Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells


Abstract
1. Introduction
2. Results
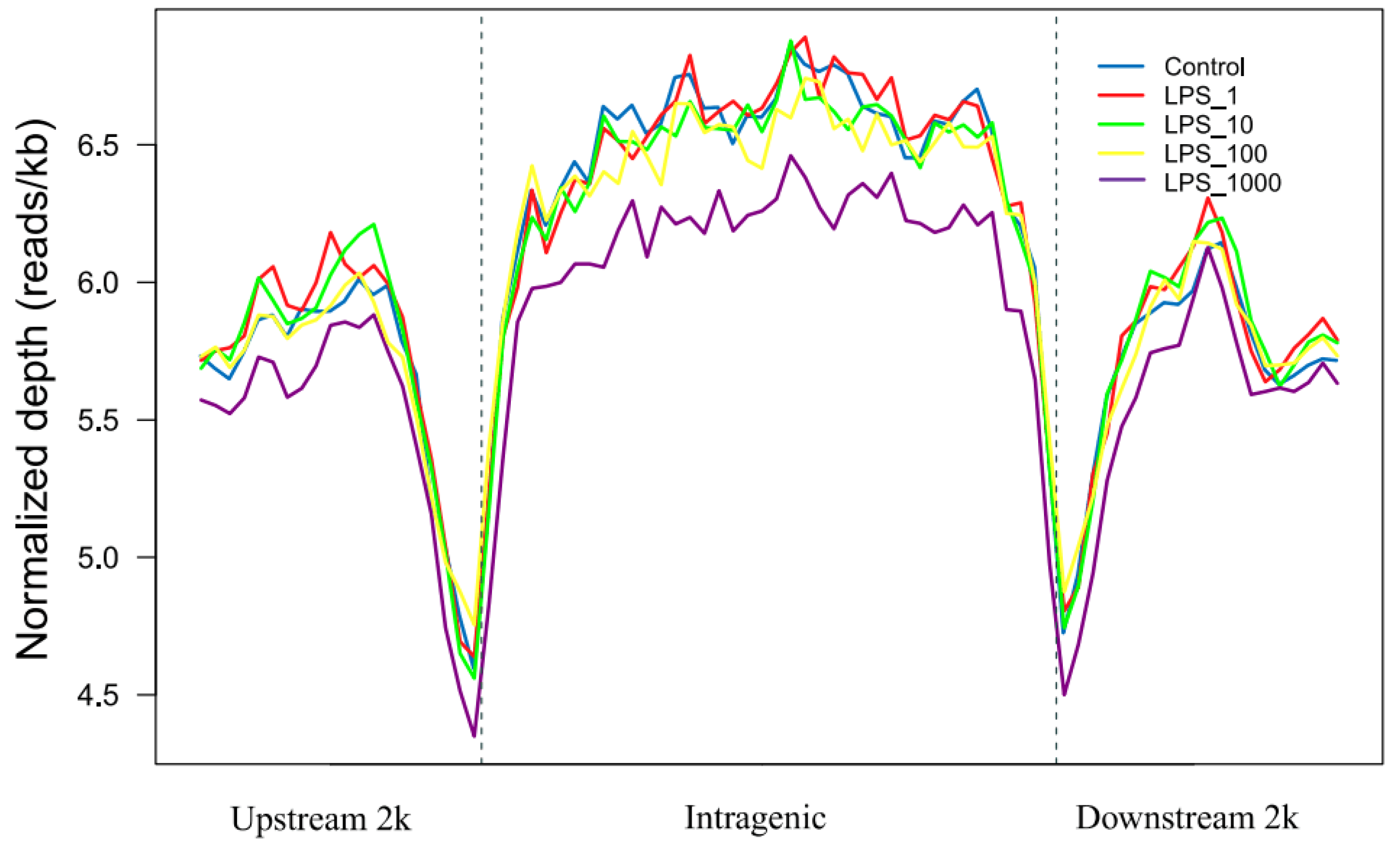
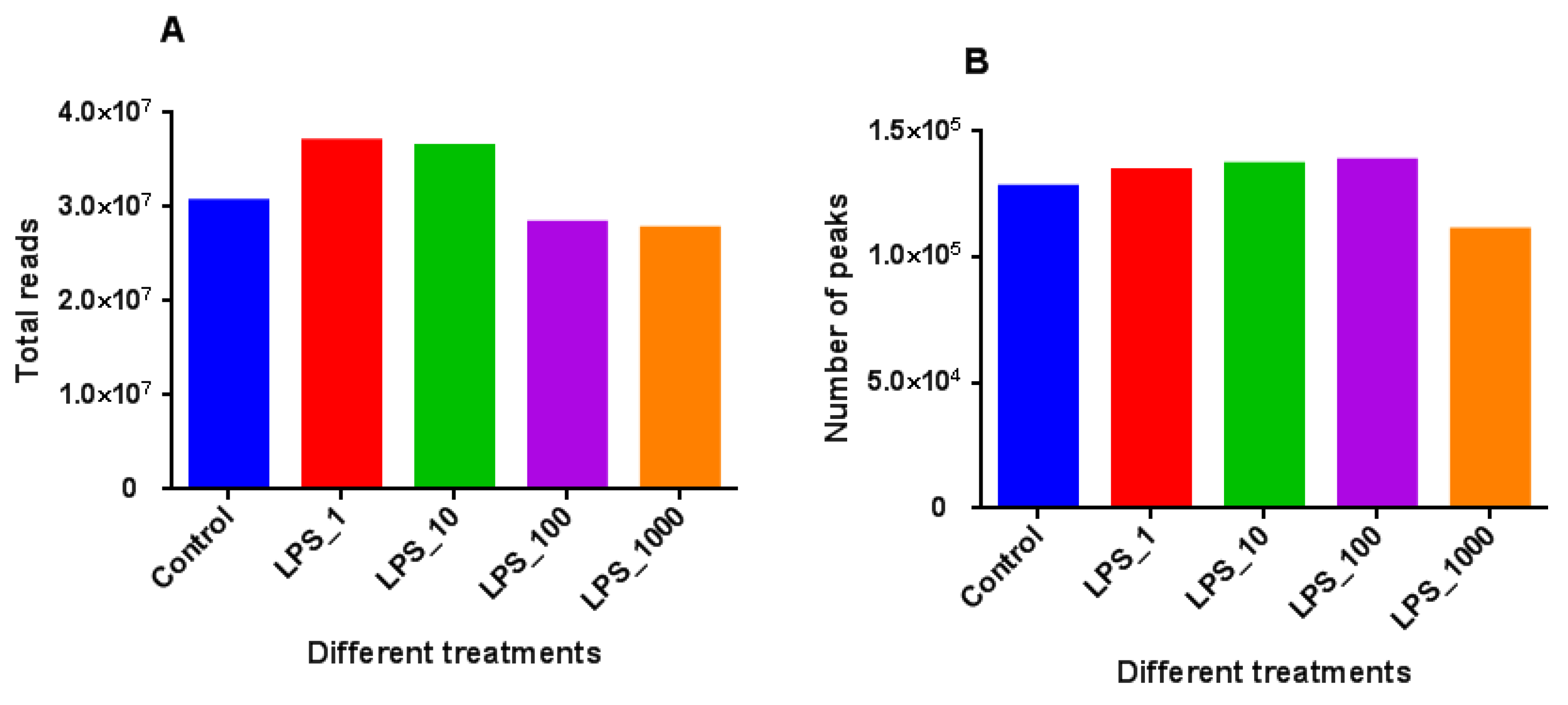
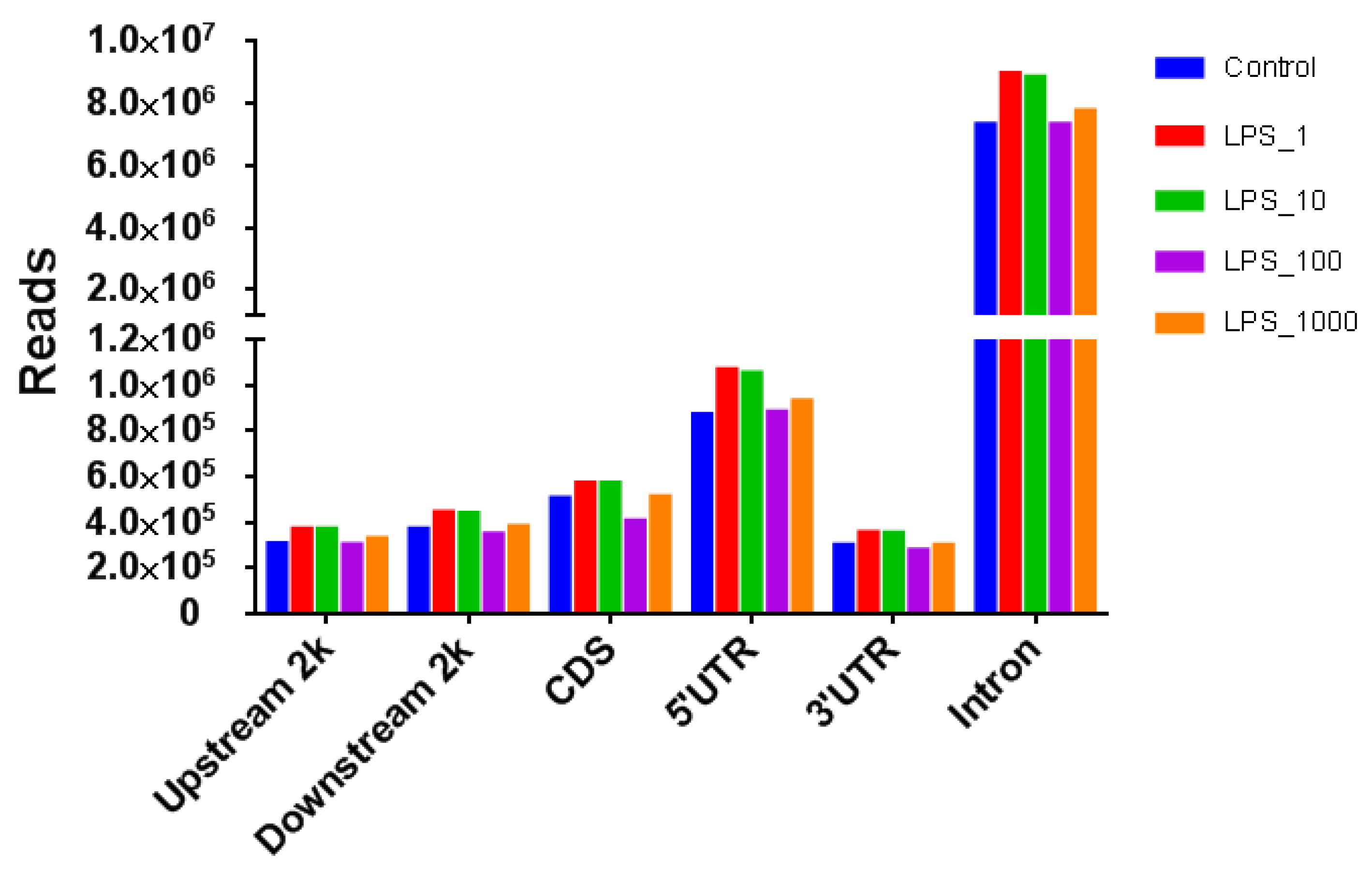
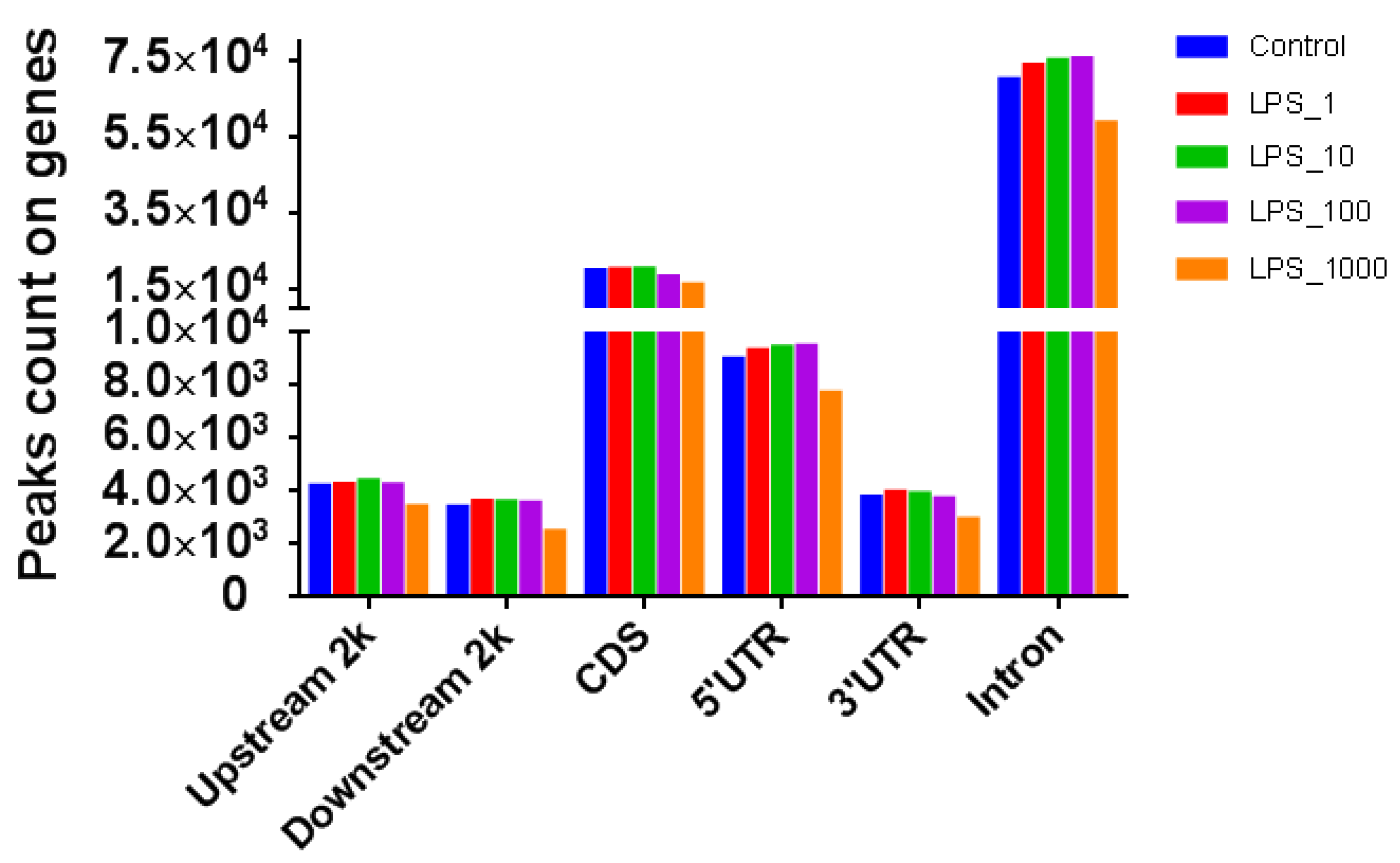
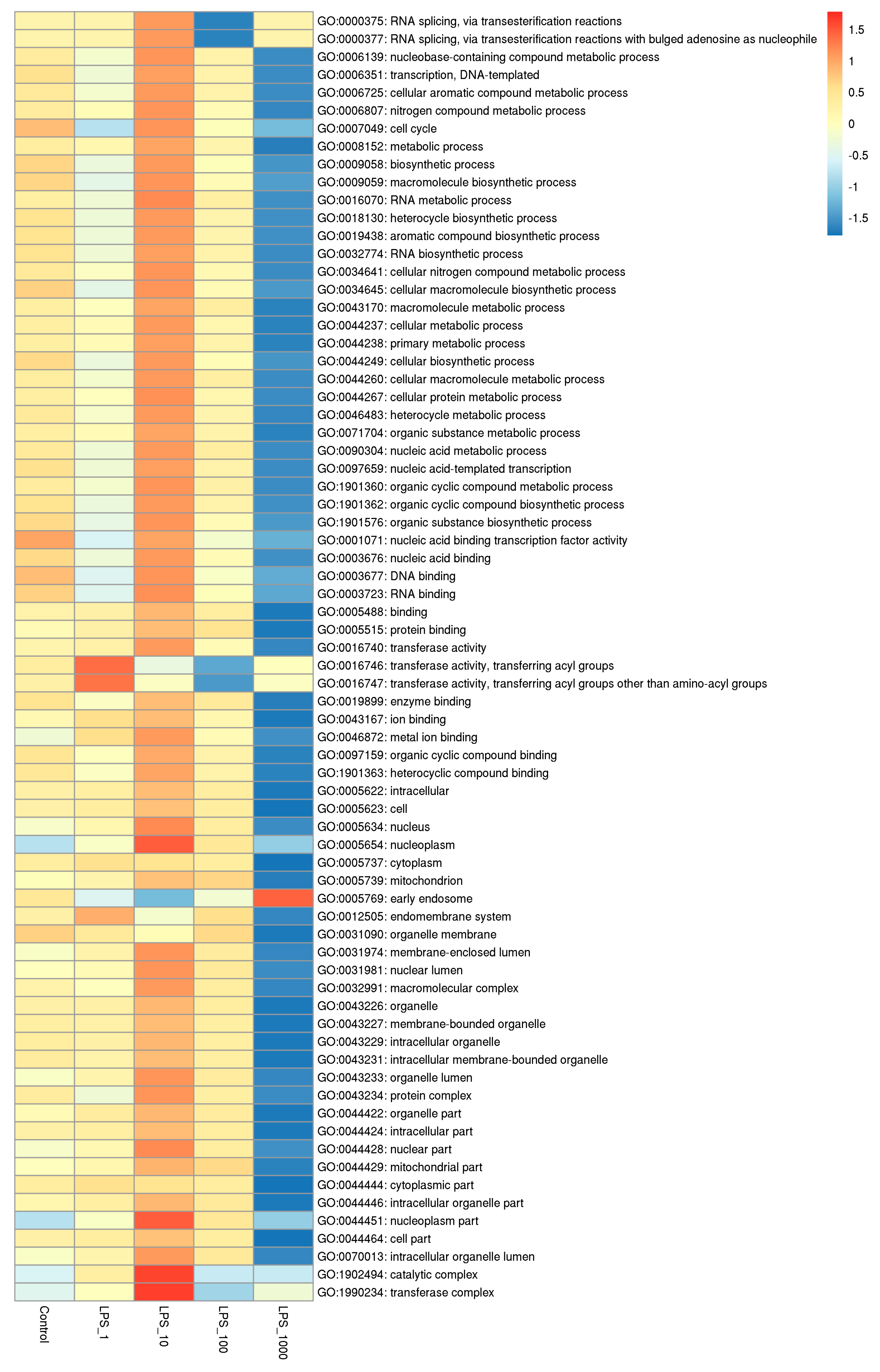
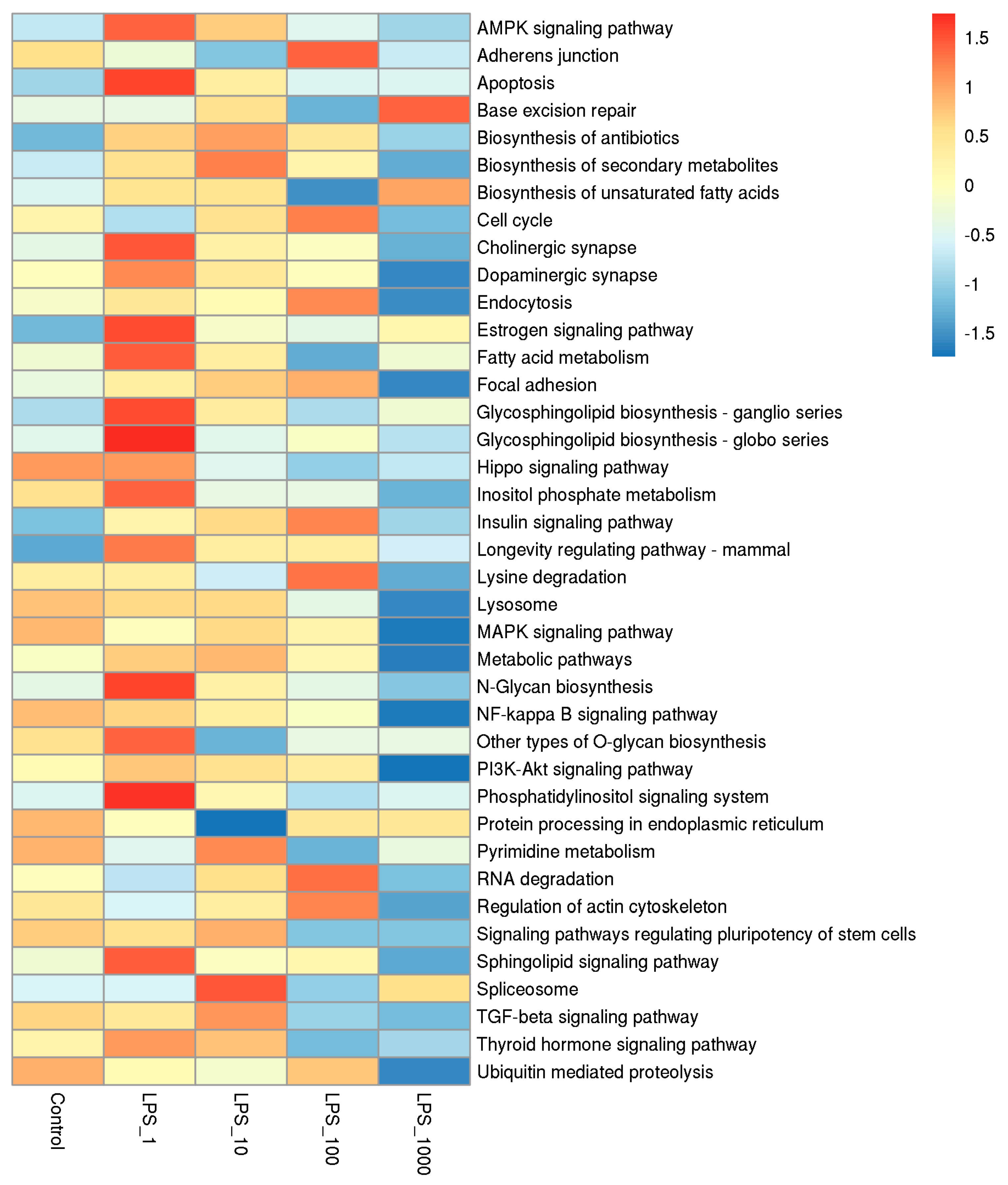
2.1. Effect of LPS on Genome-Wide DNA Methylation in Bovine Mammary Epithelial Cells
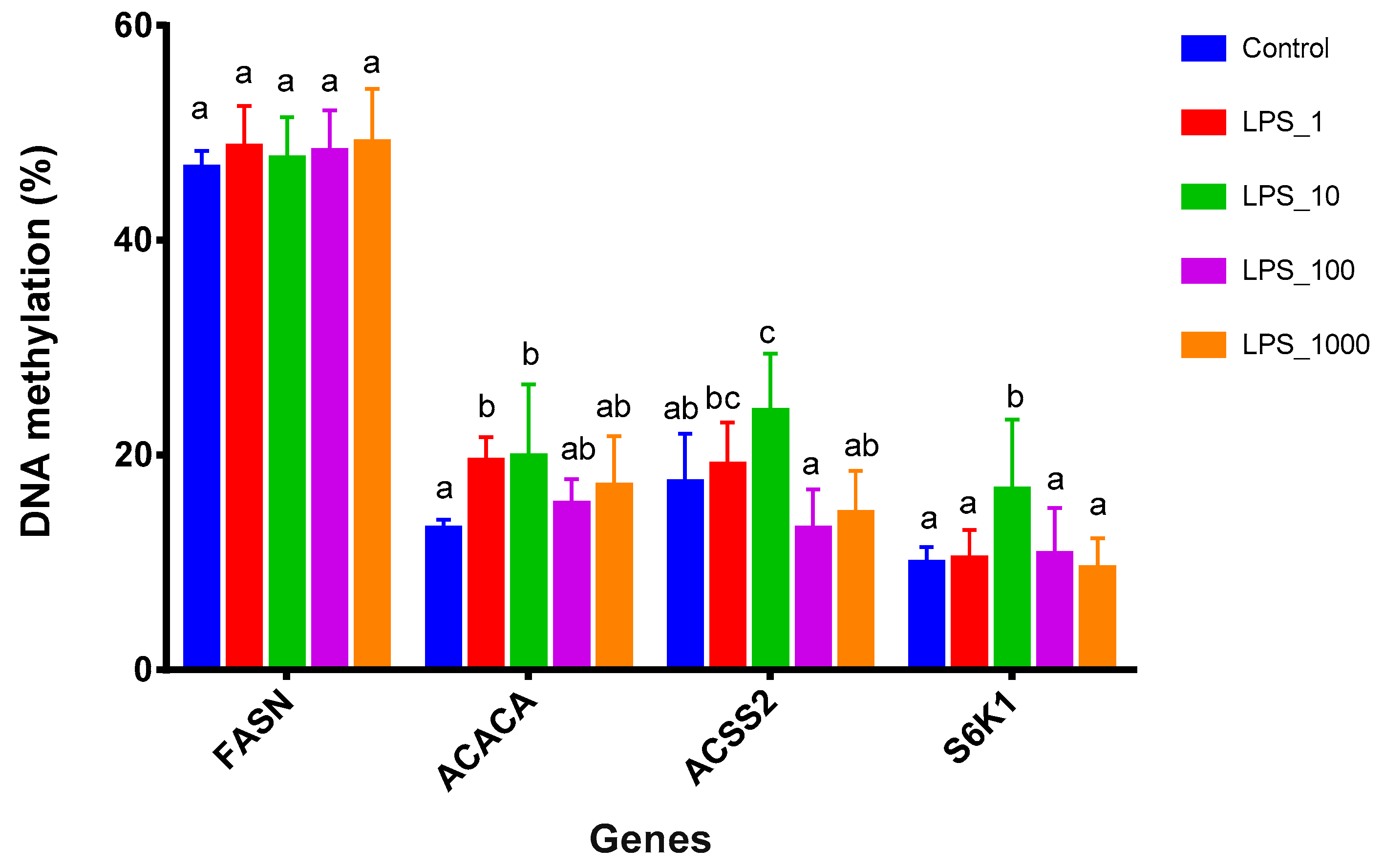
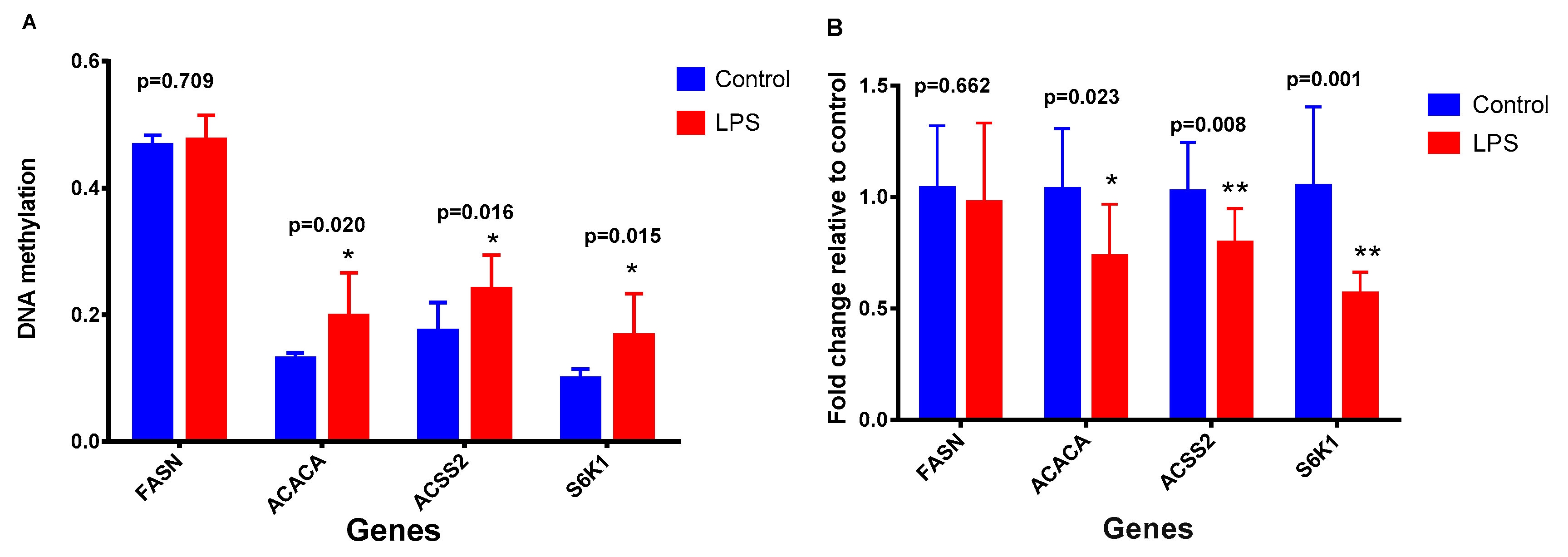
2.2. Effects of LPS on Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells
2.3. Effects of DNA Methylation on Gene Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Research Ethics
5.2. Cell Culture and Treatments
5.3. DNA Extraction and Evaluation
5.4. Assay for Genome-Wide Methylation
5.5. Assay for Promoter Methylation of Milk Genes
5.6. RNA Isolation and cDNA Synthesis
5.7. Quantitative Real-Time PCR
5.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, G.Z.; Liu, S.M.; Wu, Y.X.; Lei, C.L.; Zhou, J.; Zhang, S. Diet-induced bacterial immunogens in the gastrointestinal tract of dairy cows: impacts on immunity and metabolism. Acta. Vet. Scand. 2011, 53, 48. [Google Scholar] [CrossRef] [PubMed]
- Plaizier, J.C.; Khafipour, E.; Li, S.; Gozho, G.N.; Krause, D.O. Subacute ruminal acidosis (SARA), endotoxins and health consequences. Anim. Feed. Sci. Tech. 2012, 72, 9–21. [Google Scholar] [CrossRef]
