Lebetin 2, a Snake Venom-Derived B-Type Natriuretic Peptide, Provides Immediate and Prolonged Protection against Myocardial Ischemia-Reperfusion Injury via Modulation of Post-Ischemic Inflammatory Response

 ,
,
Abstract
1. Introduction
2. Results
2.1. L2 Effect on Blood Pressure and Heart Rate
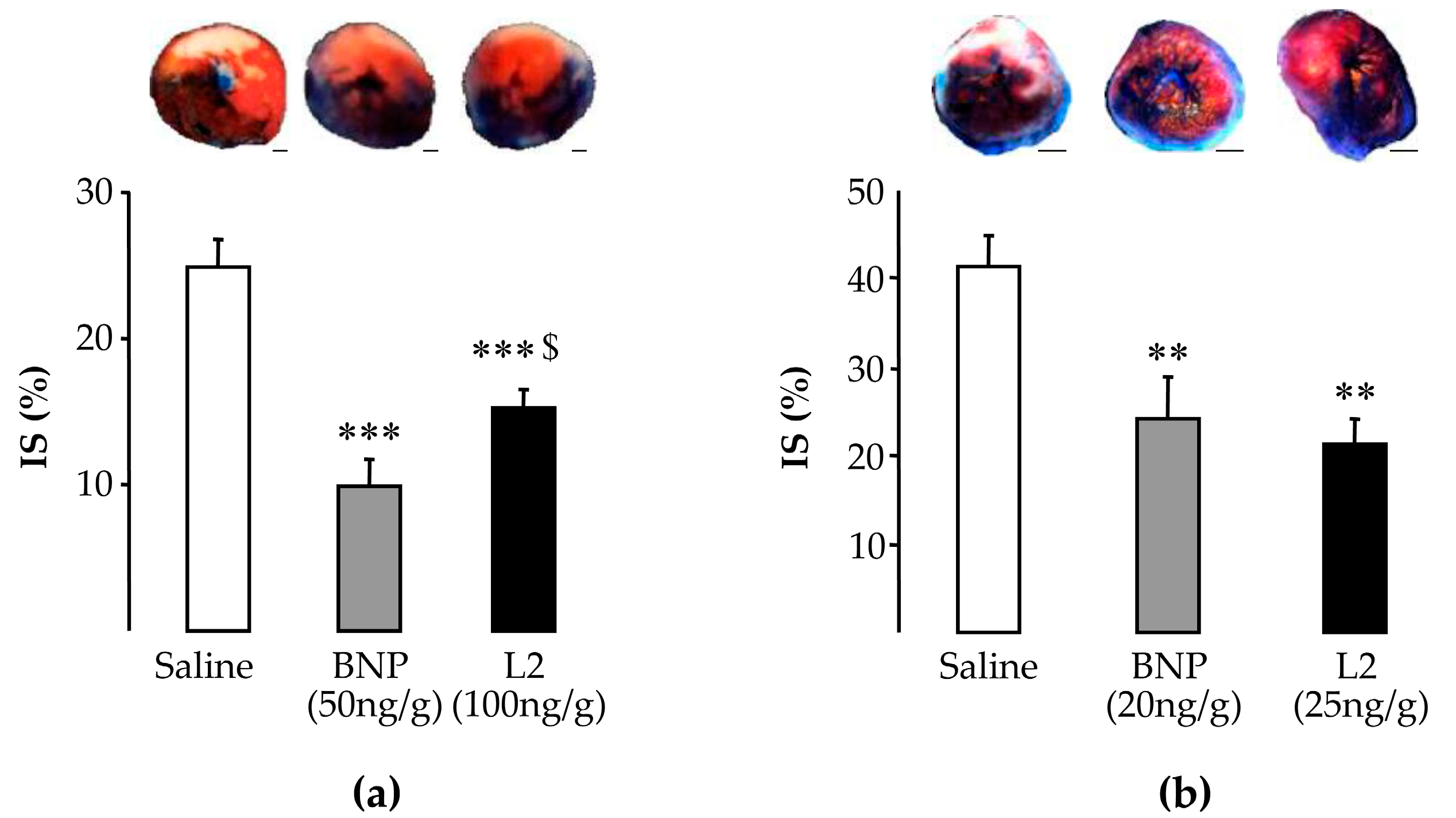
2.2. L2 Decreases LV Infarct Size Following IR Injury
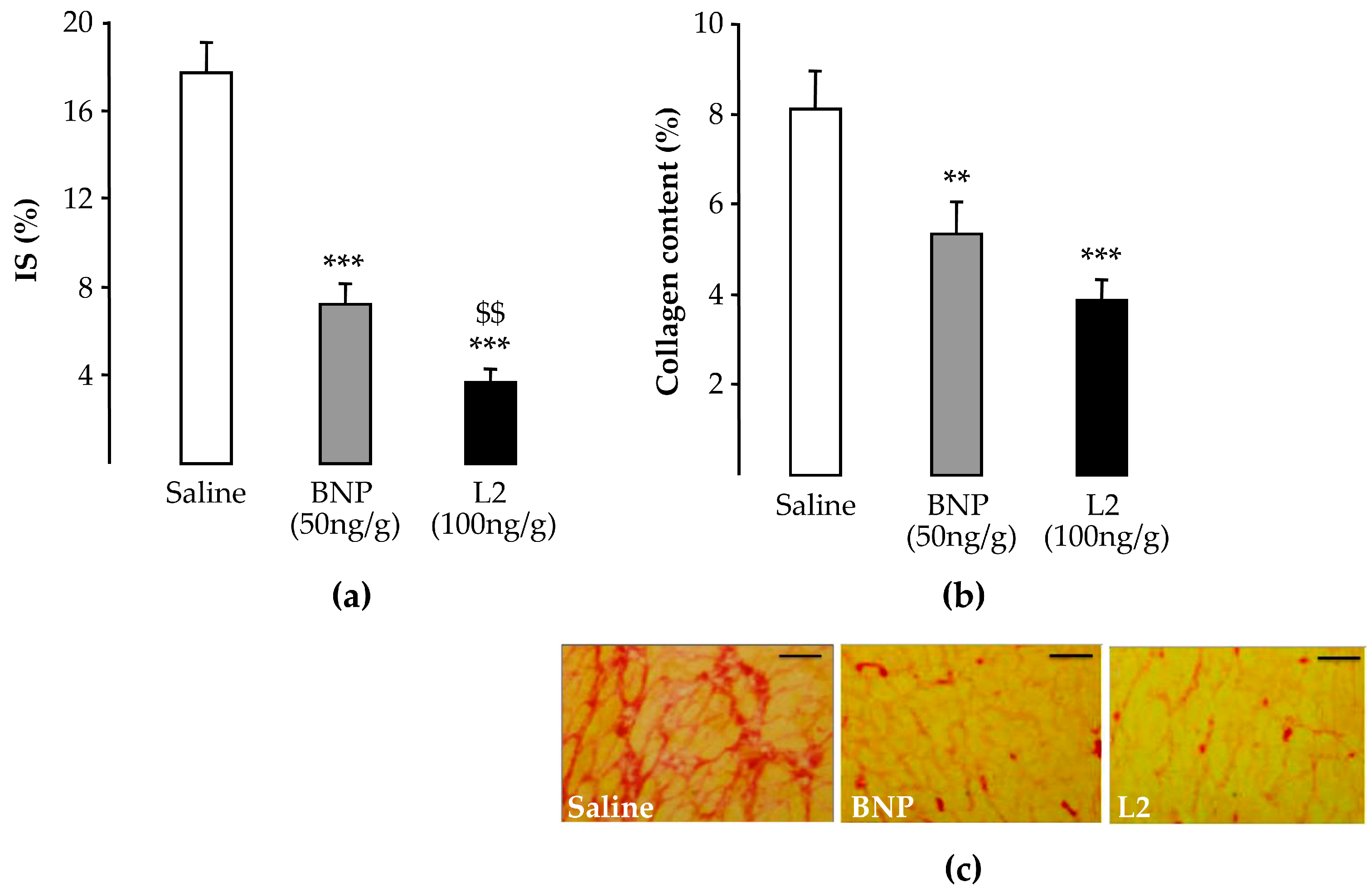
2.3. L2 Exerts Post-Ischemic Anti-Fibrotic Effect
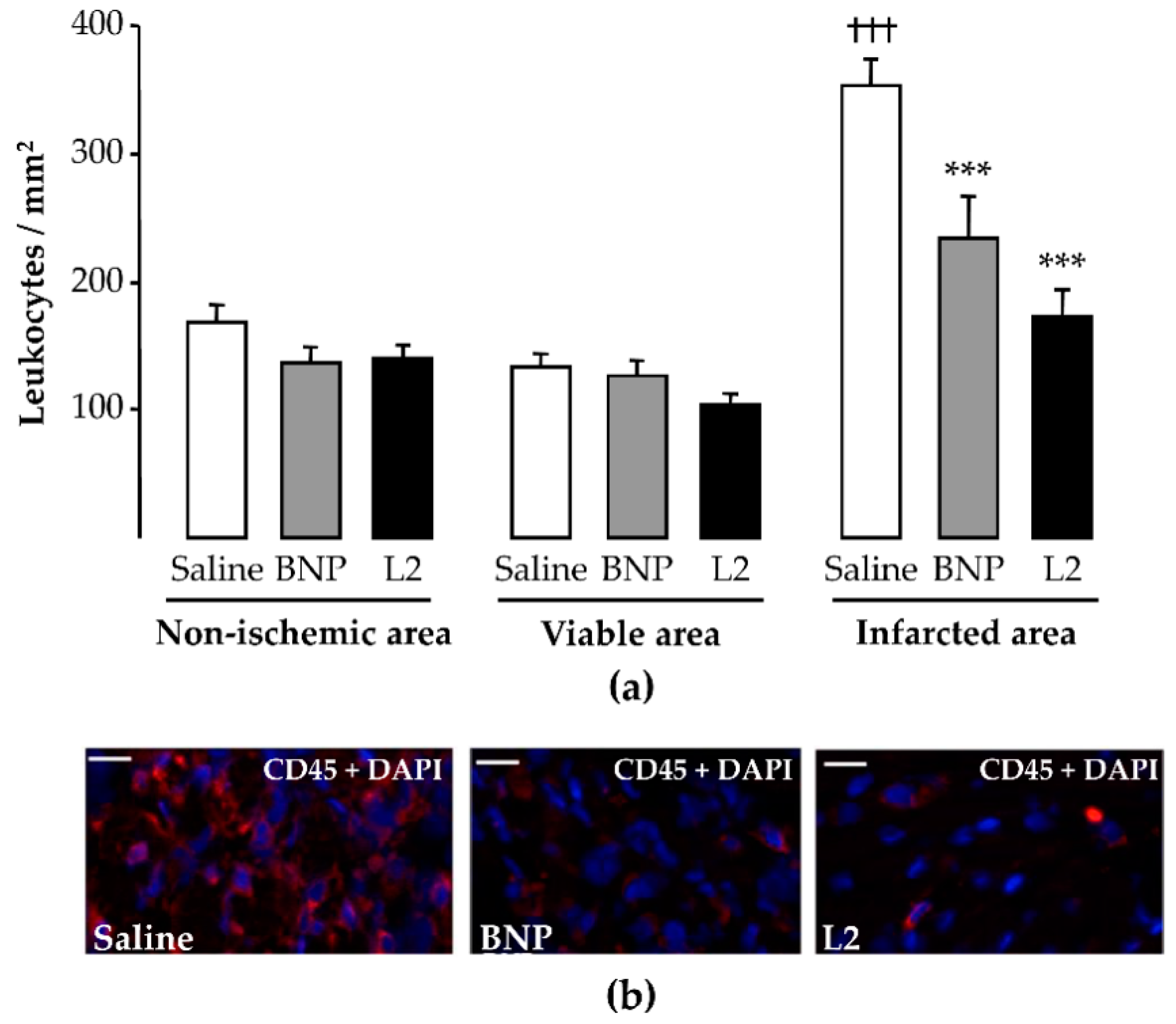
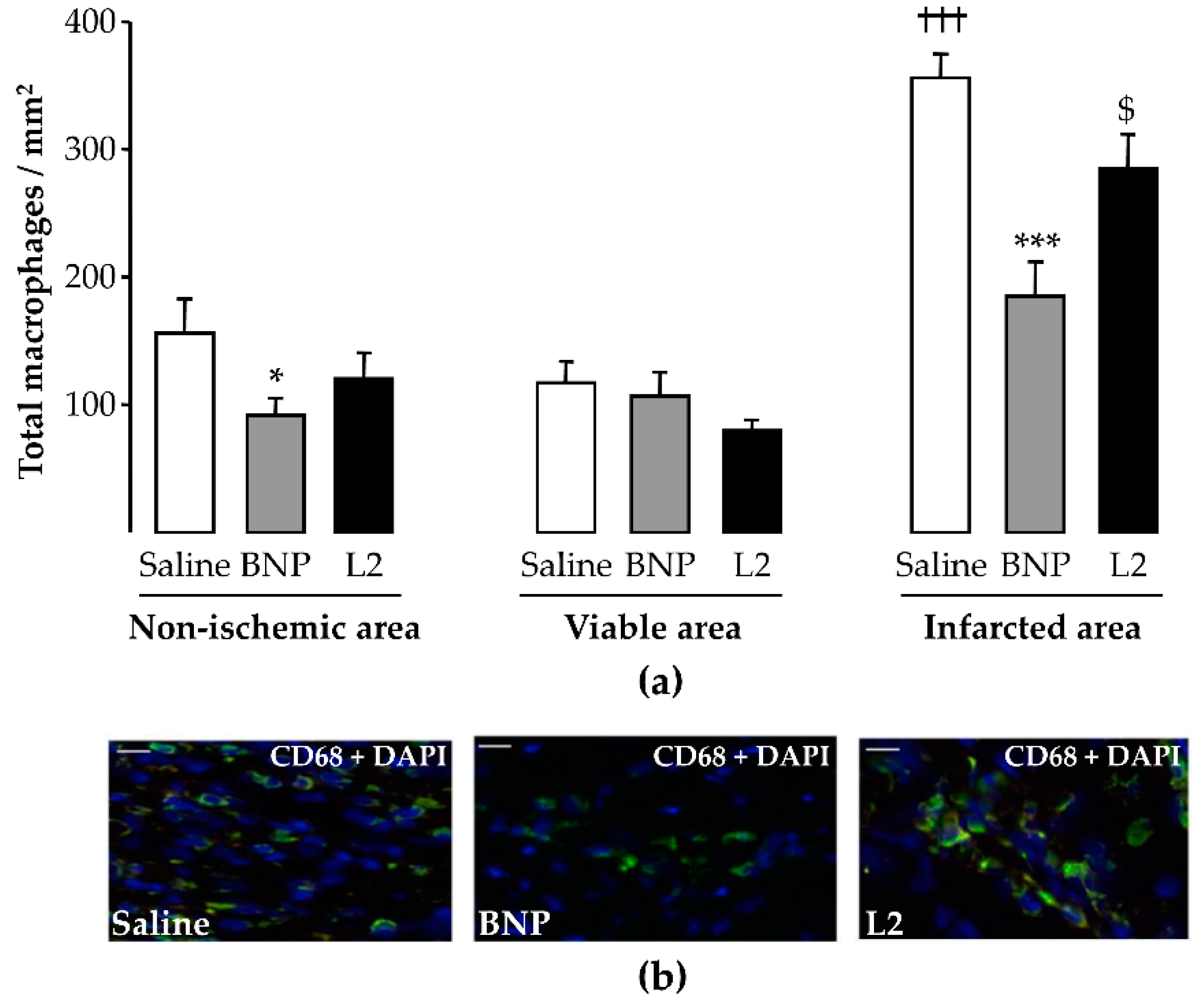
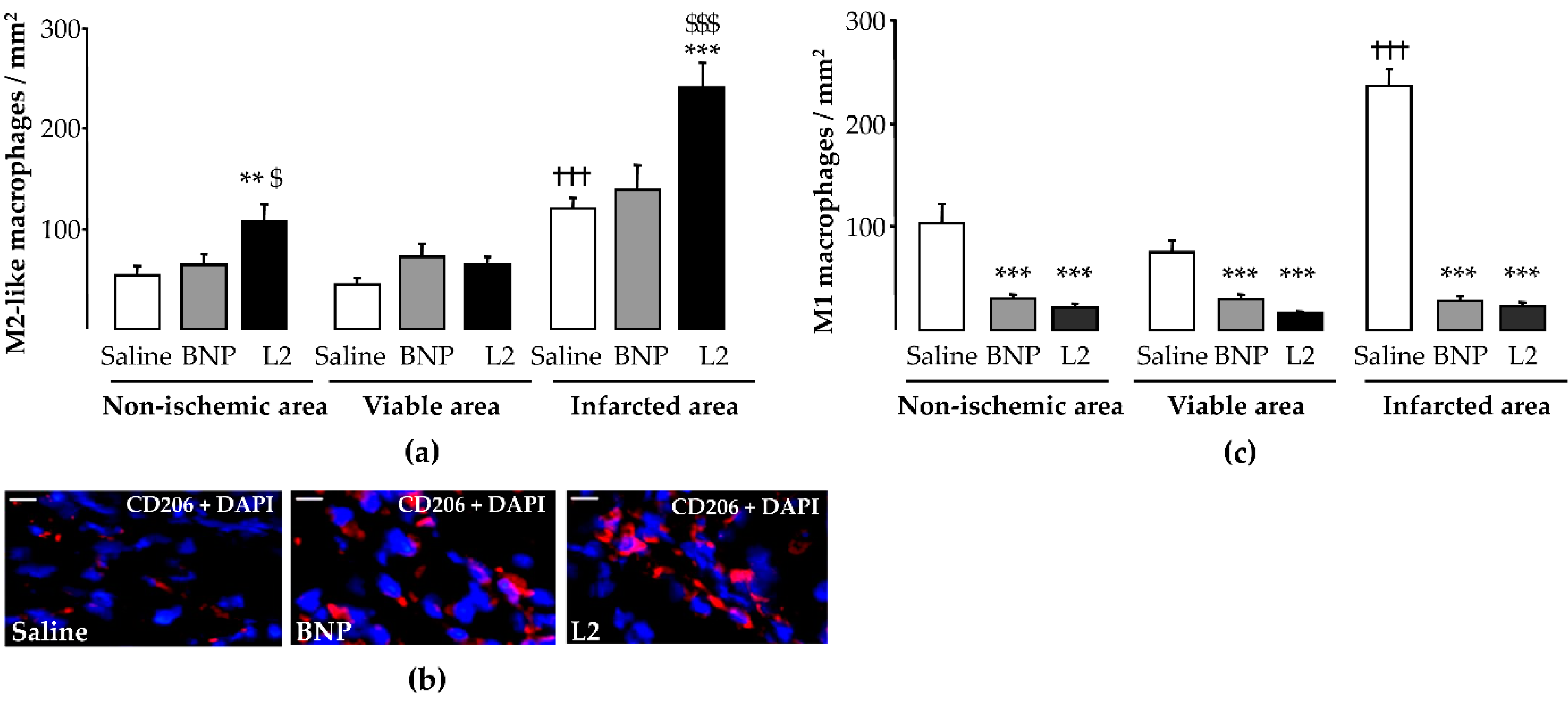
2.4. L2 Modulates the Post-Ischemic Inflammatory Response
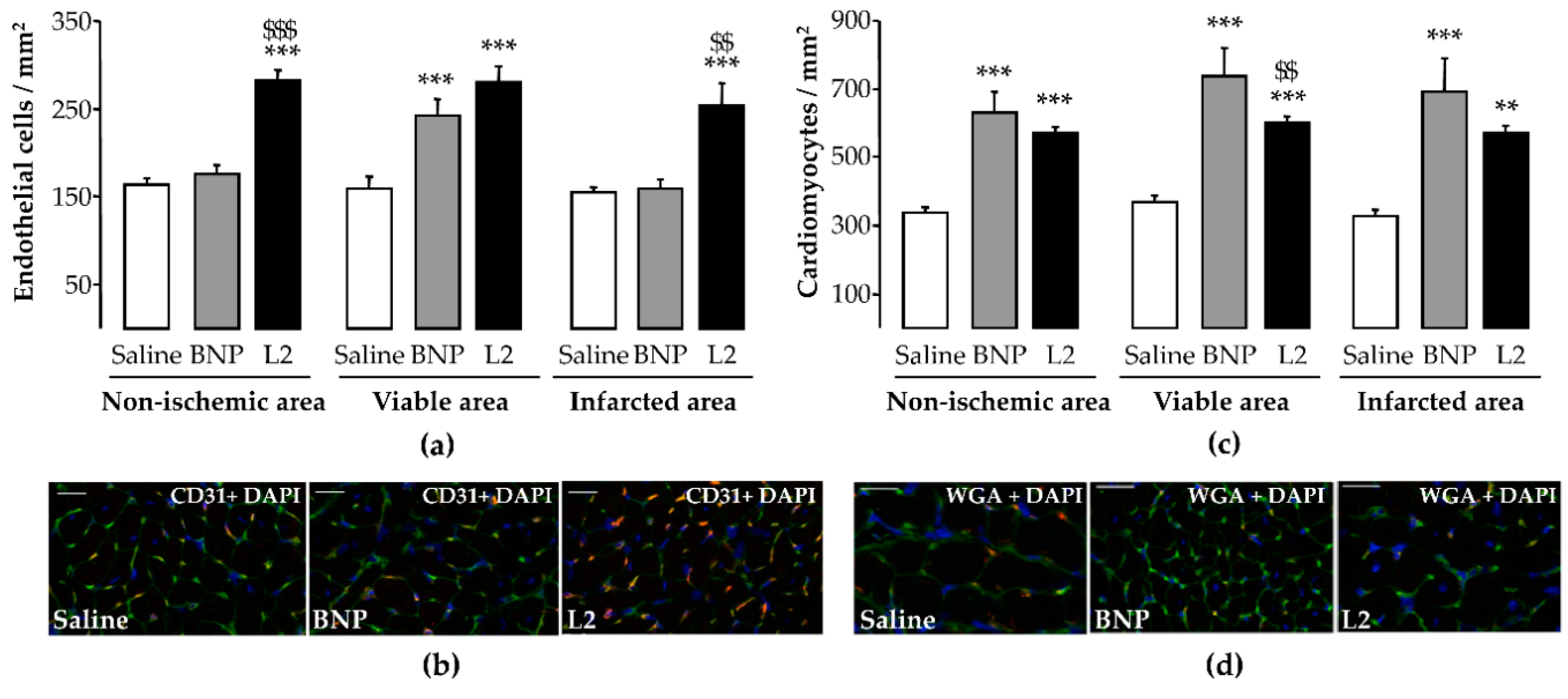
2.5. L2 Enhances Endothelial Cell and Cardiomyocyte Densities upon MI
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Drugs Used in the Study
5.2. Animals
5.3. In Vivo Dose-Response Effect on Blood Pressure
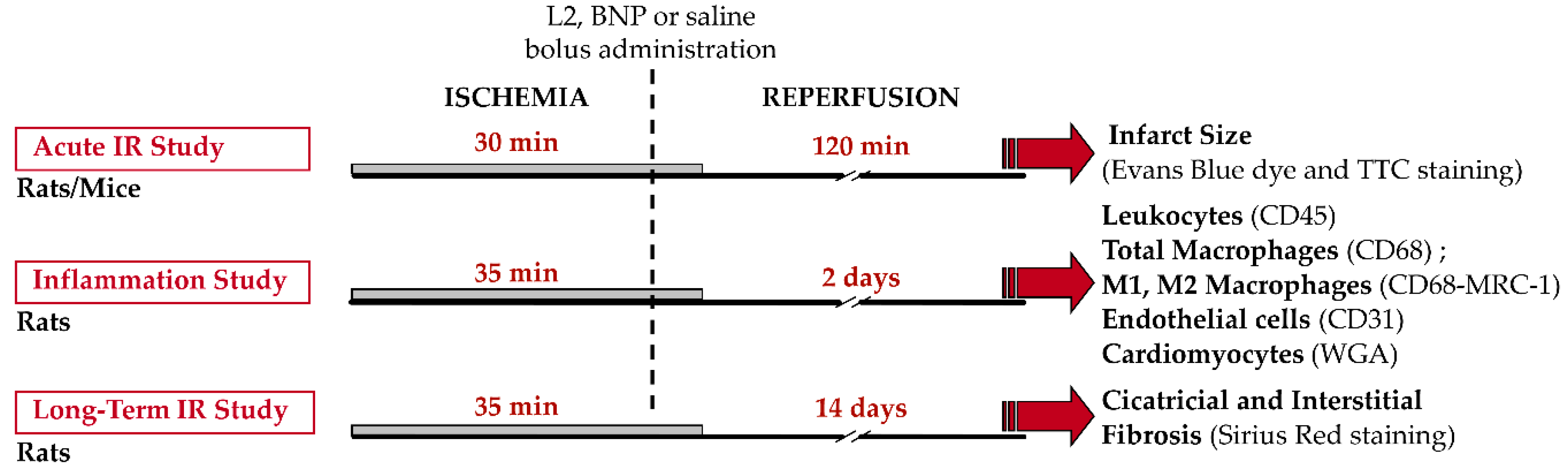
5.4. In Vivo Murine Models of Myocardial Ischemia-Reperfusion
5.4.1. Surgical Preparation
5.4.2. Experimental Protocols
5.5. Histological Analysis
5.5.1. Acute Infarct Size
5.5.2. Cardiac Morphometry: Infarct Size and Fibrosis
5.6. Immunohistochemistry
5.6.1. Post-Ischemic Inflammation
5.6.2. Endothelial Cell and Cardiomyocyte Densities
5.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AR | Area at Risk |
| AUC | Area under curve |
| BNP | B-type natriuretic peptide |
| cGMP | Cyclic guanosine monophosphate |
| DNP | Dendroaspis natriuretic peptide |
| HF | Heart failure |
| HR | Heart rate |
| IR | Ischemia-reperfusion |
| IS | Infarct size |
| L2 | Lebetin 2 |
| LV | Left ventricle |
| MAP | Mean arterial blood pressure |
| MI | Myocardial infarction |
| NO | Nitric oxide |
| NP | Natriuretic peptide |
| NPR | Natriuretic peptide receptor |
References
- Richard, V.; Kaeffer, N.; Tron, C.; Thuillez, C. Ischemic preconditioning protects against coronary endothelial dysfunction induced by ischemia and reperfusion. Circulation 1994, 89, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.G.; Trikalinos, T.A.; Kent, D.M.; Antonopoulos, G.V.; Konstam, M.A.; Udelson, J.E. Quantitative evaluation of drug or device effects on ventricular remodeling as predictors of therapeutic effects on mortality in patients with heart failure and reduced ejection fraction: A meta-analytic approach. J. Am. Coll. Cardiol. 