Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats


Abstract
1. Introduction
2. Results and Discussion
2.1. Organ Weights, Body Weights, and Clinical Chemistry
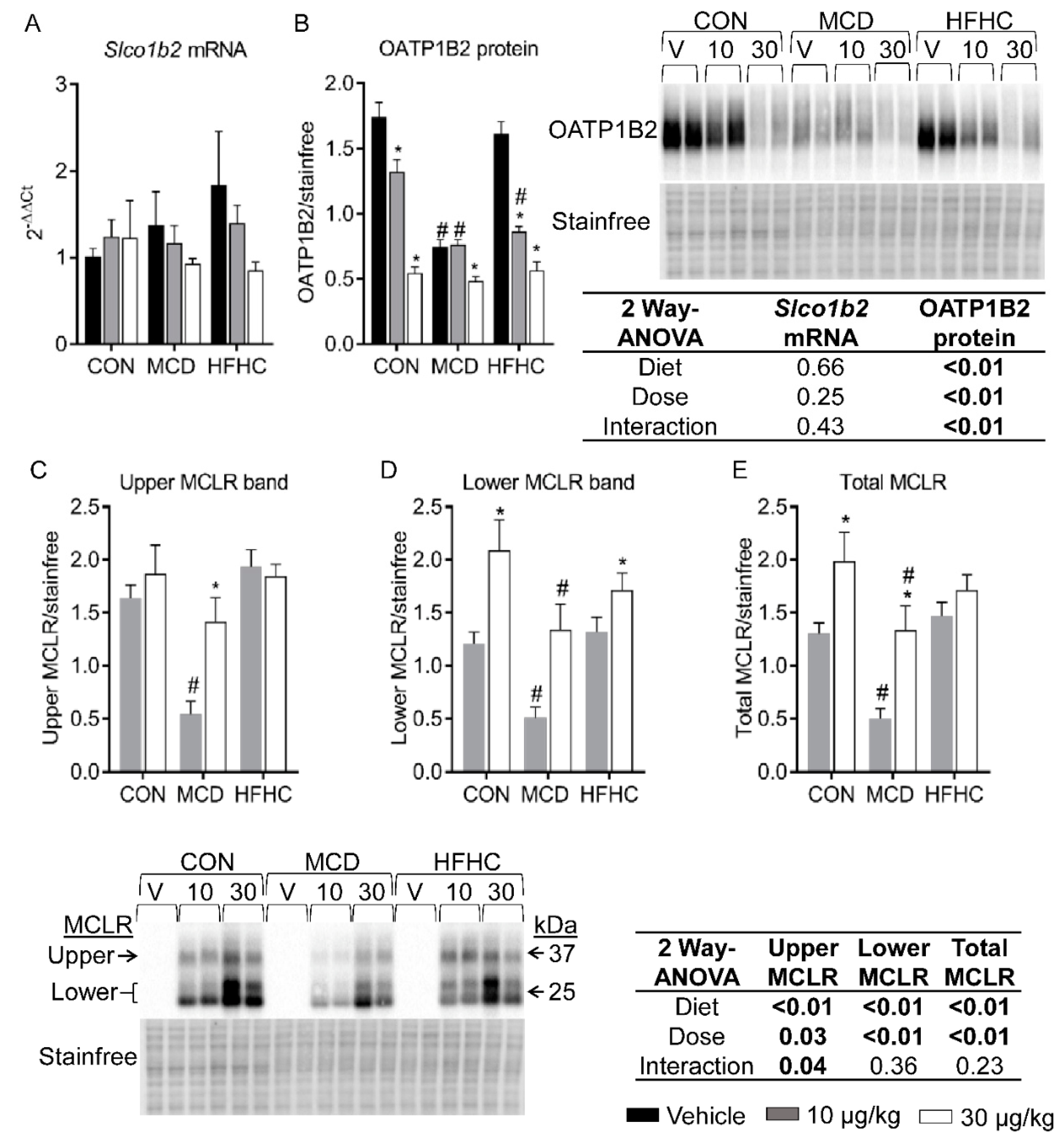
2.2. OATP1B2 and MCLR Western Blots
2.3. Lipid Homeostasis
2.3.1. Steatosis
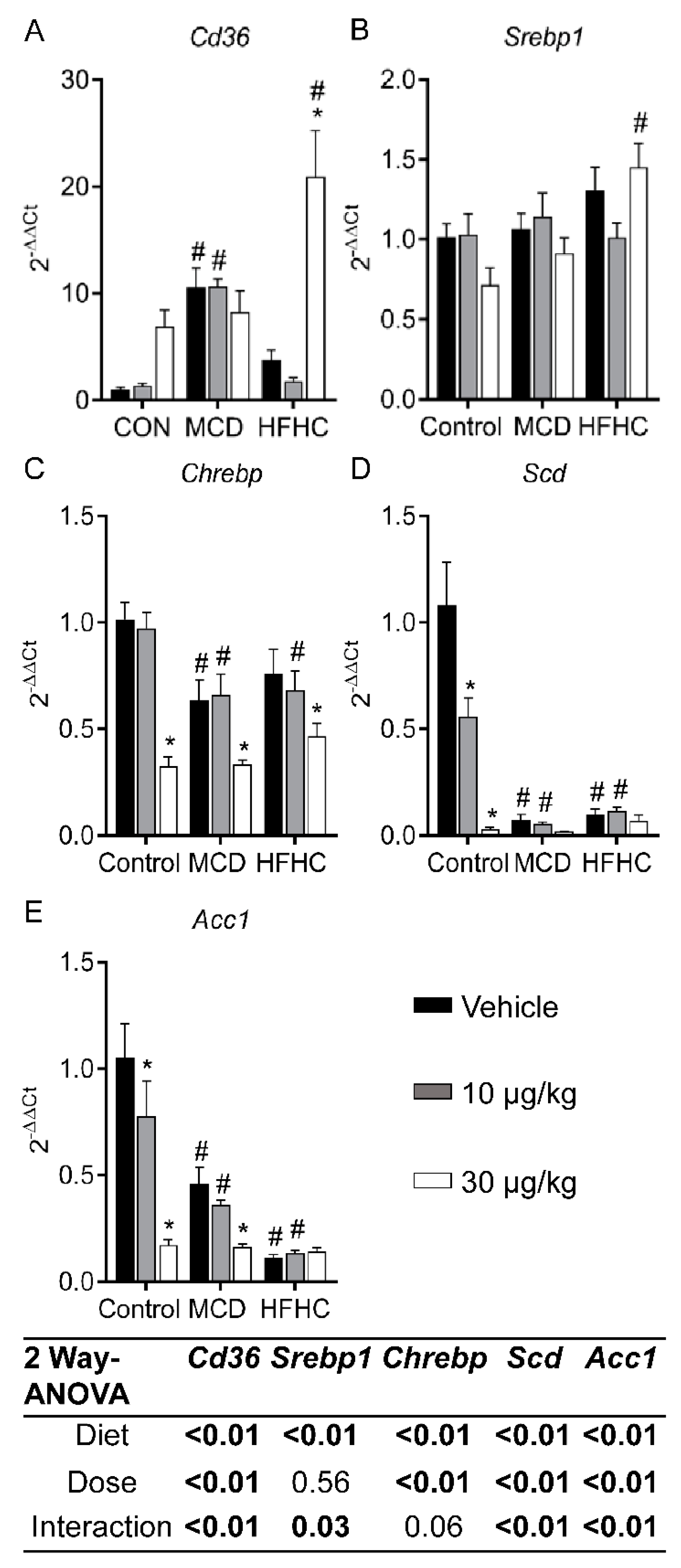
2.3.2. Fatty Acid Uptake and De Novo Lipogenesis
2.3.3. Fatty Acid Oxidation Genes
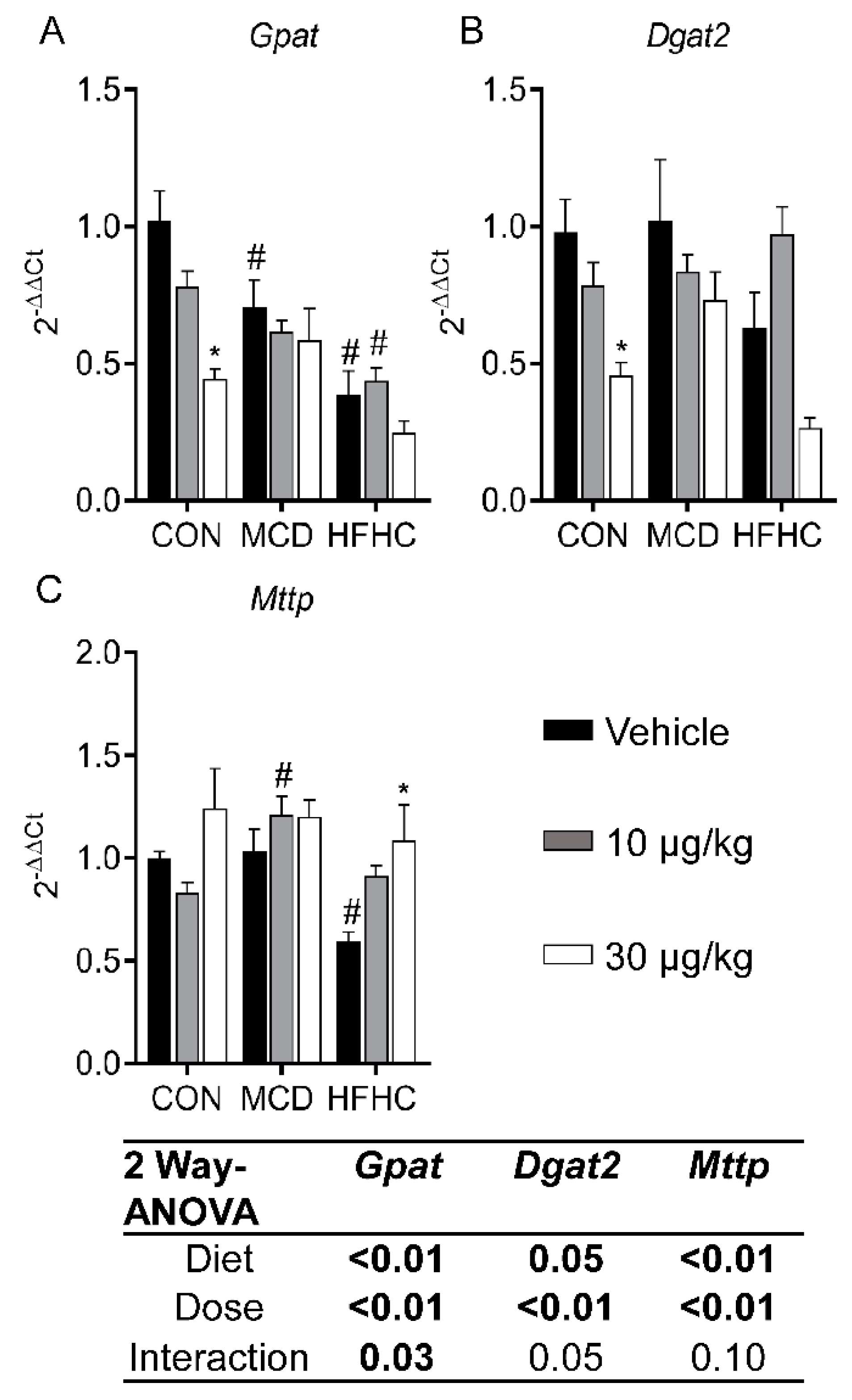
2.3.4. Fatty Acid Esterification and Lipid Export
2.4. Inflammation
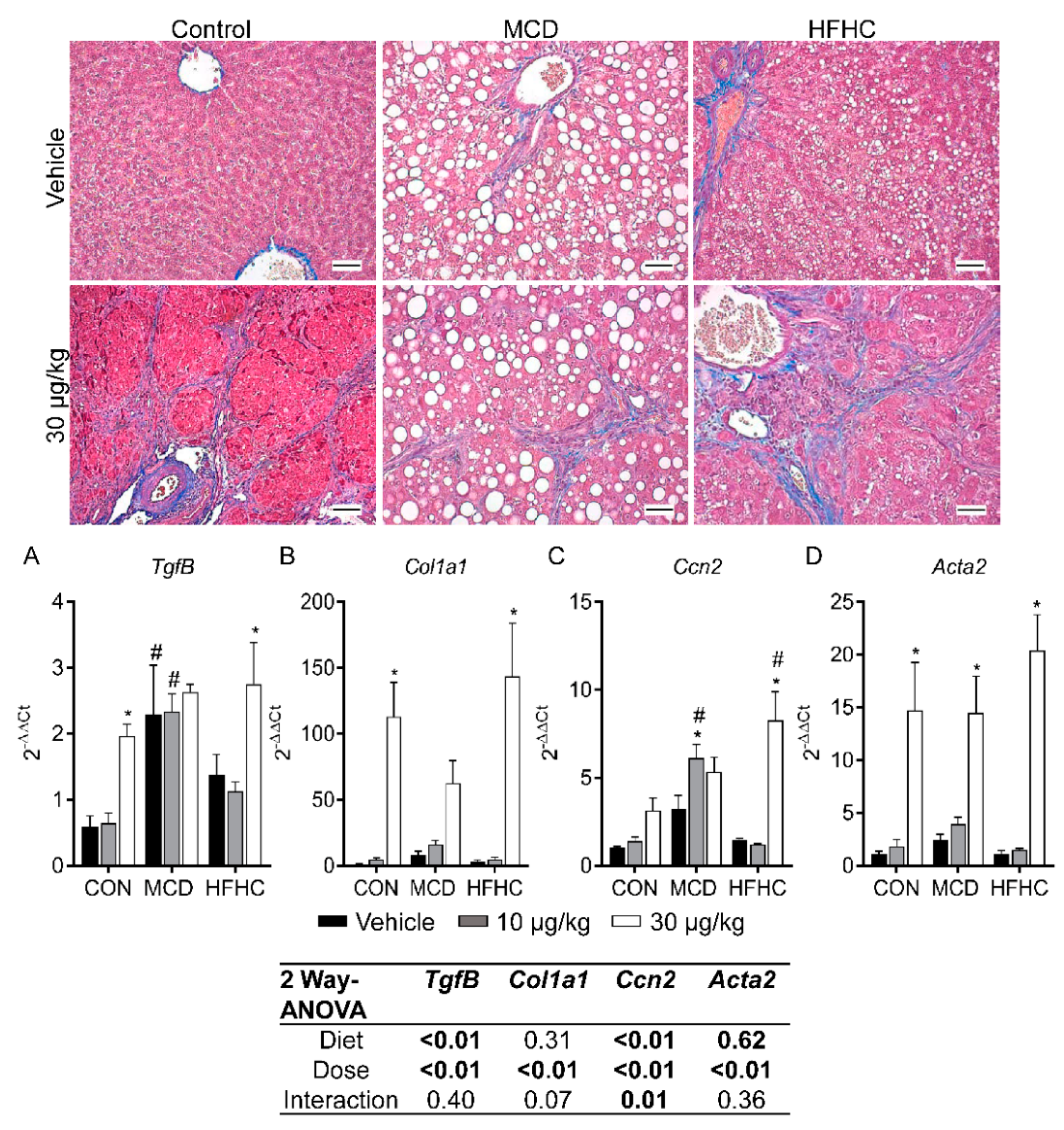
2.5. Fibrosis
3. Conclusions
4. Materials and Methods
4.1. Sub-Chronic Toxicity Study
4.1.1. Selection of Diet-Induced NASH Models
4.1.2. MCLR Exposure Protocol
4.2. mRNA Expression
4.3. Histopathological Analysis
4.4. Protein Preparations
4.5. Immunoblot Protein Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- El-Shehawy, R.; Gorokhova, E.; Fernández-Piñas, F.; del Campo, F.F. Global warming and hepatotoxin production by cyanobacteria: What can we learn from experiments? Water Res. 2012, 46, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.; Dao, T.-S.; Vo, T.-G.; Lürling, M. Warming Affects Growth Rates and Microcystin Production in Tropical Bloom-Forming Microcystis Strains. Toxins 2018, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Zimba, P.V.; Khoo, L.; Gaunt, P.S.; Brittain, S.; Carmichael, W.W. Confirmation of catfish, Ictalurus punctatus (Rafinesque), mortality from Microcystis toxins. J. Fish Dis. 2001, 24, 41–47. [Google Scholar] [CrossRef]
- de Figueiredo, D.R.; Azeiteiro, U.M.; Esteves, S.M.; Gonçalves, F.J.M.; Pereira, M.J. Microcystin-producing blooms-a serious global public health issue. Ecotoxicol. Environ. Saf. 2004, 59, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Merel, S.; Walker, D.; Chicana, R.; Snyder, S.; Baurès, E.; Thomas, O. State of knowledge and concerns on cyanobacterial blooms and cyanotoxins. Environ. Int. 2013, 59, 303–327. [Google Scholar] [CrossRef]
- Smith, J.L.; Boyer, G.L.; Zimba, P.V. A review of cyanobacterial odorous and bioactive metabolites: Impacts and management alternatives in aquaculture. Aquaculture 2008, 280, 5–20. [Google Scholar] [CrossRef]
- Rodger, H.D.; Turnbull, T.; Edwards, C.; Codd, G.A. Cyanobacterial (blue-green algal) bloom associated pathology in brown trout, Salmo trutta L., in Loch Leven, Scotland. J. Fish Dis. 1994, 17, 177–181. [Google Scholar] [CrossRef]
- Poste, A.E.; Hecky, R.E.; Guildford, S.J. Evaluating microcystin exposure risk through fish consumption. Environ. Sci. Technol. 2011, 45, 5806–5811. [Google Scholar] [CrossRef]
- Smith, J.L.; Haney, J.F. Foodweb transfer, accumulation, and depuration of microcystins, a cyanobacterial toxin, in pumpkinseed sunfish (Lepomis gibbosus). Toxicon 2006, 48, 580–589. [Google Scholar] [CrossRef]