- Eckel, E.F.; Ametaj, B.N. Invited review: Role of bacterial endotoxins in the etiopathogenesis of periparturient diseases of transition dairy cows. J. Dairy Sci. 2016, 99, 5967–5990. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Krause, D.O.; Gozho, G.N.; McBride, B.W. Subacute ruminal acidosis in dairy cows: the physiological causes, incidence and consequences. Vet. J. 2009, 176, 21–31. [Google Scholar] [CrossRef]
- Zhou, J.; Dong, G.Z.; Ao, C.J.; Zhang, S.; Qiu, M.; Wang, X. Feeding a high-concentrate corn straw diet increased the release of endotoxin in the rumen and pro-inflammatory cytokines in the mammary gland of dairy cows. BMC Vet. Res. 2014, 10, 172. [Google Scholar] [CrossRef]
- Khafipour, E.; Krause, D.O.; Plaizier, J.C. A grain-based subacute ruminal acidosis challenge causes translocation of lipopolysaccharide and triggers inflammation. J. Dairy Sci. 2009, 92, 1060–1070. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Yoon, I.; Zhu, W.Y.; Mao, S.Y. Effect of Saccharomyces cerevisiae fermentation product on lactation performance and lipopolysaccharide concentration of dairy cows. Asian Australas. J. Anim. Sci. 2013, 26, 1137–1143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, G.J.; Zhang, K.; Xu, T.L.; Jin, D.; Guo, J.F.; Zhuang, S.; Shen, X.Z. Epigenetic mechanisms contribute to the expression of immune related genes in the livers of dairy cows fed a high concentrate diet. PLoS ONE 2015, 10, e0123942. [Google Scholar] [CrossRef][Green Version]