2010, 56, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Monocyte and macrophage heterogeneity in the heart. Circ. Res. 2013, 112, 1624–1633. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Koufaki, M.; Kremastinos, D.T. Pharmacological pre- and post- conditioning agents: Reperfusion-injury of the heart revisited. Mini Rev. Med. Chem. 2008, 8, 952–959. [Google Scholar] [CrossRef]
- Colucci, W.S.; Elkayam, U.; Horton, D.P.; Abraham, W.T.; Bourge, R.C.; Johnson, A.D.; Wagoner, L.E.; Givertz, M.M.; Liang, C.S.; Neibaur, M.; et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N. Engl. J. Med. 2000, 343, 246–253. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Publication Committee for the VMAC Investigators. Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: A randomized controlled trial. JAMA 2002, 287, 1531–1540. [Google Scholar]
- Yancy, C.W.; Krum, H.; Massie, B.M.; Silver, M.A.; Stevenson, L.W.; Cheng, M.; Kim, S.S.; Evans, R.; Investigators, F.I. Safety and efficacy of outpatient nesiritide in patients with advanced heart failure: Results of the Second Follow-Up Serial Infusions of Nesiritide (FUSION II) trial. Circ. Heart Fail. 2008, 1, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Hamanaka, I.; Takahashi, N.; Kawakami, R.; Li, Y.; Takemura, G.; et al. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J. Clin. Investig. 2001, 108, 203–213. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef]
- George, I.; Morrow, B.; Xu, K.; Yi, G.H.; Holmes, J.; Wu, E.X.; Li, Z.; Protter, A.A.; Oz, M.C.; Wang, J. Prolonged effects of B-type natriuretic peptide infusion on cardiac remodeling after sustained myocardial injury. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H708–H717. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Nakagawa, Y.; Nakanishi, M.; Tanimoto, K.; Usami, S.; et al. Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation 2004, 110, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Huang, X.; Zhang, K.; Jiang, H.; Hu, X. Anti-inflammatory effect of B-type natriuretic peptide postconditioning during myocardial ischemia-reperfusion: Involvement of PI3K/Akt signaling pathway. Inflammation 2014, 37, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ng, X.W.; Lim, S.G.; Chen, H.H.; Burnett, J.C., Jr.; Boey, Y.C.; Venkatraman, S.S. In vivo Evaluation of Cenderitide-Eluting Stent (CES) II. Ann. Biomed. Eng. 2016, 44, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Lainchbury, J.G.; Burnett, J.C., Jr. Natriuretic peptide receptors and neutral endopeptidase in mediating the renal actions of a new therapeutic synthetic natriuretic peptide Dendroaspis natriuretic peptide. J. Am. Coll. Cardiol. 2002, 40, 1186–1191. [Google Scholar] [CrossRef]
- Johns, D.G.; Ao, Z.; Heidrich, B.J.; Hunsberger, G.E.; Graham, T.; Payne, L.; Elshourbagy, N.; Lu, Q.; Aiyar, N.; Douglas, S.A. Dendroaspis natriuretic peptide binds to the natriuretic peptide clearance receptor. Biochem. Biophys. Res. Commun. 2007, 358, 145–149. [Google Scholar] [CrossRef]
- St Pierre, L.; Flight, S.; Masci, P.P.; Hanchard, K.J.; Lewis, R.J.; Alewood, P.F.; de Jersey, J.; Lavin, M.F. Cloning and characterisation of natriuretic peptides from the venom glands of Australian elapids. Biochimie 2006, 88, 1923–1931. [Google Scholar] [CrossRef]
- Tourki, B.; Mateo, P.; Morand, J.; Elayeb, M.; Godin-Ribuot, D.; Marrakchi, N.; Belaidi, E.; Messadi, E. Lebetin 2, a Snake Venom-Derived Natriuretic Peptide, Attenuates Acute Myocardial Ischemic Injury through the Modulation of Mitochondrial Permeability Transition Pore at the Time of Reperfusion. PLoS ONE 2016, 11, e0162632. [Google Scholar] [CrossRef]
- Barbouche, R.; Marrakchi, N.; Mansuelle, P.; Krifi, M.; Fenouillet, E.; Rochat, H.; el Ayeb, M. Novel anti-platelet aggregation polypeptides from Vipera lebetina venom: Isolation and characterization. FEBS Lett. 1996, 392, 6–10. [Google Scholar] [CrossRef]
- Vink, S.; Jin, A.H.; Poth, K.J.; Head, G.A.; Alewood, P.F. Natriuretic peptide drug leads from snake venom. Toxicon 2012, 59, 434–445. [Google Scholar] [CrossRef]
