; Majuri, M.L.; Alenius, H.; Wolff, H.; Matikainen, S. Trichothecene Mycotoxins Activate Inflammatory Response in Human Macrophages. J. Immunol. 2009, 182, 6418–6425. [Google Scholar] [CrossRef]
- Awad, W.; Ghareeb, K.; Bohm, J.; Razzazi, E.; Hellweg, P.; Zentek, J. The Impact of the Fusarium Toxin Deoxynivalenol (DON) on Poultry. Int. J. Poult. Sci. 2008, 7, 827–842. [Google Scholar] [CrossRef]
- Mackei, M.; Molnár, A.; Nagy, S.; Pál, L.; Kővágó, C.; Gálfi, P.; Dublecz, K.; Husvéth, F.; Neogrády, Z.; Mátis, G. Effects of acute heat stress on a newly established chicken hepatocyte—Non-parenchymal cell co-culture model. Animals 2020. under review. [Google Scholar]
- Yang, L.; Yu, Z.; Hou, J.; Deng, Y.; Zhou, Z.; Zhao, Z.; Cui, J. Toxicity and oxidative stress induced by T-2 toxin and HT-2 toxin in broilers and broiler hepatocytes. Food Chem. Toxicol. 2016, 87, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.H.; Li, S.; Pestka, J.J. Down-Regulation of the Endoplasmic Reticulum Chaperone GRP78/BiP by Vomitoxin (Deoxynivalenol). Toxicol. Appl. Pharmacol. 2000, 162, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Wu, G.; He, Q.; Tang, H.; Wang, Y. The toxicity of acute exposure to T-2 toxin evaluated by the metabonomics technique. Mol. BioSyst. 2015, 11, 882–891. [Google Scholar] [CrossRef]
- Adhikari, M.; Negi, B.; Kaushik, N.; Adhikari, A.; Al-Khedhairy, A.A.; Kaushik, N.K.; Choi, E.H. T-2 mycotoxin: Toxicological effects and decontamination strategies. Oncotarget 2017, 8, 33933–33952. [Google Scholar] [CrossRef]
- Mézes, M.; Bárta, M.; Nagy, G. Comparative investigation on the effect of T-2 mycotoxin on lipid peroxidation and antioxidant status in different poultry species. Res. Vet. Sci. 1999, 66, 19–23. [Google Scholar] [CrossRef]
- Bouaziz, C.; Bouslimi, A.; Kadri, R.; Zaied, C.; Bacha, H.; Abid-Essefi, S. The in vitro effects of zearalenone and T-2 toxins on Vero cells. Exp. Toxicol. Pathol. 2013, 65, 497–501. [Google Scholar] [CrossRef]
- Maruniakova, N.; Kadasi, A.; Sirotkin, A.V.; Bulla, J.; Kolesarova, A. T-2 toxin and its metabolite HT-2 toxin combined with insulin-like growth factor-I modify progesterone secretion by porcine ovarian granulosa cells. J. Environ. Sci. Health Part A 2013, 49, 404–409. [Google Scholar] [CrossRef]
- Huang, P.X.; Akagawa, K.; Yokoyama, Y.; Nohara, K.; Kano, K.; Morimoto, K. T-2 toxin initially activates caspase-2 and induces apoptosis in U937 cells. Toxicol. Lett. 2007, 170, 1–10. [Google Scholar] [CrossRef]
- Bouaziz, C.; El Dein, O.S.; El Golli, E.; Abid-Essefi, S.; Brenner, C.; Lemaire, C.; Bacha, H. Different apoptotic pathways induced by zearalenone, T-2 toxin and ochratoxin A in human hepatoma cells. Toxicology 2008, 254, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Awad, W.A.; Ghareeb, K.; Dadak, A.; Gille, L.; Staniek, K.; Hess, M.; Böhm, J. Genotoxic effects of deoxynivalenol in broiler chickens fed low-protein feeds. Poult. Sci. 2012, 91, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Bozem, M.; Knapp, P.; Mirčeski, V.; Slowik, E.J.; Bogeski, I.; Kappl, R.; Heinemann, C.; Hoth, M. Electrochemical Quantification of Extracellular Local H2O2 Kinetics Originating from Single Cells. Antioxid. Redox Signal. 