A Review of Cardiovascular Toxicity of Microcystins



Abstract
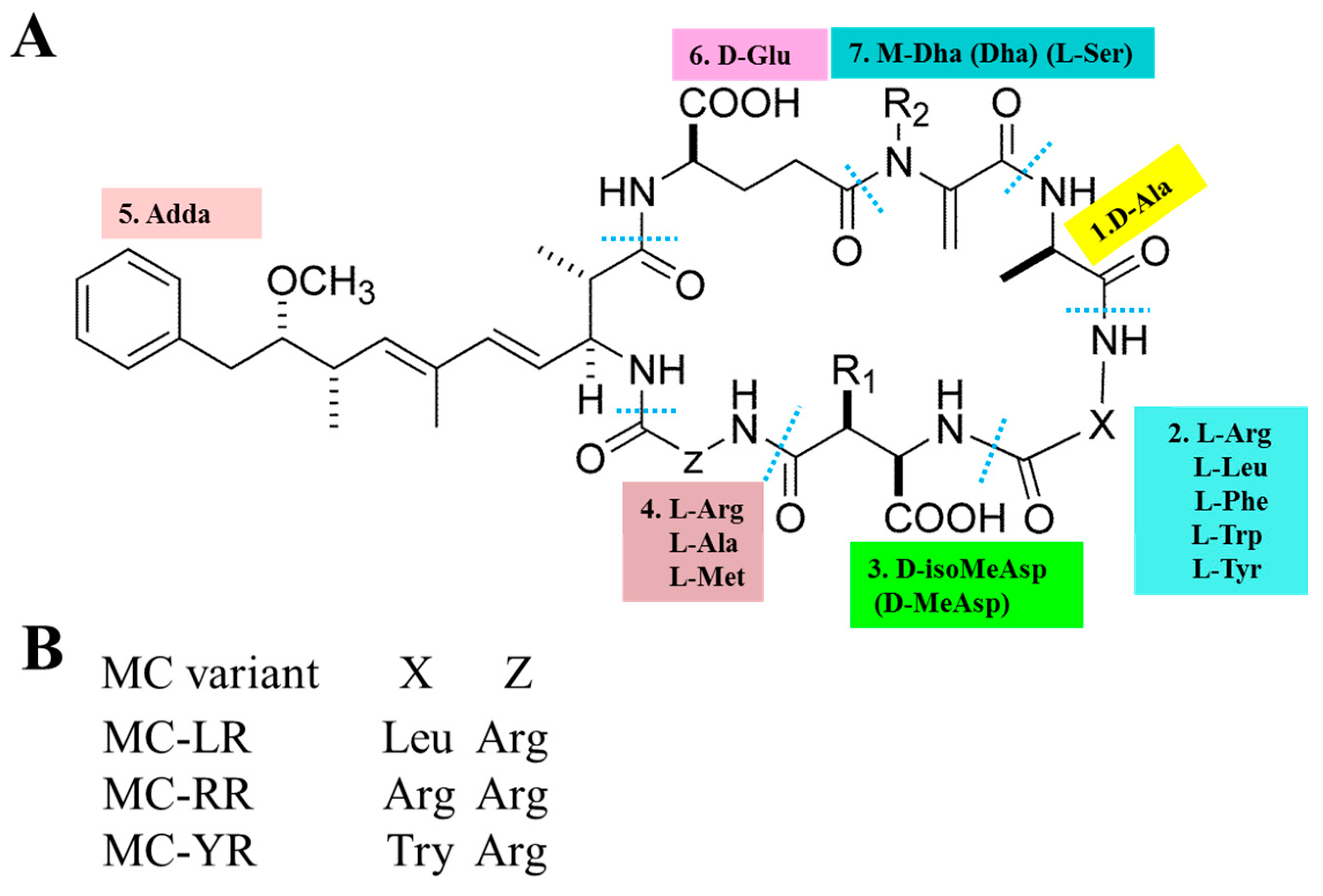
1. Introduction
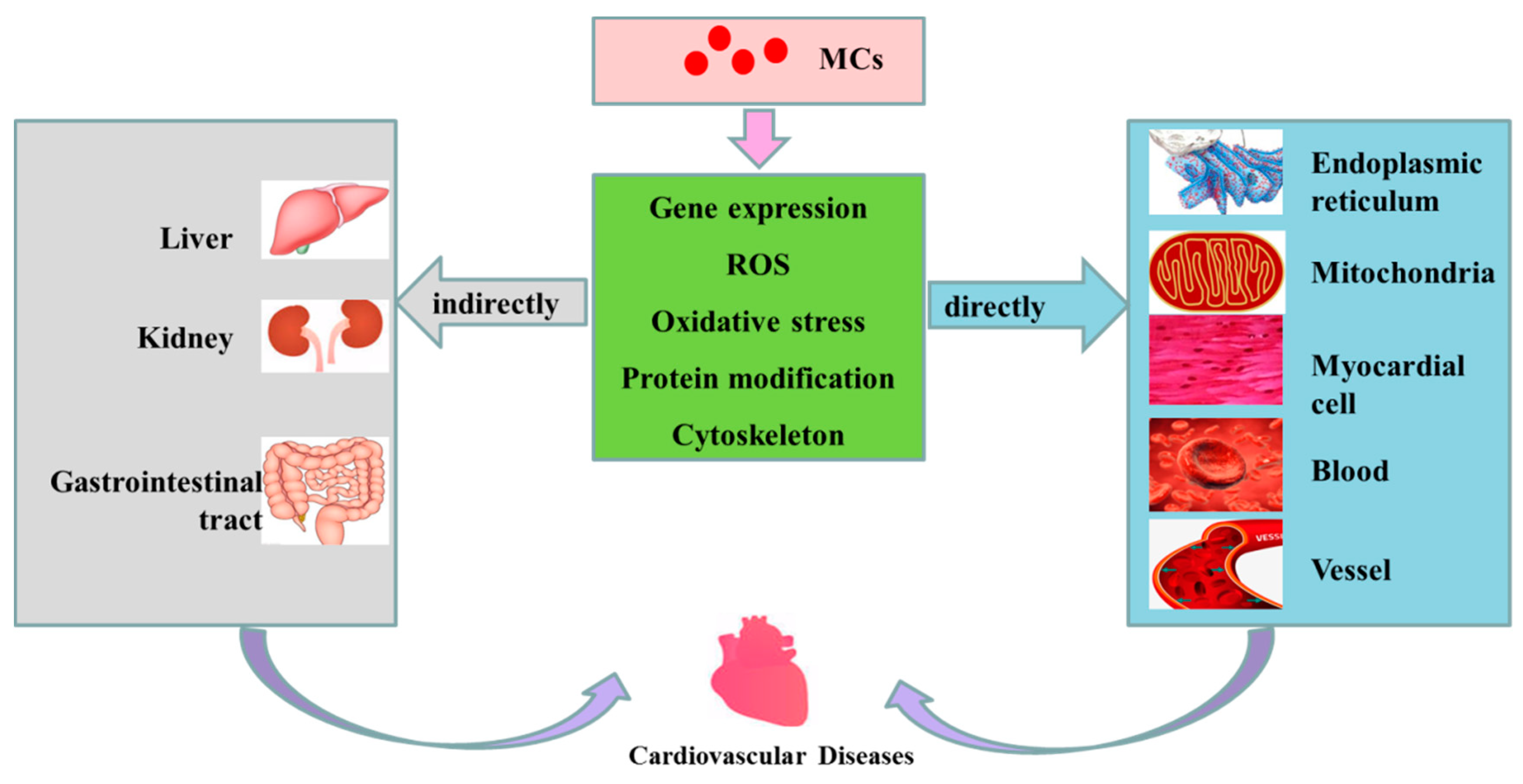
2. Cardiovascular Toxicity of MCs
2.1. Direct Cardiovascular Toxicity
2.1.1. Transportation of MCs
2.1.2. Cytoskeleton Disruption and Mitochondrial Dysfunction
2.1.3. Endoplasmic Reticulum Dysfunction
2.1.4. Inhibition of PP1 and PP2A
2.1.5. Hemodynamic Alterations and Vascular Lesions
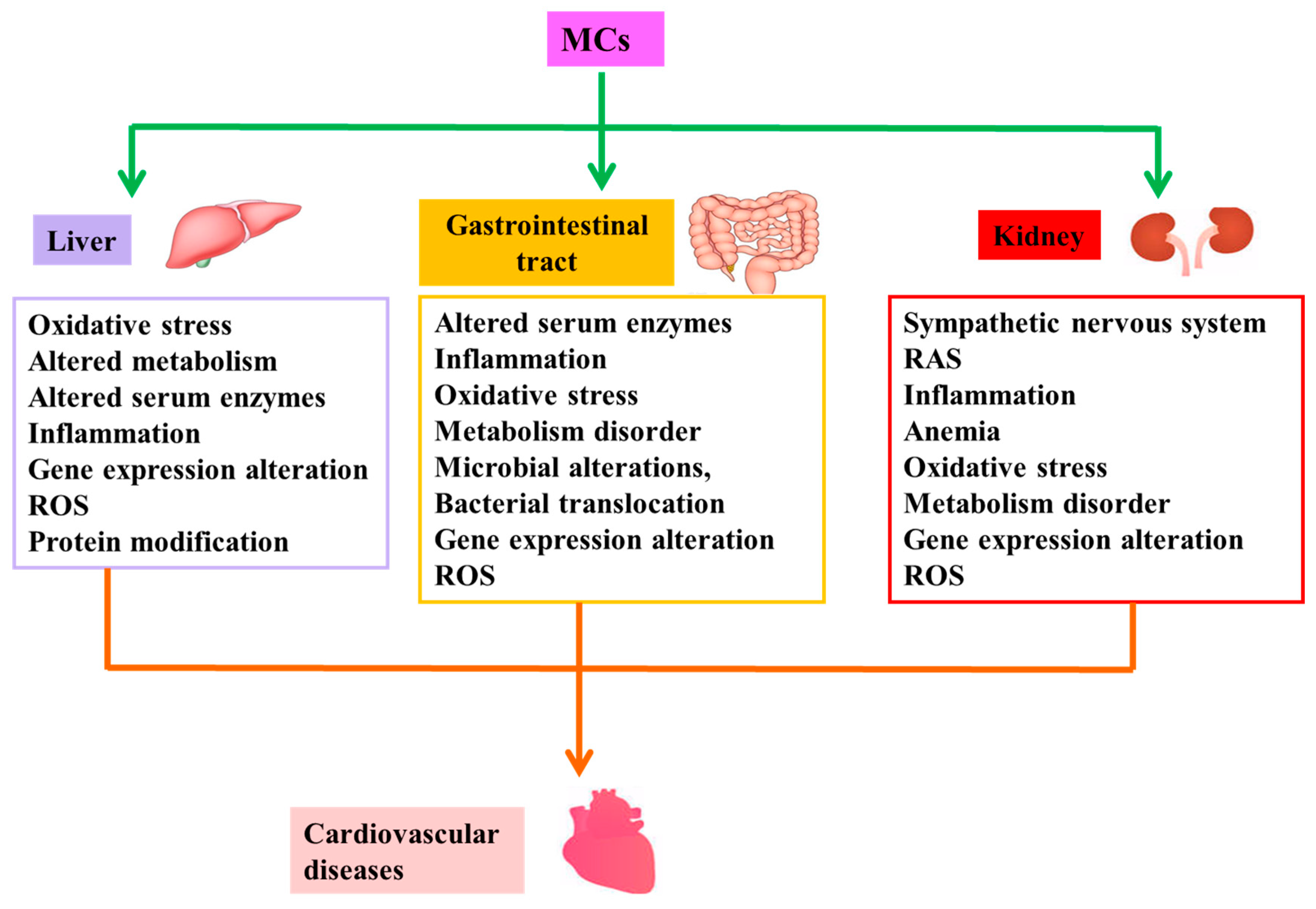
2.2. Indirect Cardiovascular Toxicity
2.2.1. Liver Diseases Induced by MCs and CVD
2.2.2. Intestinal Diseases Induced by MCs and CVD
2.2.3. Kidney Diseases Induced by MCs and CVD
3. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genom. 2014, 15, 390–399. [Google Scholar] [CrossRef]
- Hu, S.; Gao, R.; Liu, L.; Zhu, M.; Wang, W.; Wang, Y.; Wu, Z.; Li, H.; Gu, D.; Yang, Y.; et al. Summary of the 2018 Report on Cardiovascular Diseases in China. Chin. Circ. J. 2019, 34, 209. [Google Scholar]
- Merel, S.; Walker, D.; Chicana, R.; Snyder, S.; Baurès, E.; Thomas, O. State of knowledge and concerns on cyanobacterial blooms and cyanotoxins. Environ. Int. 2013, 59, 303–327. [Google Scholar] [CrossRef]
- Puddick, J.; Prinsep, M.R.; Wood, S.A.; Kaufononga, S.A.; Cary, S.C.; Hamilton, D.P. High levels of structural diversity observed in microcystins from Microcystis CAWBG11 and characterization of six new microcystin congeners. Mar. Drugs 2014, 12, 5372–5395. [Google Scholar] [CrossRef]
- Massey, I.Y.; Zhang, X.; Yang, F. Importance of bacterial biodegradation and detoxification processes of microcystins for environmental health. J. Toxicol. Environ. Health B 2018, 21, 357–369. [Google Scholar] [CrossRef]
- Yang, F.; Massey, I.Y.; Guo, J.; Yang, S.; Pu, Y.; Zeng, W.; Tan, H. Microcystin-LR degradation utilizing a novel effective indigenous bacterial community YFMCD1 from Lake Taihu. J. Toxicol. Environ Health A 2018, 81, 184–193. [Google Scholar] [CrossRef]
- Codd, G.A.; Morrison, L.F.; Metcalf, J.S. Cyanobacterial toxins: Risk management for health protection. Toxicol. Appl. Pharmacol. 2005, 203, 264–272. [Google Scholar] [CrossRef]
- Massey, I.Y.; Yang, F.; Ding, Z.; Yang, S.; Guo, J.; Tezi, C.; Al-Osman, M.; Kamegni, R.B.; Zeng, W. Exposure routes and health effects of microcystins on animals and humans: A mini-review. Toxicon 2018, 151, 156–162. [Google Scholar] [CrossRef]
- Yang, F.; Guo, J.; Huang, F.; Massey, I.Y.; Huang, R.; Li, Y.; Wen, C.; Ding, P.; Zeng, W.; Liang, G. Removal of Microcystin-LR by a Novel Native Effective Bacterial Community Designated as YFMCD4 Isolated from Lake Taihu. Toxins 2018, 10, 363. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Wen, C.; Zheng, S.; Yang, Y.; Feng, X.; Chen, J.; Luo, D.; Liu, R.; Yang, F. Regulation of Microcystin-LR-Induced DNA Damage by miR-451a in HL7702 Cells. Toxins 2019, 11, 164. [Google Scholar] [CrossRef]
- de la Cruz, A.A.; Antoniou, M.G.; Hiskia, A.; Pelaez, M.; Song, W.; O’Shea, K.E.; He, X.; Dionysiou, D.D. Can we effectively degrade microcystins?—Implications on human health. Anticancer Agents Med. Chem. 2011, 11, 19–37. [Google Scholar] [CrossRef]
- Hinojosa, M.G.; Gutierrez-Praena, D.; Prieto, A.I.; Guzman-Guillen, R.; Jos, A.; Camean, A.M. Neurotoxicity induced by microcystins and cylindrospermopsin: A review. Sci. Total Environ. 2019, 668, 547–565. [Google Scholar] [CrossRef]
- Spoof, L.; Catherine, A. Appendix 3. In Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; Meriluto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 526–537. [Google Scholar]
- Ufelmann, H.; Kruger, T.; Luckas, B.; Schrenk, D. Human and rat hepatocyte toxicity and protein phosphatase 1 and 2A inhibitory activity of naturally occurring desmethyl-microcystins and nodularins. Toxicology 2012, 293, 59–67. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fan, H.; Xie, P.; He, J. A review of neurotoxicity of microcystins. Environ. Sci. Pollut. Res. Int. 2016, 23, 7211–7219. [Google Scholar] [CrossRef]
- Hinojosa, M.G.; Prieto, A.I.; Gutierrez-Praena, D.; Moreno, F.J.; Camean, A.M.; Jos, A. Neurotoxic assessment of Microcystin-LR, cylindrospermopsin and their combination on the human neuroblastoma SH-SY5Y cell line. Chemosphere 2019, 224, 751–764. [Google Scholar] [CrossRef]
- Meriluoto, J.A.; Spoof, L.E. Cyanotoxins: Sampling, sample processing and toxin uptake. Adv. Exp. Med. Biol. 2008, 619, 483–499. [Google Scholar]
- Pearson, L.; Mihali, T.; Moffitt, M.; Kellmann, R.; Neilan, B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar. Drugs 2010, 8, 1650–1680. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.; Zhang, X.; Xie, P. A review of reproductive toxicity of microcystins. J. Hazard. Mater. 2016, 301, 381–399. [Google Scholar] [CrossRef]