- Breivik, L.; Jensen, A.; Guvag, S.; Aarnes, E.K.; Aspevik, A.; Helgeland, E.; Hovland, S.; Brattelid, T.; Jonassen, A.K. B-type natriuretic peptide expression and cardioprotection is regulated by Akt dependent signaling at early reperfusion. Peptides 2015, 66, 43–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burley, D.S.; Baxter, G.F. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res. Cardiol. 2007, 102, 529–541. [Google Scholar] [CrossRef]
- Ren, B.; Shen, Y.; Shao, H.; Qian, J.; Wu, H.; Jing, H. Brain natriuretic peptide limits myocardial infarct size dependent of nitric oxide synthase in rats. Clin. Chim. Acta 2007, 377, 83–87. [Google Scholar] [CrossRef]
- Costello-Boerrigter, L.C.; Boerrigter, G.; Cataliotti, A.; Harty, G.J.; Burnett, J.C., Jr. Renal and anti-aldosterone actions of vasopressin-2 receptor antagonism and B-type natriuretic peptide in experimental heart failure. Circ. Heart Fail. 2010, 3, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Kawakami, R.; Nakagawa, Y.; Tanimoto, K.; Yasuno, S.; et al. Role of natriuretic peptide receptor guanylyl cyclase-A in myocardial infarction evaluated using genetically engineered mice. Hypertension 2005, 46, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Messadi-Laribi, E.; Griol-Charhbili, V.; Pizard, A.; Vincent, M.P.; Heudes, D.; Meneton, P.; Alhenc-Gelas, F.; Richer, C. Tissue kallikrein is involved in the cardioprotective effect of AT1-receptor blockade in acute myocardial ischemia. J. Pharmacol. Exp. Ther. 2007, 323, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Messadi, E.; Aloui, Z.; Belaidi, E.; Vincent, M.P.; Couture-Lepetit, E.; Waeckel, L.; Decorps, J.; Bouby, N.; Gasmi, A.; Karoui, H.; et al. Cardioprotective effect of VEGF and venom VEGF-like protein in acute myocardial ischemia in mice: Effect on mitochondrial function. J. Cardiovasc. Pharmacol. 2014, 63, 274–281. [Google Scholar] [CrossRef]
- Moilanen, A.M.; Rysa, J.; Mustonen, E.; Serpi, R.; Aro, J.; Tokola, H.; Leskinen, H.; Manninen, A.; Levijoki, J.; Vuolteenaho, O.; et al. Intramyocardial BNP gene delivery improves cardiac function through distinct context-dependent mechanisms. Circ. Heart Fail. 2011, 4, 483–495. [Google Scholar] [CrossRef]