- Bilal, M.S.; Abaker, J.A.; Aabdin, Z.; Xu, T.L.; Dai, H.Y.; Zhang, K.; Liu, X.X.; Shen, X.Z. Lipopolysaccharide derived from the digestive tract triggers an inflammatory response in the uterus of mid-lactating dairy cows during SARA. BMC Vet. Res. 2016, 12, 284. [Google Scholar] [CrossRef]
- Abaker, J.A.; Xu, T.L.; Jin, D.; Chang, G.J.; Zhang, K.; Shen, X.Z. Lipopolysaccharide derived from the digestive tract provokes oxidative stress in the liver of dairy cows fed a high-grain diet. J. Dairy Sci. 2017, 100, 666–678. [Google Scholar] [CrossRef]
- Guo, J.F.; Chang, G.J.; Zhang, K.; Xu, L.; Jin, D.; Bilal, M.S.; Shen, X.Z. Rumen-derived lipopolysaccharide provoked inflammatory injury in the liver of dairy cows fed a high-concentrate diet. Oncotarget 2017, 8, 46769–46780. [Google Scholar] [CrossRef]
- Zhang, K.; Chang, G.J.; Xu, T.L.; Xu, L.; Guo, J.F.; Jin, D.; Shen, X.Z. Lipopolysaccharide derived from the digestive tract activates inflammatory gene expression and inhibits casein synthesis in the mammary glands of lactating dairy cows. Oncotarget 2016, 7, 9652–9665. [Google Scholar] [CrossRef]
- Dong, G.Z.; Qiu, M.; Ao, C.; Zhou, J.; Erdene, K.; Wang, X.; Zhang, Z.; Yang, Y. Feeding a high-concentrate corn straw diet induced epigenetic alterations in the mammary tissue of dairy cows. PLoS ONE 2014, 9, e107659. [Google Scholar] [CrossRef]