- WHO. Cyanobacterial Toxins: Microcystin-LR. In Guidelines for Drinking Water Quality; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Yang, F.; Wen, C.; Zheng, S.; Yang, S.; Chen, J.; Feng, X. Involvement of MAPK/ERK1/2 pathway in microcystin-induced microfilament reorganization in HL7702 hepatocytes. J. Toxicol. Environ. Health Part A 2018, 81, 1135–1141. [Google Scholar] [CrossRef]
- Yang, S.; Chen, L.; Wen, C.; Zhang, X.; Feng, X.; Yang, F. MicroRNA expression profiling involved in MC-LR-induced hepatotoxicity using high-throughput sequencing analysis. J. Toxicol. Environ. Health A 2018, 81, 89–97. [Google Scholar] [CrossRef]
- Hou, J.; Li, L.; Xue, T.; Long, M.; Su, Y.; Wu, N. Hepatic positive and negative antioxidant responses in zebrafish after intraperitoneal administration of toxic microcystin-LR. Chemosphere 2015, 120, 729–736. [Google Scholar] [CrossRef]
- Chen, L.; Hu, Y.; He, J.; Chen, J.; Giesy, J.P.; Xie, P. Responses of the Proteome and Metabolome in Livers of Zebrafish Exposed Chronically to Environmentally Relevant Concentrations of Microcystin-LR. Environ. Sci. Technol. 2017, 51, 596–607. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Liu, F.; Hou, D.X.; Xu, J.; Zhao, X.; Yang, F.; Feng, X. Prodigiosin Promotes Nrf2 Activation to Inhibit Oxidative Stress Induced by Microcystin-LR in HepG2 Cells. Toxins 2019, 11, 403. [Google Scholar] [CrossRef]
- Wen, C.; Yang, S.; Zheng, S.; Feng, X.; Chen, J.; Yang, F. Analysis of long non-coding RNA profiled following MC-LR-induced hepatotoxicity using high-throughput sequencing. J. Toxicol. Environ. Health A 2018, 81, 1165–1172. [Google Scholar] [CrossRef]
- Piyathilaka, M.A.; Pathmalal, M.M.; Tennekoon, K.H.; De Silva, B.G.; Samarakoon, S.R.; Chanthirika, S. Microcystin-LR-induced cytotoxicity and apoptosis in human embryonic kidney and human kidney adenocarcinoma cell lines. Microbiology 2015, 161, 819–828. [Google Scholar] [CrossRef]
- Caban-Holt, A.; Mattingly, M.; Cooper, G.; Schmitt, F.A. Neurodegenerative memory disorders: A potential role of environmental toxins. Neurol. Clin. 2005, 23, 485–521. [Google Scholar] [CrossRef]
- Feurstein, D.; Kleinteich, J.; Heussner, A.H.; Stemmer, K.; Dietrich, D.R. Investigation of microcystin congener-dependent uptake into primary murine neurons. Environ. Health Perspect. 2010, 118, 1370–1375. [Google Scholar] [CrossRef]
- Ding, J.; Wang, J.; Xiang, Z.; Diao, W.; Su, M.; Shi, W.; Wan, T.; Han, X. The organic anion transporting polypeptide 1a5 is a pivotal transporter for the uptake of microcystin-LR by gonadotropin-releasing hormone neurons. Aquat. Toxicol. 2017, 182, 1–10. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, R.; Ye, J.; Wu, X.; Zhang, Y.; Wu, C. Monitoring and research of microcystins and environmental factors in a typical artificial freshwater aquaculture pond. Environ. Sci. Pollut. Res. Int. 2018, 25, 5921–5933. [Google Scholar] [CrossRef]
- Ito, E.; Kondo, F.; Harada, K. First report on the distribution of orally administered microcystin-LR in mouse tissue using an immunostaining method. Toxicon 2000, 38, 37–48. [Google Scholar] [CrossRef]
- Cao, L.; Huang, F.; Massey, I.Y.; Wen, C.; Zheng, S.; Xu, S.; Yang, F. Effects of Microcystin-LR on the Microstructure and Inflammation-Related Factors of Jejunum in Mice. Toxins 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, S.; Liu, C.; Wu, J.; Wang, Y.; Yuan, L.; Du, X.; Wang, R.; Marwa, P.W.; Zhuang, D. Resveratrol Ameliorates Microcystin-LR-Induced Testis Germ Cell Apoptosis in Rats via SIRT1 Signaling Pathway Activation. Toxins 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhuang, H.; Yang, H.; Xue, W.; Wang, L.; Wei, W. Microcystin-LR disturbs testicular development of giant freshwater prawn Macrobrachium rosenbergii. Chemosphere 2019, 222, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, P.; Tang, R.; Zhang, X.; Li, L.; Li, D. In vivo studies on the toxic effects of microcystins on mitochondrial electron transport chain and ion regulation in liver and heart of rabbit. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2008, 148, 204–210. [Google Scholar] [CrossRef]
- Zhang, Z.; Kang, S.; Chen, C.; Wei, G.; Yu, S. The acute toxic effects of microcystin LR in SD rats. Zhonghua Yu Fang Yi Xue Za Zhi 2002, 36, 295–297. [Google Scholar]
- Liu, Y.; Song, L.; Li, X.; Liu, T. The toxic effects of microcystin-LR on embryo-larval and juvenile development of loach, Misguruns mizolepis Gunthe. Toxicon 2002, 40, 395–399. [Google Scholar] [CrossRef]
- Milutinovic, A.; Zorc-Pleskovic, R.; Petrovic, D.; Zorc, M.; Suput, D. Microcystin-LR induces alterations in heart muscle. Folia Biol. (Praha) 2006, 52, 116–118. [Google Scholar]