- Cao, L.; Gardner, D.G. Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts. Hypertension 1995, 25, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Tsuruda, T.; Boerrigter, G.; Huntley, B.K.; Noser, J.A.; Cataliotti, A.; Costello-Boerrigter, L.C.; Chen, H.H.; Burnett, J.C., Jr. Brain natriuretic Peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ. Res. 2002, 91, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Messadi, E.; Vincent, M.P.; Griol-Charhbili, V.; Mandet, C.; Colucci, J.; Krege, J.H.; Bruneval, P.; Bouby, N.; Smithies, O.; Alhenc-Gelas, F.; et al. Genetically determined angiotensin converting enzyme level and myocardial tolerance to ischemia. Faseb J. 2010, 24, 4691–4700. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Ji, Q.; Wu, B.; Yu, K.; Zeng, Q.; Xin, S.; Liu, J.; Zhou, Y. Chemerin15-Ameliorated Cardiac Ischemia-Reperfusion Injury Is Associated with the Induction of Alternatively Activated Macrophages. Mediat. Inflamm. 2015, 2015, 563951–563959. [Google Scholar] [CrossRef] [PubMed]
- Padilla, F.; Garcia-Dorado, D.; Agullo, L.; Barrabes, J.A.; Inserte, J.; Escalona, N.; Meyer, M.; Mirabet, M.; Pina, P.; Soler-Soler, J. Intravenous administration of the natriuretic peptide urodilatin at low doses during coronary reperfusion limits infarct size in anesthetized pigs. Cardiovasc. Res. 2001, 51, 592–600. [Google Scholar] [CrossRef][Green Version]
- Manivasagam, S.; Subramanian, V.; Tumala, A.; Vellaichamy, E. Differential expression and regulation of anti-hypertrophic genes Npr1 and Npr2 during beta-adrenergic receptor activation-induced hypertrophic growth in rats. Mol. Cell. Endocrinol. 2016, 433, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Burley, D.S.; Cox, C.D.; Zhang, J.; Wann, K.T.; Baxter, G.F. Natriuretic peptides modulate ATP-sensitive K(+) channels in rat ventricular cardiomyocytes. Basic Res. Cardiol. 2014, 109, 402. [Google Scholar] [CrossRef] [PubMed]
- Dayan, V.; Yannarelli, G.; Billia, F.; Filomeno, P.; Wang, X.H.; Davies, J.E.; Keating, A. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Res. Cardiol. 2011, 106, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, K.; Kaikita, K.; Hayasaki, T.; Honda, T.; Kobayashi, H.; Sakashita, N.; Suzuki, H.; Kodama, T.; Ogawa, H.; Takeya, M. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation 2007, 115, 1904–1911. [Google Scholar] [CrossRef]