- Merel, S.; Villarín, M.C.; Chung, K.; Snyder, S. Spatial and thematic distribution of research on cyanotoxins. Toxicon 2013, 76, 118–131. [Google Scholar] [CrossRef]
- Fawell, J.K.; Mitchell, R.E.; Hill, R.E.; Everett, D.J. The toxicity of cyanobacterial toxins in the mouse: I Microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, P.; Li, L.; Xu, J. First identification of the hepatotoxic microcystins in the serum of a chronically exposed human population together with indication of hepatocellular damage. Toxicol. Sci. 2009, 108, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zeng, H.; Lin, H.; Wang, J.; Feng, X.; Qiu, Z.; Chen, J.A.; Luo, J.; Luo, Y.; Huang, Y.; et al. Serum microcystin levels positively linked with risk of hepatocellular carcinoma: A case-control study in southwest China. Hepatology 2017, 66, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Nagata, S.; Tsutsumi, T.; Hasegawa, A.; Watanabe, M.F.; Park, H.D.; Chen, G.C.; Chen, G.; Yu, S.Z. Detection of microcystins, a blue-green algal hepatotoxin, in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis 1996, 17, 1317–1321. [Google Scholar] [CrossRef]
- Svirčev, Z.; Krstić, S.; Miladinov-Mikov, M.; Baltić, V.; Vidović, M. Freshwater cyanobacterial blooms and primary liver cancer epidemiological studies in Serbia. J. Environ. Sci. Health Part C 2009, 27, 36–55. [Google Scholar] [CrossRef] [PubMed]
- Fleming, L.; Rivero, C.; Burns, J.; Williams, C.; Bean, J.; Shea, K.; Stinn, J. Blue green algal (cyanobacterial) toxins, surface drinking water, and liver cancer in Florida. Harmful Algae 2002, 1, 157–168. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Streba, L.A.M.; Vere, C.C.; Rogoveanu, I.; Streba, C.T. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: An open question. World J. Gastroenterol. 2015, 21, 4103–4110. [Google Scholar] [CrossRef]
- Ahmed, M. Non-alcoholic fatty liver disease in 2015. World J. Hepatol. 2015, 7, 1450–1459. [Google Scholar] [CrossRef]
- Van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; Mcleod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: Are alterations in serum adiponectin the cause? Hepatology 2013, 57, 2180–2188. [Google Scholar] [CrossRef]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in american adults: NHANES 2003–2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Falkner, K.C.; Gregory, B.; Ansert, D.; Young, D.; Conklin, D.J.; Bhatnagar, A.; McClain, C.J.; Cave, M. Polychlorinated biphenyl 153 is a diet-dependent obesogen that worsens nonalcoholic fatty liver disease in male C57BL6/J mice. J. Nutr. Biochem. 2013, 24, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.K.; Kumar, A.; Das, S.