- Suput, D.; Zorc-Pleskovic, R.; Petrovic, D.; Milutinovic, A. Cardiotoxic injury caused by chronic administration of microcystin-YR. Folia Biol. (Praha) 2010, 56, 14–18. [Google Scholar]
- Qiu, T.; Xie, P.; Liu, Y.; Li, G.; Xiong, Q.; Hao, L.; Li, H. The profound effects of microcystin on cardiac antioxidant enzymes, mitochondrial function and cardiac toxicity in rat. Toxicology 2009, 257, 86–94. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Guo, J.; Lin, S.; Wang, Y.; Yin, T.; Gregersen, H.; Hu, T.; Wang, G. Microcystin-LR induces angiodysplasia and vascular dysfunction through promoting cell apoptosis by the mitochondrial signaling pathway. Chemosphere 2019, 218, 438–448. [Google Scholar] [CrossRef]
- Martins, N.D.; Yunes, J.S.; McKenzie, D.J.; Rantin, F.T.; Kalinin, A.L.; Monteiro, D.A. Microcystin-LR exposure causes cardiorespiratory impairments and tissue oxidative damage in trahira, Hoplias malabaricus. Ecotoxicol. Environ. Saf. 2019, 173, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.R.; Frenkel, A.; Harke, M.J.; Jankowiak, J.G.; Gobler, C.J.; McElroy, A.E. Effects of Microcystis on development of early life stage Japanese medaka (Oryzias latipes): Comparative toxicity of natural blooms, cultured Microcystis and microcystin-LR. Aquat. Toxicol. 2018, 194, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Dang, Y.; Xu, Q.; Yu, L.; Liu, C.; Yuan, Y.; Wang, J. Microcystin-LR induced developmental toxicity and apoptosis in zebrafish (Danio rerio) larvae by activation of ER stress response. Chemosphere 2016, 157, 166–173. [Google Scholar] [CrossRef] [PubMed]
- LeClaire, R.D.; Parker, G.W.; Franz, D.R. Hemodynamic and calorimetric changes induced by microcystin-LR in the rat. J. Appl. Toxicol. 1995, 15, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, Y.; Xiao, W.; Ye, X.; Zhong, Q.; Gu, K. Comparison of response indices to toxic microcystin-LR in blood of mice. Chemosphere 2013, 92, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Palikova, M.; Ondrackova, P.; Mares, J.; Adamovsky, O.; Pikula, J.; Kohoutek, J.; Navratil, S.; Blaha, L.; Kopp, R. In vivo effects of microcystins and complex cyanobacterial biomass on rats (Rattus norvegicus var. alba): Changes in immunological and haematological parameters. Toxicon 2013, 73, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, X.; Jiang, S.; Men, C.; Xu, D.; Guo, Y.; Wu, J. Microcystin-LR regulates circadian clock and antioxidant gene expression in cultured rat cardiomyocytes. Cell. Mol. Biol. Lett. 2018, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, J.; Zhang, M. Microcystins Induces Vascular Inflammation in Human Umbilical Vein Endothelial Cells via Activation of NF-kappaB. Mediat. Inflamm. 2015, 2015, 942159. [Google Scholar] [CrossRef]
- Shi, J.; Deng, H.; Pan, H.; Xu, Y.; Zhang, M. Epigallocatechin-3-gallate attenuates microcystin-LR induced oxidative stress and inflammation in human umbilical vein endothelial cells. Chemosphere 2017, 168, 25–31. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, M.; Zhang, L.; Deng, H. Epigallocatechin-3-gallate attenuates microcystin-LR-induced apoptosis in human umbilical vein endothelial cells through activation of the NRF2/HO-1 pathway. Environ. Pollut. 2018, 239, 466–472. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, H.; Zhang, X. Erythrocyte damage of crucian carp (Carassius auratus) caused by microcystin-LR: In vitro study. Fish Physiol. Biochem. 2012, 38, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Krag, A.; Bendtsen, F.; Mortensen, C.; Henriksen, J.H.; Moller, S. Effects of a single terlipressin administration on cardiac function and perfusion in cirrhosis. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.H.; Gotze, J.P.; Fuglsang, S.; Christensen, E.; Bendtsen, F.; Moller, S. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: Relation to cardiovascular dysfunction and severity of disease. Gut 2003, 52, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Haapamaki, J.; Roine, R.P.; Turunen, U.; Farkkila, M.A.; Arkkila, P.E. Increased risk for coronary heart disease, asthma, and connective tissue diseases in inflammatory bowel disease. J. Crohns Colitis 2011, 5, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Chen, G.; Cai, D.; Zhao, S.; Cheng, J.; Shen, H. Inflammatory Bowel Disease and Risk of Ischemic Heart Disease: An Updated Meta-Analysis of Cohort Studies. J. Am. Heart Assoc. 2017, 6, e005892. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Benedetto, F.A.; Mallamaci, F.; Tripepi, G.; Giacone, G.; Cataliotti, A.; Seminara, G.; Stancanelli, B.; Malatino, L.S. Prognostic value of echocardiographic indicators of left ventricular systolic function in asymptomatic dialysis patients. J. Am. Soc. Nephrol. 