- Talha, S.; Bouitbir, J.; Charles, A.L.; Zoll, J.; Goette-Di Marco, P.; Meziani, F.; Piquard, F.; Geny, B. Pretreatment with brain natriuretic peptide reduces skeletal muscle mitochondrial dysfunction and oxidative stress after ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 172–179. [Google Scholar] [CrossRef]
- Kook, H.; Itoh, H.; Choi, B.S.; Sawada, N.; Doi, K.; Hwang, T.J.; Kim, K.K.; Arai, H.; Baik, Y.H.; Nakao, K. Physiological concentration of atrial natriuretic peptide induces endothelial regeneration in vitro. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1388–H1397. [Google Scholar] [CrossRef]
- Barbay, V.; Houssari, M.; Mekki, M.; Banquet, S.; Edwards-Levy, F.; Henry, J.P.; Dumesnil, A.; Adriouch, S.; Thuillez, C.; Richard, V.; et al. Role of M2-like macrophage recruitment during angiogenic growth factor therapy. Angiogenesis 2015, 18, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Suga, S.; Nakao, K.; Hosoda, K.; Mukoyama, M.; Ogawa, Y.; Shirakami, G.; Arai, H.; Saito, Y.; Kambayashi, Y.; Inouye, K.; et al. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology 1992, 130, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Stress-activated cytokines and the heart: From adaptation to maladaptation. Annu. Rev. Physiol. 2003, 65, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F.; Jadhav, A. Hemin therapy improves kidney function in male streptozotocin-induced diabetic rats: Role of the heme oxygenase/atrial natriuretic peptide/adiponectin axis. Endocrinology 2014, 155, 215–229. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Izzi, V.; D’Aquilio, F.; Carotenuto, F.; Di Nardo, P.; Baldini, P.M. Brain Natriuretic Peptide (BNP) regulates the production of inflammatory mediators in human THP-1 macrophages. Regul. Pept. 2008, 148, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; Xu, L.; Li, X.; Yu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. 2019, 133, 1705–1720. [Google Scholar] [CrossRef]
- Sontia, B.; Montezano, A.C.; Paravicini, T.; Tabet, F.; Touyz, R.M. Downregulation of renal TRPM7 and increased inflammation and fibrosis in aldosterone-infused mice: Effects of magnesium. Hypertension 2008, 51, 915–921. [Google Scholar] [CrossRef]