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci. 2013, 134, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, J.A.; Furuya, S.; Luo, Y.S.; Iwata, Y.; Konganti, K.; Chiu, W.A.; Threadgill, D.W.; Pogribny, I.P.; Rusyn, I. Nonalcoholic fatty liver disease is a susceptibility factor for perchloroethylene-induced liver effects in mice. Toxicol. Sci. 2017, 159, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Song, M.; Beier, J.I.; Cameron Falkner, K.; Al-Eryani, L.; Clair, H.B.; Prough, R.A.; Osborne, T.S.; Malarkey, D.E.; Christopher States, J.; et al. Evaluation of Aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol. Appl. Pharmacol. 2014, 279, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.C.; Yeo, H.; Kaelin, B.R.; Lang, A.L.; Bushau, A.M.; Douglas, A.N.; Cave, M.; Arteel, G.E.; McClain, C.J.; Beier, J.I. Role of dietary fatty acids in liver injury caused by vinyl chloride metabolites in mice. Toxicol. Appl. Pharmacol. 2016, 311, 34–41. [Google Scholar] [CrossRef]
- Zhang, F.; Lee, J.; Liang, S.; Shum, C.K. Cyanobacteria blooms and non-alcoholic liver disease: Evidence from a county level ecological study in the United States. Environ. Health 2015, 14, 41. [Google Scholar] [CrossRef]
- Zhao, Y.; Xue, Q.; Su, X.; Xie, L.; Yan, Y.; Wang, L.; Steinman, A.D. First Identification of the Toxicity of Microcystins on Pancreatic Islet Function in Humans and the Involved Potential Biomarkers. Environ. Sci. Technol. 2016, 50, 3137–3144. [Google Scholar] [CrossRef]
- He, J.; Li, G.; Chen, J.; Lin, J.; Zeng, C.; Chen, J.; Deng, J.; Xie, P. Prolonged exposure to low-dose microcystin induces nonalcoholic steatohepatitis in mice: A systems toxicology study. Arch. Toxicol. 2017, 91, 465–480. [Google Scholar] [CrossRef]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, J.; Lu, C.; Wang, J.; Ge, J.; Huang, Y.; Zhang, L.; Wang, Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006, 79, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig. Dis. Sci. 2016, 61, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Andrinolo, D.; Sedan, D.; Telese, L.; Aura, C.; Masera, S.; Giannuzzi, L.; Marra, C.A.; de Alaniz, M.J. Hepatic recovery after damage produced by sub-chronic intoxication with the cyanotoxin microcystin LR. Toxicon 2008, 51, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Sedan, D.; Andrinolo, D.; Telese, L.; Giannuzzi, L.; de Alaniz, M.J.; Marra, C.A. Alteration and recovery of the antioxidant system induced by sub-chronic exposure to microcystin-LR in mice: Its relation to liver lipid composition. Toxicon 2010, 55, 333–342. [Google Scholar] [CrossRef]
- Sedan, D.; Giannuzzi, L.; Rosso, L.; Marra, C.A.; Andrinolo, D. Biomarkers of prolonged exposure to microcystin-LR in mice. Toxicon 2013, 68, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, P.; Fan, H. Genomic profiling of microRNAs and proteomics reveals an early molecular alteration associated with tumorigenesis induced by MC-LR in mice. Environ. Sci. Technol. 2012, 46, 34–41. [Google Scholar] [CrossRef]