2004, 15, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Dubin, R.F.; Deo, R.; Bansal, N.; Anderson, A.H.; Yang, P.; Go, A.S.; Keane, M.; Townsend, R.; Porter, A.; Budoff, M. Associations of Conventional Echocardiographic Measures with Incident Heart Failure and Mortality: The Chronic Renal Insufficiency Cohort. Clin. J. Am. Soc. Nephrol. 2017, 12, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.P.; Chandra, A.; Navaneethan, S.D. Blood pressure parameters and morbid and mortal outcomes in nondialysis-dependent chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 16–22. [Google Scholar] [CrossRef]
- Hassan, M.O.; Duarte, R.; Dix-Peek, T.; Vachiat, A.; Naidoo, S.; Dickens, C.; Grinter, S.; Manga, P.; Naicker, S. Correlation between volume overload, chronic inflammation, and left ventricular dysfunction in chronic kidney disease patients. Clin. Nephrol. 2016, 86, 131–135. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kimura, T.; Kuragano, T. The Hepcidin-Anemia Axis: Pathogenesis of Anemia in Chronic Kidney Disease. Contrib. Nephrol. 2019, 198, 124–134. [Google Scholar]
- Steiner, K.; Zimmermann, L.; Hagenbuch, B.; Dietrich, D. Zebrafish Oatp-mediated transport of microcystin congeners. Arch. Toxicol. 2016, 90, 1129–1139. [Google Scholar] [CrossRef]
- Hausner, E.A.; Elmore, S.A.; Yang, X. Overview of the Components of Cardiac Metabolism. Drug Metab. Dispos. 2019, 47, 673–688. [Google Scholar] [CrossRef]
- Fujiwara, K.; Adachi, H.; Nishio, T.; Unno, M.; Tokui, T.; Okabe, M.; Onogawa, T.; Suzuki, T.; Asano, N.; Tanemoto, M. Identification of thyroid hormone transporters in humans: Different molecules are involved in a tissue-specific manner. Endocrinology 2001, 142, 2005–2012. [Google Scholar] [CrossRef]
- Lu, R.; Kanai, N.; Bao, Y.; Schuster, V.L. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J. Clin. Investig. 1996, 98, 1142–1149. [Google Scholar] [CrossRef]
- Grube, M.; Kock, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Bohm, M.; Felix, S.B.; Vogelgesang, S.; Jedlitschky, G. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar] [CrossRef]
- Huber, R.D.; Gao, B.; Sidler Pfandler, M.A.; Zhang-Fu, W.; Leuthold, S.; Hagenbuch, B.; Folkers, G.; Meier, P.J.; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Cell Physiol. 2007, 292, C795–C806. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, P.; Chen, J.; Liang, G. Distribution of microcystins in various organs (heart, liver, intestine, gonad, brain, kidney and lung) of Wistar rat via intravenous injection. Toxicon 2008, 52, 721–727. [Google Scholar] [CrossRef]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef]
- Varga, I.; Kyselovic, J.; Galfiova, P.; Danisovic, L. The Non-cardiomyocyte Cells of the Heart. Their Possible Roles in Exercise-Induced Cardiac Regeneration and Remodeling. Adv. Exp. Med. Biol. 2017, 999, 117–136. [Google Scholar]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N.; Li, T.; Kumar, D. Adriamycin cardiomyopathy: Pathophysiology and prevention. FASEB J. 1997, 11, 931–936. [Google Scholar] [CrossRef]
- Xu, M.F.; Tang, P.L.; Qian, Z.M.; Ashraf, M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci. 2001, 68, 889–901. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef]
- Moreno, I.M.; Mate, A.; Repetto, G.; Vazquez, C.M.; Camean, A.M. Influence of microcystin-LR on the activity of membrane enzymes in rat intestinal mucosa. J. Physiol. Biochem. 2003, 59, 293–299. [Google Scholar] [CrossRef]
- Moreno, I.; Pichardo, S.; Jos, A.; Gomez-Amores, L.; Mate, A.; Vazquez, C.M.; Camean, A.M. Antioxidant enzyme activity and lipid peroxidation in liver and kidney of rats exposed to microcystin-LR administered intraperitoneally. Toxicon 2005, 45, 395–402. [Google Scholar] [CrossRef]
- Botha, N.; van de Venter, M.; Downing, T.G.; Shephard, E.G.; Gehringer, M.M. The effect of intraperitoneally administered microcystin-LR on the gastrointestinal tract of Balb/c mice. Toxicon 2004, 43, 251–254. [Google Scholar] [CrossRef]
- Ding, W.X.; Shen, H.M.; Ong, C.N. Critical role of reactive oxygen species formation in microcystin-induced cytoskeleton disruption in primary cultured hepatocytes. J. Toxicol. Environ. Health A 2001, 64, 507–519. [Google Scholar] [CrossRef]
- Ding, W.X.; Nam Ong, C. Role of oxidative stress and mitochondrial changes in cyanobacteria-induced apoptosis and hepatotoxicity. FEMS Microbiol. Lett. 2003, 220, 1–7. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef]
- Choy, K.W.; Murugan, D.; Mustafa, M.R. Natural products targeting ER stress pathway for the treatment of cardiovascular diseases. Pharmacol. Res. 2018, 132, 119–129. [Google Scholar] [CrossRef]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef]