- Van Linthout, S.; Riad, A.; Dhayat, N.; Spillmann, F.; Du, J.; Dhayat, S.; Westermann, D.; Hilfiker-Kleiner, D.; Noutsias, M.; Laufs, U.; et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia 2007, 50, 1977–1986. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Wang, X.; Lau, W.; Wang, Y.; Xing, Y.; Zhang, X.; Ma, X.; Gao, F. Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3beta signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS ONE 2013, 8, e70956. [Google Scholar]
- Mulder, P.; Devaux, B.; Richard, V.; Henry, J.P.; Wimart, M.C.; Thibout, E.; Mace, B.; Thuillez, C. Early versus delayed angiotensin-converting enzyme inhibition in experimental chronic heart failure. Effects on survival, hemodynamics, and cardiovascular remodeling. Circulation 1997, 95, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Besnier, M.; Galaup, A.; Nicol, L.; Henry, J.P.; Coquerel, D.; Gueret, A.; Mulder, P.; Brakenhielm, E.; Thuillez, C.; Germain, S.; et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014, 28, 3351–3361. [Google Scholar] [CrossRef] [PubMed]
- Contard, F.; Glukhova, M.; Sabri, A.; Marotte, F.; Sartore, S.; Narcisse, G.; Schatz, C.; Guez, D.; Rappaport, L.; Samuel, J.L. Comparative effects of indapamide and hydrochlorothiazide on cardiac hypertrophy and vascular smooth-muscle phenotype in the stroke-prone, spontaneously hypertensive rat. J. Cardiovasc. Pharmacol. 1993, 22, S29–S34. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemodynamic Study | IR Study | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Group | MAP (mmHg) | HR (bpm) | AR/LV (%) | ||||||
| n | Before | After | Before | After | n | R, 120 min | n | R, 14 days | |
| Rats | |||||||||
| Control (saline 1 µL/g) | 7 | 108 ± 5 | 96 ± 2 | 321 ± 6 | 310 ± 7 | 5 | 65.3 ± 3.0 | 10 | 82.4 ± 1.8 |
| BNP (10 ng/g) | 6 | 105 ± 7 | 92 ± 4 | 323 ± 6 | 315 ± 6 | ||||
| BNP (50 ng/g) | 6 | 104 ± 6 | 59 ± 6 * | 335 ± 5 | 326 ± 6 | 5 | 66.6 ± 3.2 | 6 | 85.6 ± 1.3 |
| L2 (100 ng/g) | 7 | 105 ± 5 | 63 ± 3 * | 326 ± 6 | 317 ± 6 | 6 | 66.4 ± 3.5 | 7 | 84.8 ± 1.1 |
| L2 (200 ng/g) | 7 | 106 ± 5 | 34 ± 7 * | 330 ± 7 | 320 ± 4 | ||||