- Clark, S.P.; Davis, M.A.; Ryan, T.P.; Searfoss, G.H.; Hooser, S.B. Hepatic gene expression changes in mice associated with prolonged sublethal microcystin exposure. Toxicol. Pathol. 2007, 35, 594–605. [Google Scholar] [CrossRef]
- Guzman, R.E.; Solter, P.F. Hepatic Oxidative Stress Following Prolonged Sublethal Microcystin LR Exposure. Toxicol. Pathol. 1999, 27, 582–588. [Google Scholar] [CrossRef]
- Canet, M.J.; Hardwick, R.N.; Lake, A.D.; Dzierlenga, A.L.; Clarke, J.D.; Cherrington, N.J. Modeling human nonalcoholic steatohepatitis-associated changes in drug transporter expression using experimental rodent models. Drug Metab. Dispos. 2014, 42, 586–595. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.L.; Sun, B.J.; Nie, P. Ultrastructural alteration of lymphocytes in spleen and pronephros of grass carp (Ctenopharyngodon idella) experimentally exposed to microcystin-LR. Aquaculture 2008, 280, 270–275. [Google Scholar] [CrossRef]
- Yea, S.S.; Kim, H.M.; Oh, H.M.; Paik, K.H.; Yang, K.H. Microcystin-induced down-regulation of lymphocyte functions through reduced IL-2 mRNA stability. Toxicol. Lett. 2001, 122, 21–31. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Bolognesi, M.; Gatta, A.; Nava, V.; Merkel, C. Role of spleen enlargement in cirrhosis with portal hypertension. Dig. Liver Dis. 2003, 34, 144–150. [Google Scholar]
- Rabelo, F.; Oliveira, C.P.M.S.; Faintuch, J.; Mazo, D.F.C.; Lima, V.M.R.; Stefano, J.T.; Barbeiro, H.V.; Soriano, F.G.; Ferreira Alves, V.A.; Carrilho, F.J. Pro-and anti-inflammatory cytokines in Steatosis and steatohepatitis. Obes. Surg. 2010, 20, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.K.C.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Beier, J.I.; Clair, H.B.; Bellis-Jones, H.J.; Falkner, K.C.; McClain, C.J.; Cave, M.C. Toxicant-associated Steatohepatitis. Toxicol. Pathol. 2013, 41, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, H.; Yamamoto, Y.; Takeshita, E.; Tokumoto, Y.; Tada, F.; Miyake, T.; Hirooka, M.; Abe, M.; Kumagi, T.; Matsuura, B.; et al. Upregulated absorption of dietary palmitic acids with changes in intestinal transporters in non-alcoholic steatohepatitis (NASH). J. Gastroenterol. 2017, 52, 940–954. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, K.; Su, X.; Xie, L.; Yan, Y. Microcystin-LR induces dysfunction of insulin secretion in rat insulinoma (INS-1) cells: Implications for diabetes mellitus. J. Hazard. Mater. 2016, 314, 11–21. [Google Scholar] [CrossRef]