- Kim, A.J.; Shi, Y.; Austin, R.C.; Werstuck, G.H. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J. Cell Sci. 2005, 118, 89–99. [Google Scholar] [CrossRef]
- Takada, A.; Miki, T.; Kuno, A.; Kouzu, H.; Sunaga, D.; Itoh, T.; Tanno, M.; Yano, T.; Sato, T.; Ishikawa, S. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PLoS ONE 2012, 7, e39893. [Google Scholar] [CrossRef]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef]
- Young, C.N.; Cao, X.; Guruju, M.R.; Pierce, J.P.; Morgan, D.A.; Wang, G.; Iadecola, C.; Mark, A.L.; Davisson, R.L. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J. Clin. Investig. 2012, 122, 3960–3964. [Google Scholar] [CrossRef]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.A.; Gotteland, J.P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef]
- Cai, F.; Liu, J.; Li, C.; Wang, J. Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR. Int. J. Mol. Sci. 2015, 16, 28077–28086. [Google Scholar] [CrossRef]
- Cai, F.; Liu, J.; Li, C.; Wang, J. Intracellular Calcium Plays a Critical Role in the Microcystin-LR-Elicited Neurotoxicity through PLC/IP3 Pathway. Int. J. Toxicol. 2015, 34, 551–558. [Google Scholar] [CrossRef]
- Zhao, S.; Li, G.; Chen, J. A proteomic analysis of prenatal transfer of microcystin-LR induced neurotoxicity in rat offspring. J. Proteom. 2015, 114, 197–213. [Google Scholar] [CrossRef]
- Zhou, M.; Tu, W.W.; Xu, J. Mechanisms of microcystin-LR-induced cytoskeletal disruption in animal cells. Toxicon 2015, 101, 92–100. [Google Scholar] [CrossRef]
- Lubbers, E.R.; Mohler, P.J. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J. Mol. Cell. Cardiol. 2016, 101, 127–133. [Google Scholar] [CrossRef]
- Xu, H.; Ginsburg, K.S.; Hall, D.D.; Zimmermann, M.; Stein, I.S.; Zhang, M.; Tandan, S.; Hill, J.A.; Horne, M.C.; Bers, D. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1.2. Biochemistry 2010, 49, 10298–10307. [Google Scholar] [CrossRef]
- Bhasin, N.; Cunha, S.R.; Mudannayake, M.; Gigena, M.S.; Rogers, T.B.; Mohler, P.J. Molecular basis for PP2A regulatory subunit ubunit B56alpha targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H109–H119. [Google Scholar] [CrossRef]
- Meyer-Roxlau, S.; Lammle, S.; Opitz, A.; Kunzel, S.; Joos, J.P.; Neef, S.; Sekeres, K.; Sossalla, S.; Schondube, F.; Alexiou, K. Differential regulation of protein phosphatase 1 (PP1) isoforms in human heart failure and atrial fibrillation. Basic Res. Cardiol. 2017, 112, 43. [Google Scholar] [CrossRef]
- Fontanillo, M.; Kohn, M. Microcystins: Synthesis and structure-activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef]
- Heijman, J.; Ghezelbash, S.; Wehrens, X.H.; Dobrev, D. Serine/Threonine Phosphatases in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2017, 103, 110–120. [Google Scholar] [CrossRef]
- Hall, D.D.; Feekes, J.A.; Arachchige Don, A.S.; Shi, M.; Hamid, J.; Chen, L.; Strack, S.; Zamponi, G.W.; Horne, M.C.; Hell, J.W. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 2006, 45, 3448–3459. [Google Scholar] [CrossRef]
- Kimura, T.; Han, W.; Pagel, P.; Nairn, A.C.; Caplan, M.J. Protein phosphatase 2A interacts with the Na,K-ATPase and modulates its trafficking by inhibition of its association with arrestin. PLoS ONE 2011, 6, e29269. [Google Scholar] [CrossRef]
- Lecuona, E.; Dada, L.A.; Sun, H.; Butti, M.L.; Zhou, G.; Chew, T.L.; Sznajder, J.I. Na,K-ATPase alpha1-subunit dephosphorylation by protein phosphatase 2A is necessary for its recruitment to the plasma membrane. FASEB J. 2006, 20, 2618–2620. [Google Scholar] [CrossRef]
- DeGrande, S.T.; Little, S.C.; Nixon, D.J.; Wright, P.; Snyder, J.; Dun, W.; Murphy, N.; Kilic, A.; Higgins, R.; Binkley, P.F. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J. Biol. Chem. 2013, 288, 1032–1046. [Google Scholar] [CrossRef]
- Gergs, U.; Boknik, P.; Buchwalow, I.; Fabritz, L.; Matus, M.; Justus, I.; Hanske, G.; Schmitz, W.; Neumann, J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 2004, 279, 40827–40834. [Google Scholar] [CrossRef]
- Hoehn, M.; Zhang, Y.; Xu, J.; Gergs, U.; Boknik, P.; Werdan, K.; Neumann, J.; Ebelt, H. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of beta-catenin by glycogen synthase kinase 3beta. Int. J. Cardiol. 2015, 183, 39–46. [Google Scholar] [CrossRef]
- Baskaran, R.; Velmurugan, B.K. Protein phosphatase 2A as therapeutic targets in various disease models. Life Sci. 2018, 210, 40–46. [Google Scholar] [CrossRef]