- Dutkiewicz, J.; Pomorski, Z.J.H.; Sitkowska, J.; Krysińska-Traczyk, E.; Skórska, C.; Prażmo, Z.; Cholewa, G.; Wójtowicz, H. Airborne microorganisms and endotoxin in animal houses. Grana 1993, 33, 85–90. [Google Scholar] [CrossRef]
- Zucker, B.A.; Müller, W. Concentrations of airborne endotoxin in cow and calf stables. J. Aerosol Sci. 1998, 29, 217–221. [Google Scholar] [CrossRef]
- Hakogi, E.; Tamura, H.; Tanaka, S.; Kohata, A.; Shimada, Y.; Tabuchi, K. Endotoxin levels in milk and plasma of mastitisaffected cows measured with a chromogenic limulus test. Vet. Microbiol. 1989, 20, 267–274. [Google Scholar] [CrossRef]
- Mateus, L.; da Costa, L.L.; Diniz, P.; Ziecik, A.J. Relationship between endotoxin and prostaglandin (PGE2 and PGFM) concentrations and ovarian function in dairy cows with puerperal endometritis. Anim. Reprod. Sci. 2003, 76, 143–154. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Raizin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef]
- Davis, C.D.; Uthus, E.O. DNA methylation, cancer susceptibility, and nutrient interactions. Expl. Biol. Med. 2004, 229, 988–995. [Google Scholar] [CrossRef]
- Pfalzer, A.C.; Choi, S.W.; Tammen, S.A.; Bottiglier, T.; Parnell, L.D.; Lamon-Fava, S. S-adenosylmethionine mediates inhibition of inflammatory responses and changes in DNA methylation in human macrophages. Physiol. Genom. 2014, 46, 617–623. [Google Scholar] [CrossRef]