2018, 29, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Spolarics, Z.; Stein, D.S.; Garcia, Z.C. Endotoxin Stimulates Hydrogen Peroxide Detoxifying Activity in Rat Hepatic Endothelial Cells. Hepatology 1996, 24, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Polla, B.S.; Kantengwa, S.; Francois, D.; Salvioli, S.; Franceschi, C.; Marsac, C.; Cossarizza, A. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc. Natl. Acad. Sci. USA 1996, 93, 6458–6463. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Ihsan, A.; Huang, L.; Dai, M.; Hao, H.; Cheng, G.; Liu, Z.; Wang, Y.; Yuan, Z. JAK/STAT Pathway Plays a Critical Role in the Proinflammatory Gene Expression and Apoptosis of RAW264.7 Cells Induced by Trichothecenes as DON and T-2 Toxin. Toxicol. Sci. 2012, 127, 412–424. [Google Scholar] [CrossRef]
- Wu, Q.H.; Wang, X.; Wan, D.; Li, J.; Yuan, Z.H. Crosstalk of JNK1-STAT3 is critical for RAW264.7 cell survival. Cell. Signal. 2014, 26, 2951–2960. [Google Scholar] [CrossRef]
- Zhou, X.R.; Wang, Z.L.; Chen, J.H.; Wang, W.; Song, D.Q.; Li, S.Y.; Yang, H.J.; Xue, S.H.; Chen, C. Increased levels of IL-6, IL-1β, and TNF-α in Kashin–Beck disease and rats induced by T-2 toxin and selenium deficiency. Rheumatol. Int. 2013, 34, 995–1004. [Google Scholar] [CrossRef]
- Yang, F.; Wang, L.; Zhao, B.C.; Gong, Q. The effect of T-2 toxin on IL-1β and IL-6 secretion in human fetal chondrocytes. Int. Orthop. 2001, 25, 199–201. [Google Scholar]
- Kruber, P.; Trump, S.; Behrens, J.; Lehmann, I. T-2 toxin is a cytochrome P450 1A1 inducer and leads to MAPK/p38-but not aryl hydrocarbon receptor-dependent interleukin-8 secretion in the human intestinal epithelial cell line Caco-2. Toxicology 2011, 284, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Aviagen. Ross Broiler Management Handbook; Aviagen: Scotland, UK, 2018; pp. 1–148. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackei, M.; Orbán, K.; Molnár, A.; Pál, L.; Dublecz, K.; Husvéth, F.; Neogrády, Z.; Mátis, G. Cellular Effects of T-2 Toxin on Primary Hepatic Cell Culture Models of Chickens. Toxins 2020, 12, 46. https://doi.org/10.3390/toxins12010046
Mackei M, Orbán K, Molnár A, Pál L, Dublecz K, Husvéth F, Neogrády Z, Mátis G. Cellular Effects of T-2 Toxin on Primary Hepatic Cell Culture Models of Chickens. Toxins. 2020; 12(1):46. https://doi.org/10.3390/toxins12010046
Chicago/Turabian StyleMackei, Máté, Kata Orbán, Andor Molnár, László Pál, Károly Dublecz, Ferenc Husvéth, Zsuzsanna Neogrády, and Gábor Mátis. 2020. "Cellular Effects of T-2 Toxin on Primary Hepatic Cell Culture Models of Chickens" Toxins 12, no. 1: 46. https://doi.org/10.3390/toxins12010046
APA StyleMackei, M., Orbán, K., Molnár, A., Pál, L., Dublecz, K., Husvéth, F., Neogrády, Z., & Mátis, G. (2020). Cellular Effects of T-2 Toxin on Primary Hepatic Cell Culture Models of Chickens. Toxins, 12(1), 46. https://doi.org/10.3390/toxins12010046