- Weber, S.; Meyer-Roxlau, S.; Wagner, M.; Dobrev, D.; El-Armouche, A. Counteracting Protein Kinase Activity in the Heart: The Multiple Roles of Protein Phosphatases. Front. Pharmacol. 2015, 6, 270. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Zhong, T.P. Zebrafish genetics and formation of embryonic vasculature. Curr. Top. Dev. Biol. 2005, 71, 53–81. [Google Scholar]
- Curry, F.R.; Adamson, R.H. Vascular permeability modulation at the cell, microvessel, or whole organ level: Towards closing gaps in our knowledge. Cardiovasc. Res. 2010, 87, 218–229. [Google Scholar] [CrossRef]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Sturtzel, C. Endothelial Cells. In The Immunology of Cardiovascular Homeostasis and Pathology; Springer: Berlin, Germany, 2017; pp. 71–91. [Google Scholar]
- Komatsu, M.; Furukawa, T.; Ikeda, R.; Takumi, S.; Nong, Q.; Aoyama, K.; Akiyama, S.; Keppler, D.; Takeuchi, T. Involvement of mitogen-activated protein kinase signaling pathways in microcystin-LR-induced apoptosis after its selective uptake mediated by OATP1B1 and OATP1B3. Toxicol. Sci. 2007, 97, 407–416. [Google Scholar] [CrossRef]
- Falconer, I.R.; Yeung, D.S. Cytoskeletal changes in hepatocytes induced by microcystis toxins and their relation to hyperphosphorilation of cell proteins. Chem. Biol. Interact. 1992, 81, 181–196. [Google Scholar] [CrossRef]
- Strack, S.; Cribbs, J.T.; Gomez, L. Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J. Biol. Chem. 2004, 279, 47732–47739. [Google Scholar] [CrossRef]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef]
- Weng, D.; Lu, Y.; Wei, Y.; Liu, Y.; Shen, P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology 2007, 232, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.M.; Govender, S.; Shah, M.; Downing, T.G. An investigation of the role of vitamin E in the protection of mice against microcystin toxicity. Environ. Toxicol. 2003, 18, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.; Anand, T.; Rao, P.V. Activity and gene expression profile of certain antioxidant enzymes to microcystin-LR induced oxidative stress in mice. Toxicology 2006, 220, 136–146. [Google Scholar] [CrossRef]
- Gehringer, M.M.; Shephard, E.G.; Downing, T.G.; Wiegand, C.; Neilan, B.A. An investigation into the detoxification of microcystin-LR by the glutathione pathway in Balb/c mice. Int. J. Biochem. Cell Biol. 2004, 36, 931–941. [Google Scholar] [CrossRef]
- Mellinger, J.L.; Pencina, K.M.; Massaro, J.M.; Hoffmann, U.; Seshadri, S.; Fox, C.S.; O’Donnell, C.J.; Speliotes, E.K. Hepatic steatosis and cardiovascular disease outcomes: An analysis of the Framingham Heart Study. J. Hepatol. 2015, 63, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol. 2008, 49, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Oni, E.T.; Agatston, A.S.; Blaha, M.J. A systematic review: Burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis 2013, 230, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, S.; Perseghin, G.; Molon, G. Nonalcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in patients with type 2 diabetes. Diabetes Care 2012, 35, 389–395. [Google Scholar] [CrossRef] [PubMed]
- VanWagner, L.B.; Wilcox, J.E.; Colangelo, L.A. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: A population-based study. Hepatology 2015, 62, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Hallsworth, K.; Hollingsworth, K.G.; Thoma, C. Cardiac structure and function are altered in adults with non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 757–762. [Google Scholar] [CrossRef]
- Boddi, M.; Tarquini, R.; Chiostri, M. Nonalcoholic fatty liver in nondiabetic patients with acute coronary syndromes. Eur. J. Clin. Investig. 2013, 43, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.K.; Honar, H.; Liu, H.; ter Keurs, H.E.; Lee, S.S. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J. Hepatol. 2011, 55, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, A.M.; Barents, M.; de Jongste, M.J.; Romer, J.W.; Steward, R.N.; Muskiet, F.A. High Intraindividual Variation of N-Terminal Pro-B-Type Natriuretic Peptide in Urine of Patients with Stable Chronic Heart Failure: Comparison with Plasma. Clin. Chem. 2016, 62, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zheng, X.L. Correlation of serum brain natriuretic peptide level, severity and cardiac function of liver disease in patients with hepatitis C viral cirrhosis and nonalcoholic fatty liver. Modern Med. 2016, 44, 1675–1680. [Google Scholar]
- Kao, M.P.; Ang, D.S.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, J.; Feng, Y.; Zhang, J.; Cui, L.; Zhang, H.; Bai, Y. Extracellular acidosis suppresses calcification of vascular smooth muscle cells by inhibiting calcium influx via L-type calcium channels. Clin. Exp. Hypertens. 2018, 40, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Falconer, I.R.; Dornbusch, M.; Moran, G.; Yeung, S.K. Effect of the cyanobacterial (blue-green algal) toxins from Microcystis aeruginosa on isolated enterocytes from the chicken small intestine. Toxicon 1992, 30, 790–793. [Google Scholar] [CrossRef]