- Li, E.; Bird, A. DNA methylation in mammals. In Epigenetics; Allis, C.D., Jenuwein, T., Reinberg, D., Caparros, M.L., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007; pp. 341–356. [Google Scholar]
- Ollikainen, M.; Smith, K.R.; Joo, E.J.; Ng, H.K.; Andronikos, R.; Novakovic, B.; Abdu Aziz, N.K.; Carlin, J.B.; Morley, R.; Saffery, R.; et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 2010, 19, 4176–4188. [Google Scholar] [CrossRef]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta. Physiol. (Oxf.) 2011, 202, 103–118. [Google Scholar] [CrossRef]
- Song, M.Y.; He, Y.H.; Zhou, H.K.; Zhang, Y.; Li, X.Z.; Yu, Y. Combined analysis of DNA methylome and transcriptome reveal novel candidate genes with susceptibility to bovine Staphylococcus aureus subclinical mastitis. Scientif. Rep. 2016, 6, 29390. [Google Scholar] [CrossRef]
- Su, J.M.; Wang, Y.S.; Xing, X.P.; Liu, J.; Zhang, Y. Genome-wide analysis of DNA methylation in bovine placentas. BMC Genom. 2014, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Gou, W.Y.; Ma, J.; Zhang, H.L.; Zhang, Y.; Zhang, H. Genome methylation and regulatory functions for hypoxic adaptation in Tibetan chicken embryos. Peer J. 2017, 5, e3891. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.R. The Effects of Endotoxin on Immune Activation and the Synthesis of Milk Fat and Milk Protein and Their Control in the Mammary Tissue of Dairy Cows. Master’s Thesis, Southwest University, Chongqing, China, 2015. [Google Scholar]