- Pouria, S.; De Andrade, A.; Barbosa, J.; Cavalcanti, R.L.; Barreto, V.T.S.; Ward, C.J.; Preiser, W.; Poon, G.K.; Neild, G.H.; Codd, G.A. Fatal microcystin intoxication in haemodialysis unit in Caruaru, Brazil. Lancet 1998, 352, 21–26. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, Q.; He, Y.; Xue, Q.; Xie, L.; Yan, Y. Impairment of endoplasmic reticulum is involved in B-cell dysfunction induced by microcystin-LR. Environ. Pollut. 2017, 223, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, B.; Seo, H.S.; Lee, Y.J.; Kim, H.H.; Son, H.-H.; Choi, M.H. Cholesterol-induced non-alcoholic fatty liver disease and atherosclerosis aggravated by systemic inflammation. PLoS ONE 2014, 9, e97841. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, K.; Zhang, S.; Liu, J.; Xu, C.; Xu, L.; Huang, P.; Guo, Z. Microcystin-LR disrupts insulin signaling by hyperphosphorylating insulin receptor substrate 1 and glycogen synthase. Environ. Toxicol. 2017, 33, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.J.; Altheimer, S.; Cattori, V.; Meier, P.J.; Dietrich, D.R.; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef]
- Komatsu, M.; Furukawa, T.; Ikeda, R.; Takumi, S.; Nong, Q.; Aoyama, K.; Akiyama, S.; Keppler, D.; Takeuchi, T. Involvement of mitogen-activated protein kinase signaling pathways in microcystin-LR-induced apoptosis after its selective uptake mediated by OATP1B1 and OATP1B3. Toxicol. Sci. 2007, 97, 407–416. [Google Scholar] [CrossRef]
- Niedermeyer, T.H.J.; Daily, A.; Swiatecka-Hagenbruch, M.; Moscow, J.A. Selectivity and Potency of Microcystin Congeners against OATP1B1 and OATP1B3 Expressing Cancer Cells. PLoS ONE 2014, 9, e91476. [Google Scholar] [CrossRef]
- Daily, A.; Monks, N.R.; Leggas, M.; Moscow, J.A. Abrogation of microcystin cytotoxicity by MAP kinase inhibitors and N-acetyl cysteine is confounded by OATPIB1 uptake activity inhibition. Toxicon 2010, 55, 827–837. [Google Scholar] [CrossRef]
- Lu, H.; Choudhuri, S.; Ogura, K.; Csanaky, I.L.; Lei, X.; Cheng, X.; Song, P.Z.; Klaassen, C.D. Characterization of organic anion transporting polypeptide 1b2-null mice: Essential role in hepatic uptake/toxicity of phalloidin and microcystin-LR. Toxicol. Sci. 2008, 103, 35–45. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Johnston, C.K.; Tsakalozou, E.; Bridges, A.S.; Paine, M.F.; Jia, W.; Stewart, P.W.; Barritt, A.S.; Brouwer, K.L.R. Altered morphine glucuronide and bile acid disposition in patients with nonalcoholic steatohepatitis. Clin. Pharmacol. Ther. 2015, 97, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Cherrington, N.J. Nonalcoholic steatohepatitis in precision medicine: Unraveling the factors that contribute to individual variability. Pharmacol. Ther. 2015, 151, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hardwick, R.N.; Lake, A.D.; Canet, M.J.; Cherrington, N.J. Experimental Nonalcoholic Steatohepatitis Increases Exposure to Simvastatin Hydroxy Acid by Decreasing Hepatic Organic Anion Transporting Polypeptide Expression. J. Pharmacol. Exp. Ther. 2014, 348, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hardwick, R.N.; Lake, A.D.; Lickteig, A.J.; Goedken, M.J.; Klaassen, C.D.; Cherrington, N.J. Synergistic interaction between genetics and disease on pravastatin disposition. J. Hepatol. 