- Botha, N.; Gehringer, M.M.; Downing, T.G.; van de Venter, M.; Shephard, E.G. The role of microcystin-LR in the induction of apoptosis and oxidative stress in CaCo2 cells. Toxicon 2004, 43, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, X.; Yu, B.; Yu, G. Characterization of in vitro effects of microcystin-LR on intestinal epithelial cells. Environ. Toxicol. 2017, 32, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, J.; Huet, S.; Jarry, G.; Fessard, V. In vivo DNA damage induced by the cyanotoxin microcystin-LR: Comparison of intra-peritoneal and oral administrations by use of the comet assay. Mutat. Res. 2008, 652, 65–71. [Google Scholar] [CrossRef]
- Nobre, A.C.L.; Nunes-Monteiro, S.M.; Monteiro, M.C.; Martins, A.M.; Havt, A.; Barbosa, P.S.; Lima, A.A.; Monteiro, H.S. Microcystin-LR promote intestinal secretion of water and electrolytes in rats. Toxicon 2004, 44, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Huguet, A.; Henri, J.; Petitpas, M.; Hogeveen, K.; Fessard, V. Comparative cytotoxicity, oxidative stress, and cytokine secretion induced by two cyanotoxin variants, microcystin LR and RR, in human intestinal Caco-2 cells. J. Biochem. Mol. Toxicol. 2013, 27, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Pichardo, S.; Jos, A.; Camean, A.M. Microcystin-LR induces toxic effects in differentiated and undifferentiated Caco-2 cells. Arch. Toxicol. 2010, 84, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, C.; Zhang, T.; Liu, W.; Wang, L.; Chen, Y.; Wu, L.; Hegazy, A.M.; El-Sayed, A.F.; Zhang, X. muEvaluation of microcystin-LR absorption using an in vivo intestine model and its effect on zebrafish intestine. Aquat. Toxicol. 2019, 206, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Kondo, F.; Harada, K. Hepatic necrosis in aged mice by oral administration of microcystin-LR. Toxicon 1997, 35, 231–239. [Google Scholar] [CrossRef]
- Lin, J.; Chen, J.; He, J.; Chen, J.; Yan, Q.; Zhou, J.; Xie, P. Effects of microcystin-LR on bacterial and fungal functional genes profile in rat gut. Toxicon 2015, 96, 50–56. [Google Scholar] [CrossRef]
- Chen, J.; Xie, P.; Lin, J.; He, J.; Zeng, C.; Chen, J. Effects of microcystin-LR on gut microflora in different gut regions of mice. J. Toxicol. Sci. 2015, 40, 485–494. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Anker, S.D.; Egerer, K.R.; Volk, H.D.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am. J. Cardiol. 1997, 79, 1426–1430. [Google Scholar] [CrossRef]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M. Altered intestinal function in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef]
- Zhao, L.H.; Lu, C.L.; Sun, H.T. New progress in the study of the relationship between intestinal flora and mucous membrane immunity and cardiovascular disease. Chin. J. Immunol. 2018, 34, 1433–1436. [Google Scholar]
- Long, M.T.; Ko, D.; Arnold, L.M.; Trinquart, L.; Sherer, J.A.; Keppel, S.S.; Benjamin, E.J.; Helm, R.H. Gastrointestinal and liver diseases and atrial fibrillation: A review of the literature. Ther. Adv. Gastroenterol. 2019, 12, 1756284819832237. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.A.; Pace, J.G.; Matson, C.F.; Miura, G.A.; Lawrence, W.B. Tissue distribution, excretion and hepatic biotransformation of microcystin-LR in mice. J. Pharmacol. Exp. Ther. 1991, 256, 176–182. [Google Scholar] [PubMed]
- Hagenbuch, B.; Meier, P.J. The superfamily of organic anion transporting polypeptides. Biochim. Biophys. Acta 2003, 1609, 1–18. [Google Scholar] [CrossRef]
- Menezes, C.; Valerio, E.; Dias, E. The Kidney Bero-E6 Cell Line: A Suitable Model to Study the Toxicity of Microcystins. In New Insights into Toxicity and Drug Testing; IntechOpen: London, UK, 2013; pp. 29–48. [Google Scholar]
- Jia, J.; Luo, W.; Lu, Y.; Giesy, J.P. Bioaccumulation of microcystins (MCs) in four fish species from Lake Taihu, China: Assessment of risks to humans. Sci. Total Environ. 2014, 487, 224–232. [Google Scholar] [CrossRef]
- Wang, Z.; Li, G.; Wu, Q.; Liu, C.; Shen, J.; Yan, W. Microcystin-LR exposure induced nephrotoxicity by triggering apoptosis in female zebrafish. Chemosphere 2019, 214, 598–605. [Google Scholar] [CrossRef]
- McLellan, N.L.; Manderville, R.A. Toxic mechanisms of microcystins in mammals. Toxicol. Res. 2017, 6, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Dias, E.; Matos, P.; Pereira, P.; Batoreu, M.C.; Silva, M.J.; Jordan, P. Microcystin-LR activates the ERK1/2 kinases and stimulates the proliferation of the monkey kidney-derived cell line Vero-E6. Toxicol. In Vitro 2010, 24, 1689–1695. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Animals | Exposure | Toxicant | Dose | Time Point | Toxic Effects | References |