- Zebeli, Q.; Ametaj, B.N. Relationships between rumen lipopolysaccharide and mediators of inflammatory response with milk fat production and efficiency in dairy cows. J. Dairy Sci. 2009, 92, 3800–3809. [Google Scholar] [CrossRef]
- Choi, Y.J.; Jang, K.; Yim, D.S.; Baik, M.G.; Myung, K.H.; Kim, Y.S.; Lee, H.J.; Kim, J.S.; Han, I.K. Effects of compensatory growth on the expression of milk protein gene and biochemical changes of the mammary gland in Holstein cows. J. Nutr. Biochem. 1998, 9, 380–387. [Google Scholar] [CrossRef]
- Nguyen, M.; Boutinaud, M.; Petridou, B.; Gabory, A.; Pannetier, M.; Chat, S.; Bouet, S.; Jouneau, L.; Jaffrezic, F.; Laloe, D.; et al. DNA Methylation and transcription in a distal region upstream from the bovine alphaS1 casein gene after once or twice daily milking. PLoS ONE 2014, 9, e111556. [Google Scholar] [CrossRef]
- Vanselow, J.; Yang, W.; Herrmann, J.; Zerbe, H.; Schuberth, H.J.; Petzl, W.; Tomek, W.; Seyfert, H.M. DNA-remethylation around a STAT5-binding enhancer in the aS1-casein promoter is associated with abrupt shutdown of aS1-casein synthesis during acute mastitis. J. Mol. Endocr. 2006, 37, 463–477. [Google Scholar] [CrossRef]