2014, 61, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of Protein Phosphatase 2A Core Enzyme Bound to Tumor-Inducing Toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Kong, S.; Berndt, N. Protein phosphatase inhibition and in vivo hepatotoxicity of microcystins. Am. J. Physiol. Liver Physiol. 2017, 265, G224–G230. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, J.Y.; Xu, Z.; Kho, D.H.; Zhuang, Z.; Raz, A.; Wu, G.S. The role of cullin3-mediated ubiquitination of the catalytic subunit of PP2A in TRAIL signaling. Cell Cycle 2014, 13, 3750–3758. [Google Scholar] [CrossRef]
- Li, T.; Huang, P.; Liang, J.; Fu, W.; Guo, Z.-L.; Xu, L.-H. Microcystin-LR (MCLR) induces a compensation of PP2A activity mediated by α4 protein in HEK293 cells. Int. J. Biol. Sci. 2011, 7, 740–752. [Google Scholar] [CrossRef]
- Toivola, D.M.; Eriksson, J.E.; Brautigan, D.L. Identification of protein phosphatase 2A as the primary target for microcystin-LR in rat liver homogenates. FEBS Lett. 1994, 344, 175–180. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Hames, K.C.; Vella, A.; Kemp, B.J.; Jensen, M.D. Free fatty acid uptake in humans with CD36 deficiency. Diabetes 2014, 63, 3606–3614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Ducheix, S.; Vegliante, M.C.; Villani, G.; Napoli, N.; Sabbà, C.; Moschetta, A. Is hepatic lipogenesis fundamental for NAFLD/NASH? A focus on the nuclear receptor coactivator PGC-1β. Cell. Mol. Life Sci. 2016, 73, 3809–3822. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-Coa desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. CEG-62831-molecular-pathways-in-non-alcoholic-fatty-liver-disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar]
- Iizuka, K.; Horikawa, Y. ChREBP: A Glucose-activated Transcription Factor Involved in the Development of Metabolic Syndrome. Endocr. J. 2008, 55, 617–624. [Google Scholar] [CrossRef]
- Postic, C.; Dentin, R.; Denechaud, P.-D.; Girard, J. ChREBP, a Transcriptional Regulator of Glucose and Lipid Metabolism. Annu. Rev. Nutr. 2007, 27, 179–192. [Google Scholar] [CrossRef]