|---|---|---|---|---|---|---|
| Rabbits | I.P | MCs (mainly containing MC-RR, MC-LR | 12.5, 50 μg/kg | 1, 3, 12, 24, 48 h | Damage of mitochondrial morphology, lipid peroxidation ↑, SDH ↑, Ca2+-Mg2+-ATPase activities of mitochondria ↓, disrupted ionic homeostasis, MMP ↓ | [36] |
| SD rats | I.P | MC-LR | 122 μg/kg | 1, 7, 14 day | Myocardial cells damage including pyknosis, plasma dissolve and myofibrilla necrosis, LDH ↑, AST ↑, CK ↑ | [37] |
| loach, Misguruns mizolepis Gunthe embryos | orally | MC-LR | 1, 3, 10, 100, 1000 μg/L | 7 days | Pericardial edema, tubular heart and bradycardia | [38] |
| Wistar rats | I.P | MC-LR | 10 μg/kg | every two days for 8 months | Destruction cytoskeletons of cardiomyocytes, loss of cell crossstriations, myofibril volume fraction ↓, cardiomyocytes volume ↑,myofibrillar volume ↓, even fibrosis, infiltration of mononuclear in the interstitial tissue | [39] |
| Male Wistar rats | I.P | MC-YR | 10 μg/kg | every two days for 8 months | Volume density of cardiac muscle tissue ↓, fibrous proliferation, lymphocyte infiltration, enlarged and often bizarre-shaped nuclei, myofibril volume fraction ↓ | [40] |
| Male Wistar rats | I.P | MC-LR | 0.16 LD50 (14 g/kg), 1 LD50 (87 g/kg) | 24 h | Myocardial infarction in dead rats, CK ↑, troponin I ↑, GSH ↑, lipid peroxides ↑, antioxidant enzymes ↑, heart rate ↓, blood pressure ↓, loss of adhesion between cardiomyocytes, swelling or rupture of mitochondria, complex I/III ↓, electron flow along the mitochondrial respiratory chain ↓ | [41] |
| Zebrafish | orally | MC-LR | 0.1, 1 μM | 24 h | Angiodysplasia, damaged vascular structures, lumen size ↓, blood flow ↓, vascular dysfunction | [42] |
| Trahira, hoplias malabaricus | orally | MC-LR | 100 μg/kg | 48 h | GPx ↑, GSH ↓ | [43] |
| Japanese medaka (Oryzias latipes) | orally | MC-LR | 600, 6300 μg/L | 15 days | Heart rate ↓, bradycardia | [44] |
| zebrafish juvenile fish | orally | MC-LR | 4.0 mM | 96h | Heart rate ↓, apoptosis ↑, morphological deformity, stunted growth | [45] |
| Male Fischer 344 Rats | orally | MC-LR | 100 μg/kg | Cardiac output and stroke volume ↓, an acute hypotension responsive to volume, expansion with whole blood; heart rate ↓, oxygen consumption ↓, carbon dioxide production ↓, metabolic rate ↓, progressive hypothermia, acid-base balance changes | [46] | |
| KM (Kunming) male mice | I.P | MC-LR | 0, 3.125, 6.25, 12.5, 25 μg/kg/day | 7 days | ALP ↑, LDH ↑, AST ↑, ALT ↑, RBC↓, HGB↓, HCT ↓, MCV -, MCH -, MCHC -, WBC ↑, Mon ↑, Gran ↑ | [47] |
| Male Wistar albino rats | orally | MCs (mainly containing MC-LR, MC-YR, MC-RR, MC-LF, MC-LW) | 25,000 μg/kg of food | 28 days | RBC ↓, MCV ↑, MCH ↓, MCHC ↑ | [48] |
| Cells | Toxicant | Dose | Time Point | Toxic Effects | References |
|---|---|---|---|---|---|
| H9C2 (rat cardiomyocytes) | MC-LR | 10 μM | 0, 4, 8, 12, 16, 20, 24 h | Expression levels of rhythmic genes ↓, antioxidant genes ↑ | [49] |
| Human umbilical vein endothelial cells (HUVECs) | MC-LR | 0.1, 1 μM | 24 h | Apoptosis ↑, caspase3/9 ↑, mitochondrial ROS ↑, MMP ↓, p53 ↑, PCNA ↓ | [42] |
| HUVECs | MC-LR | 40 μM | 24 h | Proliferation ↓, apoptosis ↑, migration ↓, capillary-like structure formation ↓, ROS ↑, NF-κB ↑, TNF-α ↑, VCAM-1 ↑, ICAM-1 ↑ | [50] |
| HUVECs | MC-LR | 40 μM | 24 h | Cell death ↑, cell viability ↓, migration ↓, tube formation ↓, ROS ↑, NF-κB ↑, TNF-α ↑, IL6 ↑, SOD ↓, GSH ↓, MDA ↑ | [51] |
| HUVECs | MC-LR | 40 μM | 24 h | MMP ↓, decreased levels of cytochrome c ↓, caspase-3/-9 ↑, ROS ↑, apoptosis ↑, NRF 2/ HO 1 ↓ | [52] |
| Crucian carp erythrocytes | MC-LR | 0, 1, 10, 100, 1000 nM. | 1, 3, 12, 24, 48 h | GSH ↑, SOD ↑, CAT ↑, GPx ↑, GST ↑, GLT ↓, lipid peroxidate ↑, hemolysis ↑, AchE ↓, Ca2+-Mg2+-ATPase ↓, Na2+-k+-ATPase ↓ | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Massey, I.Y.; Feng, H.; Yang, F. A Review of Cardiovascular Toxicity of Microcystins. Toxins 2019, 11, 507. https://doi.org/10.3390/toxins11090507
Cao L, Massey IY, Feng H, Yang F. A Review of Cardiovascular Toxicity of Microcystins. Toxins. 2019; 11(9):507. https://doi.org/10.3390/toxins11090507
Chicago/Turabian StyleCao, Linghui, Isaac Yaw Massey, Hai Feng, and Fei Yang. 2019. "A Review of Cardiovascular Toxicity of Microcystins" Toxins 11, no. 9: 507. https://doi.org/10.3390/toxins11090507
APA StyleCao, L., Massey, I. Y., Feng, H., & Yang, F. (2019). A Review of Cardiovascular Toxicity of Microcystins. Toxins, 11(9), 507. https://doi.org/10.3390/toxins11090507