| Mice | R, 120 min | ||||||||
| Control (saline 1 µL/g) | 11 | 72 ± 3 | 70 ± 4 | 447 ± 27 | 422 ± 16 | 6 | 25.8 ± 2.5 | ||
| BNP (1.5 ng/g) | 10 | 70 ± 3 | 62 ± 2 * | 436 ± 14 | 467 ± 21 | ||||
| BNP (5 ng/g) | 5 | 67 ± 2 | 57 ± 3 * | 432 ± 32 | 415 ± 36 | ||||
| BNP (20 ng/g) | 7 | 71 ± 6 | 54 ± 5 * | 469 ± 35 | 473 ± 32 | 5 | 31.7 ± 1.2 | ||
| L2 (25 ng/g) | 5 | 75 ± 4 | 64 ± 3 | 518 ± 9 | 414 ± 21 | 5 | 31.0 ± 2.6 | ||
| L2 (50 ng/g) | 4 | 79 ± 4 | 63 ± 3 * | 497 ± 42 | 412 ± 19 | ||||
| L2 (100 ng/g) | 5 | 69 ± 1 | 36 ± 8 * | 511 ± 19 | 454 ± 7 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourki, B.; Dumesnil, A.; Belaidi, E.; Ghrir, S.; Godin-Ribuot, D.; Marrakchi, N.; Richard, V.; Mulder, P.; Messadi, E. Lebetin 2, a Snake Venom-Derived B-Type Natriuretic Peptide, Provides Immediate and Prolonged Protection against Myocardial Ischemia-Reperfusion Injury via Modulation of Post-Ischemic Inflammatory Response. Toxins 2019, 11, 524. https://doi.org/10.3390/toxins11090524
Tourki B, Dumesnil A, Belaidi E, Ghrir S, Godin-Ribuot D, Marrakchi N, Richard V, Mulder P, Messadi E. Lebetin 2, a Snake Venom-Derived B-Type Natriuretic Peptide, Provides Immediate and Prolonged Protection against Myocardial Ischemia-Reperfusion Injury via Modulation of Post-Ischemic Inflammatory Response. Toxins. 2019; 11(9):524. https://doi.org/10.3390/toxins11090524
Chicago/Turabian StyleTourki, Bochra, Anais Dumesnil, Elise Belaidi, Slim Ghrir, Diane Godin-Ribuot, Naziha Marrakchi, Vincent Richard, Paul Mulder, and Erij Messadi. 2019. "Lebetin 2, a Snake Venom-Derived B-Type Natriuretic Peptide, Provides Immediate and Prolonged Protection against Myocardial Ischemia-Reperfusion Injury via Modulation of Post-Ischemic Inflammatory Response" Toxins 11, no. 9: 524. https://doi.org/10.3390/toxins11090524
APA StyleTourki, B., Dumesnil, A., Belaidi, E., Ghrir, S., Godin-Ribuot, D., Marrakchi, N., Richard, V., Mulder, P., & Messadi, E. (2019). Lebetin 2, a Snake Venom-Derived B-Type Natriuretic Peptide, Provides Immediate and Prolonged Protection against Myocardial Ischemia-Reperfusion Injury via Modulation of Post-Ischemic Inflammatory Response. Toxins, 11(9), 524. https://doi.org/10.3390/toxins11090524