- Koo, S.-H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of lipotoxicity in NAFLD and clinical implications. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 2009, 1791, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Xing, X.Y.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Neschen, S.; Morino, K.; Hammond, L.E.; Zhang, D.; Liu, Z.X.; Romanelli, A.J.; Cline, G.W.; Pongratz, R.L.; Zhang, X.M.; Choi, C.S.; et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005, 2, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Wen, F.; Zhou, S.; Su, Z.; Liu, G.; Wang, M.; Zhou, J.; He, F. Association of MTTP gene variants with pediatric NAFLD: A candidate-gene-based analysis of single nucleotide variations in obese children. PLoS ONE 2017, 12, e0185396. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Xia, Z. Microcystin-LR Exhibits Immunomodulatory Role in Mouse Primary Hepatocytes Through Activation of the NF-κB and MAPK Signaling Pathways. Toxicol. Sci. 2013, 136, 86–96. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dzierlenga, A.; Arman, T.; Toth, E.; Li, H.; Lynch, K.D.; Tian, D.-D.; Goedken, M.; Paine, M.F.; Cherrington, N. Nonalcoholic fatty liver disease alters microcystin-LR toxicokinetics and acute toxicity. Toxicon 2019, 162, 1–8. [Google Scholar] [CrossRef]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746. [Google Scholar] [CrossRef]
- Rocha, M.F.G.; Sidrim, J.J.C.; Soares, A.M.; Jimenez, G.C.; Guerrant, R.L.; Ribeiro, R.A.; Lima, A.A.M. Supernatants from macrophages stimulated with microcystin-LR induce electrogenic intestinal response in rabbit ileum. Pharmacol. Toxicol. 2000, 87, 46–51. [Google Scholar] [CrossRef]
- Frangez, R.; Kosec, M.; Sedmak, B.; Beravs, K.; Demsar, F.; Juntes, P.; Pogacnik, M.; Suput, D. Subchronic liver injuries caused by microcystins. Pflügers Arch. Eur. J. Physiol. 2000, 440, R103–R104. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef]
- Heinze, R. Toxicity of the cyanobacterial toxin microcystin-LR to rats after 28 days intake with the drinking water. Environ. Toxicol. 1999, 14, 57–60. [Google Scholar] [CrossRef]
- Ito, E.; Kondo, F.; Harada, K.I. Hepatic necrosis in aged mice by oral administration of microcystin-LR. Toxicon 1997, 35, 231–239. [Google Scholar] [CrossRef]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Landaverde, C.; Jennings, L.; Goldstein, R.M.; Davis, G.L. Patients With NASH and Cryptogenic Cirrhosis Are Less Likely Than Those With Hepatitis C to Receive Liver Transplants. Clin. Gastroenterol. Hepatol. 2011, 9, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; De Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Haque, J.A.; McMahan, R.S.; Campbell, J.S.; Shimizu-Albergine, M.; Wilson, A.M.; Botta, D.; Bammler, T.K.; Beyer, R.P.; Montine, T.J.; Yeh, M.M.; et al. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab. Investig. 2010, 90, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. Total protein STAIN. J. Neurosci. Methods 2009, 172, 250–254. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Body (g) | Liver (g) | Spleen (g) | Liver: Body | Spleen: Body |
|---|---|---|---|---|---|
| Control | |||||