- Huynh, H.T.; Robitaille, G.; Turner, J.D. Establishment of bovine mammary epithelial cells (MAC-T): An in vitro model for bovine lactation. Exp. Cell Res. 1991, 197, 191–199. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2−△△CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Methylation | Control vs. LPS_1 | Control vs. LPS_10 | Control vs. LPS_100 | Control vs. LPS_1000 |
|---|---|---|---|---|---|
| Lipid metabolism | up | 30 | 22 | 13 | 24 |
| down | 16 | 16 | 20 | 28 | |
| Amino acid metabolism | up | 20 | 18 | 10 | 10 |
| down | 9 | 12 | 20 | 27 | |
| Carbohydrate metabolism | up | 17 | 20 | 23 | 17 |
| down | 14 | 11 | 18 | 22 | |
| Immune response | up | 44 | 41 | 42 | 44 |
| down | 34 | 34 | 46 | 62 | |
| Cell growth | up | 18 | 23 | 20 | 15 |
| down | 18 | 16 | 22 | 20 |
| Gene | Primer | Product Size (bp) |
|---|---|---|
| FASN | F: GTATTTGGGTTATTTTGGGGGTTAT R: CCAAAAAAACTCCTTTATTCAAACC | 482 |
| SCD | F: TGAATTTTTTGAAGGTAAGGATTATG R: AAACACCTAACAATTAAAATTCCCC | 560 |
| STAT5A | F: TTTTTTGATAGATGAGAAAATTGAGG R: ACAACAACAACAACAACAACAAAAT | 532 |
| S6K1 | F: GAGGATTTGGTTTTTAGGTGTGAG R: AAAAAATACAACAAAACCCCCTTAC | 451 |
| ACSL1 | F: AGGAAAGTTTTTAGGATTTTAGGGA R: CTAAAAAAAACACTCACCTCCTCC | 568 |
| ACACA | F: TTTTGTAGAGAGGTTTGAGAGGTTG R: TAACATCAAAATAAACCACCACATC | 478 |
| ACSS1 | F: GATTAGTTGGTTTTTGGGAGGTTAG R: AAAAATCCCACTACTACTTCCTTCC | 529 |
| ACSS2 | F: AATTTTGGTTAATGGGTTTTTTTGT R: ATAAACCCCCACTTCTCTCCTAACTA | 453 |