| Vehicle | 379 ± 7.7 | 15.38 ± 0.4 | 0.75 ± 0.03 | 0.041 ± 0.001 | 0.002 ± 0.0001 |
| 10 µg/kg | 357 ± 9 | 12.08 ± 0.62 * | 0.69 ± 0.02 | 0.034 ± 0.002 * | 0.002 |
| 30 µg/kg | 375 ± 11.8 | 12.07 ± 0.52 * | 1.11 ± 0.07 * | 0.032 ± 0.002 * | 0.003 ± 0.0001 * |
| MCD | |||||
| Vehicle | 179 ± 5.6# | 8.39 ± 0.33# | 0.36 ± 0.01# | 0.047 ± 0.001# | 0.002 |
| 10 µg/kg | 187 ± 4.4# | 8.40 ± 0.19# | 0.40 ± 0.02# | 0.045 ± 0.001# | 0.002 ± 0.0001 |
| 30 µg/kg | 177 ± 3.6# | 7.21 ± 0.2# | 0.42 ± 0.03# | 0.041 ± 0.001 *# | 0.002 ± 0.0001 |
| HFHC | |||||
| Vehicle | 408 ± 6.8# | 18.49 ± 1.1# | 1.13 ± 0.1# | 0.045 ± 0.003 | 0.003 ± 0.0002# |
| 10 µg/kg | 420 ± 6.8# | 18.07 ± 0.39# | 0.89 ± 0.05 * | 0.043 ± 0.001# | 0.002 ± 0.0001 * |
| 30 µg/kg | 406 ± 10.3# | 20.20 ± 1.51# | 2.15 ± 0.16 *# | 0.050 ± 0.003# | 0.005 ± 0.0004 *# |
| Two-way ANOVA | |||||
| Diet | <0.01 (97%) | <0.01 (84%) | <0.01 (55%) | <0.01 (44%) | <0.01 (26%) |
| Dose | 0.91 (<1%) | 0.10 (1%) | <0.01 (21%) | 0.04 (6%) | <0.01 (36%) |
| Interaction | 0.15 (<1%) | <0.01 (4%) | <0.01 (16%) | <0.01 (14%) | <0.01 (24%) |
| Group | Steatosis | Inflammation | Fibrosis | Biliary Hyperplasia |
|---|---|---|---|---|
| Control | ||||
| Vehicle | 0 | 0 | 0 | 0 |
| 10 µg/kg | 0 | 0.2 ± 0.2 | 0 | 0 |
| 30 µg/kg | 0 | 2.2 ± 0.2 * | 3.2 ± 0.3 * | 1.7 ± 0.2 * |
| MCD | ||||
| Vehicle | 3.5 ± 0.3# | 1# | 0 | 0.2 ± 0.2 |
| 10 µg/kg | 3.3 ± 0.2# | 2 *# | 1 *# | 1 *# |
| 30 µg/kg | 2.2 ± 0.2 *# | 1.8 ± 0.2 * | 1.7 ± 0.2 *# | 1.8 ± 0.2 * |
| HFHC | ||||
| Vehicle | 2.6 ± 0.2# | 1.3 ± 0.2# | 0.2 ± 0.2 | 0.3 ± 0.2 |
| 10 µg/kg | 2.3 ± 0.3# | 0.5 ± 0.3 * | 0 | 0.2 ± 0.2 |
| 30 µg/kg | 1.8 ± 0.2 *# | 2.2 ± 0.2 * | 2.3 ± 0.2 *# | 2.0 ± 0.2 * |
| Two-way ANOVA | ||||
| Diet | <0.01 (82%) | <0.01 (15%) | 0.19 (1%) | <0.01 (4%) |
| Dose | <0.01 (5%) | <0.01 (41%) | <0.01 (77%) | <0.01 (72%) |
| Interaction | 0.02 (3%) | <0.01 (26%) | <0.01 (14%) | 0.01 (6%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arman, T.; Lynch, K.D.; Montonye, M.L.; Goedken, M.; Clarke, J.D. Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats. Toxins 2019, 11, 398. https://doi.org/10.3390/toxins11070398
Arman T, Lynch KD, Montonye ML, Goedken M, Clarke JD. Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats. Toxins. 2019; 11(7):398. https://doi.org/10.3390/toxins11070398
Chicago/Turabian StyleArman, Tarana, Katherine D. Lynch, Michelle L. Montonye, Michael Goedken, and John D. Clarke. 2019. "Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats" Toxins 11, no. 7: 398. https://doi.org/10.3390/toxins11070398
APA StyleArman, T., Lynch, K. D., Montonye, M. L., Goedken, M., & Clarke, J. D. (2019). Sub-Chronic Microcystin-LR Liver Toxicity in Preexisting Diet-Induced Nonalcoholic Steatohepatitis in Rats. Toxins, 11(7), 398. https://doi.org/10.3390/toxins11070398