| FADS2 | F: TTTGTTGATTATTGTGGAAATTTAGG R: AAAAAACCCCAAACCCTTACC | 595 |
| Gene | Primer | Product Size (bp) | Accession Number |
|---|---|---|---|
| ACACA | F: GATCCAGGCCATGCTAAG R: CTGTTTCTCCAGCCACTC | 103 | XM_024979607.1 |
| ACSS2 | F: GGGCGAATGCCTCTACTGC R: GCTGGGTGATGATGGATGG | 254 | NM_001105339.1 |
| S6K1 | F: GGACATGGCAGGGGTGTTT R: GGTATTTGCTCCTGTTACTTTTCG | 283 | NM_205816.1 |
| FASN | F: AGGACCTCGTGAAGGCTGTGA R: CCAAGGTCTGAAAGCGAGCTG | 85 | XM_005220997.3 |
| GAPDH | F: GGGTCATCATCTCTGCACCT R: GGTCATAAGTCCCTCCACGA | 177 | NM_001034034.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Wu, Y.; Sun, Y.; Dong, X.; Wang, Z.; Zhang, Z.; Xiao, Y.; Dong, G. Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells. Toxins 2019, 11, 298. https://doi.org/10.3390/toxins11050298
Chen J, Wu Y, Sun Y, Dong X, Wang Z, Zhang Z, Xiao Y, Dong G. Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells. Toxins. 2019; 11(5):298. https://doi.org/10.3390/toxins11050298
Chicago/Turabian StyleChen, Jingbo, Yongjiang Wu, Yawang Sun, Xianwen Dong, Zili Wang, Zhu Zhang, Yanli Xiao, and Guozhong Dong. 2019. "Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells" Toxins 11, no. 5: 298. https://doi.org/10.3390/toxins11050298
APA StyleChen, J., Wu, Y., Sun, Y., Dong, X., Wang, Z., Zhang, Z., Xiao, Y., & Dong, G. (2019). Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells. Toxins, 11(5), 298. https://doi.org/10.3390/toxins11050298